

Abstract
Dihydropteroate synthase (DHPS) is a key enzyme in bacterial folate synthesis and the target of the sulfonamide class of antibacterials. Resistance and toxicities associated with sulfonamides have led to a decrease in their clinical use. Compounds that bind to the pterin binding site of DHPS, as opposed to the p-amino benzoic acid (pABA) binding site targeted by the sulfonamide agents, are anticipated to bypass sulfonamide resistance. To identify such inhibitors and map the pterin binding pocket, we have performed virtual screening, synthetic, and structural studies using Bacillus anthracis DHPS. Several compounds with inhibitory activity have been identified, and crystal structures have been determined that show how the compounds engage the pterin site. The structural studies identify the key binding elements and have been used to generate a structure-activity based pharmacophore map that will facilitate the development of the next generation of DHPS inhibitors which specifically target the pterin site.
INTRODUCTION
There is an urgent need for novel antibacterial agents for treating infections caused by resistant organisms. 1 The emergence of bacterial resistance is a pressing concern and has led to a significant decrease in the clinical utility of many antibacterial agents. One approach to this problem is to identify new classes of antibacterial agents with novel mechanisms of action, but this has proven to be extremely difficult in practice leading to high failure rates. 2 An alternative approach is to characterize the mechanism of resistance in traditional antibacterial drug targets and to design new agents that can bypass these mechanisms. This approach has proven to be more productive in recent years, for example, with the successful development of glycylcycline and ketolide antibiotics. 3, 4 There are several advantages to this approach. First, the target would be pre-validated by the prior clinical use of the earlier generation agents. Second, key biochemical information about the target and the mechanisms of resistance are typically already available to guide the design of the next generation agents. Finally, clinical experience with the earlier generation agents can also provide valuable information for the design and development of the next generation agents.
The sulfonamide class of antibacterial drugs has been used clinically since the 1930’s, and it was the first class of synthetic antibacterial agents to be used successfully. 5 Sulfonamides target the enzyme dihydropteroate synthase (DHPS) which catalyzes the addition of p-aminobenzoic acid (pABA) to dihydropterin pyrophosphate (DHPP) (Figure 1, panel a) to form pteroic acid as a key step in bacterial folate biosynthesis. The folate biosynthetic pathway has a key role in nucleic acid synthesis, and inhibition by the sulfonamides prevents bacterial growth and cell division. The absence of the pathway in higher organisms makes it a particularly attractive target for antibacterial drug design. Historically, the sulfonamides have been successfully used for a variety of Gram-positive and Gram-negative bacterial infections, and combinations with inhibitors of dihydrofolate reductase (DHFR) which catalyzes a subsequent step in folate synthesis have proven to be particularly effective. For example, co-trimoxazole is a commonly-used sulfamethoxazole-trimethoprim combination. However, drug resistance has emerged as an important factor that now severely limits the use of the sulfonamides. 6 For example, previously considered to be a first-line agent, co-trimoxazole has now been relegated to a 2nd or 3rd line option for a broad variety of infections. Resistance can be caused by altered drug uptake or efflux, but the predominant mechanism is mutation of the FolP gene that encodes DHPS. However, several emerging pathogens have shown universal susceptibility to co-trimoxazole, and this warrants further investigation of DHPS as a drug target. Notably, co-trimoxazole is a recommended agent for treating community-acquired MRSA and the recommended prophylactic agent for the prevention of Pneumocystis pneumonia (PCP) in adult HIV patients. 7, 8
Figure 1.
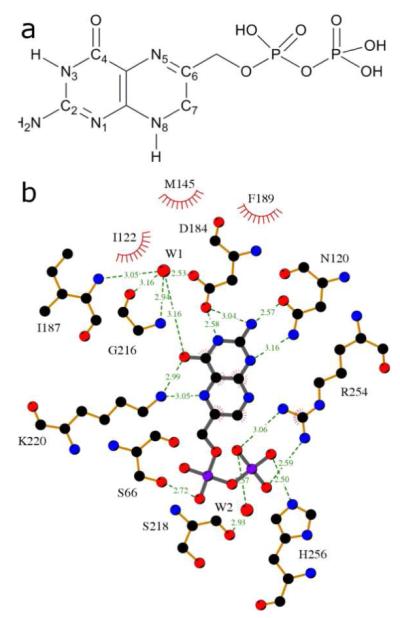
The pterin substrate binding pocket of DHPS. a) The structure of the natural substrate, DHPP, with ring numbering. b) LigPlot 18 view of the PtPP substrate analog bound in the BaDHPS active site with the key binding interactions displayed.
The first crystal structure of DHPS (from E. coli) was determined in 1997, fully 36 years after the last sulfonamide agent entered the market. Since that time, five additional crystal structures have been resolved, from S. aureus, M. tuberculosis, B. anthracis, T. thermophilus, and S. pneumoniae, and also one from the fungus S. cerevisiae. 9-15 These structures and associated mechanistic studies represent valuable new information with which to revisit DHPS as a therapeutic target. DHPS has a classic (β/α)8 TIM barrel structure in which the active site is located at the ‘C-terminal’ end of the barrel and contributed to by elements of the flexible loops that connect the β strands and α helices. The crystal structure of B. anthracis DHPS (BaDHPS) with a pteroate product analog in the active site is a key structure determined by our group because it reveals the locations of both the pterin and pABA binding sites. Although a sulfonamide has yet to be unequivocally visualized in complex with DHPS, these molecules appear to bind to the pABA sub-site and inhibit product formation and/or form “dead-end” products with pterin. Consistent with this notion, mutations that confer sulfonamide resistance all map to the pABA binding site locale. Although it has not been established how these mutations produce resistance, agents that inhibit the DHPS enzyme by binding to the distinct pterin sub-site are predicted to bypass these sulfonamide resistance sites. Another advantage of targeting the pterin site is revealed by Table 1, which reveals the high conservation of the key pterin-binding residues in several common pathogenic bacteria. This conservation reflects the severe constraints imposed on the pocket by its substrate specificity, compactness and structural integrity within the β-barrel. This contrasts with the pABA site that is comprised largely of flexible loop residues. Thus, inhibitors of the constrained pterin binding pocket would be predicted to have a broad spectrum of activity against both Gram-positive and Gram-negative bacteria, and also be less able to tolerate resistance mutations.
Table 1. DHPS pterin binding site residues.
| B. anthracis | Interaction Type | E. coli | S. aureus | M. tuberculosis | S. pneumoniae | P. aeruginosa | Y. pestis | F. tularensis |
|---|---|---|---|---|---|---|---|---|
| Asp101 | H-Acceptor | Asp96 | Asp84 | Asp86 | Asp91 | Asp82 | Asp96 | Asp254 |
| Asn120 | H-Acceptor | Asn115 | Asn103 | Asn105 | Asn110 | Asn101 | Asn115 | Asn276 |
| Ile122 | vDw | Ile117 | Gln105 | Val107 | Ile112 | Ile103 | Ile117 | Val278 |
| Ile143 | vDw | Cys137 | Val126 | Val128 | Val133 | Val123 | Cys137 | Ile299 |
| Met145 | Pi Electronic | Met139 | Met128 | Met130 | Met135 | Met125 | Met139 | His301 |
| Asp184 | H-Acceptor | Asp185 | Asp167 | Asp177 | Asp201 | Asp173 | Asp185 | Asp345 |
| Phe189 | Pi Electronic | Phe190 | Phe172 | Phe182 | Phe206 | Phe178 | Phe190 | Phe350 |
| Leu214 | vDw | Leu215 | Leu197 | Leu207 | Phe231 | Leu206 | Leu215 | Leu376 |
| Gly216 | no direct | Gly217 | Ala199 | Gly209 | Gly233 | Ser208 | Gly217 | Gly378 |
| Lys220 | H-donor | Lys221 | Lys203 | Lys213 | Lys237 | Lys212 | Lys221 | Lys382 |
| Arg254 | Pi Electronic | Arg255 | Arg239 | Arg253 | Arg282 | Arg246 | Arg255 | Arg417 |
Residues differing from B. anthracis target are colored in red.
In the mid-1980’s, a series of compounds with inhibitory activity against E. coli DHPS was disclosed by researchers at Burroughs-Wellcome, Inc. 16, 17 The compounds were pterin-like, had activity in the low micromolar range and were presumed to bind within the pterin pocket, although no structural information was reported. During our initial investigations into the structure of B. anthracis DHPS, we were able to re-synthesize and structurally analyze one of these compounds within the DHPS active site. 12 The compound, 2-amino-6-(methylamino)-5-nitropyrimidin-4(3H)-one (MANIC, but herein referred to as 1), engages the pterin pocket as predicted, and this structure has now led to the identification of similar inhibitory molecules that are presented in this report. The identification of these molecules has progressed in defined stages. The initial compounds were also derived from the Burroughs-Wellcome studies and include 2, a particularly potent inhibitor of B. anthracis DHPS that provided valuable design features for three stages of subsequent virtual screening (VS) studies. Our final cohort of 12 inhibitory molecules have been characterized by enzyme kinetics, X-ray crystallography, and antibacterial activity. This information was then combined in an initial structure-activity relationship (SAR) analysis which allowed us to develop a set of pharmacophore hypotheses with which to develop future pterin-based inhibitors.
RESULTS AND DISCUSSION
The DHPS Pterin-Binding Pocket
The pterin-binding pocket has been visualized in all the available crystal structures of DHPS and shown to be highly conserved (Table 1). 9-15 The pocket is located within the TIM barrel, directly below two flexible loops (loop1 and loop2) that are known to contain important elements of the active site, and is bounded by several key conserved residues that recognize the pterin-pyrophosphate substrate (Figure 2). In BaDHPS, Asp101, Asn120, Asp184, Lys220 and a structural water molecule provide a hydrogen bond donor/acceptor constellation that recognizes the pterin ring. Arg254 at the ‘base’ of the pocket provides a stacking platform for the pterin ring and, together with His265 and Asn27, also provides an anion-binding pocket for the β-phosphate of the substrate. A LigPlot view of this binding site, which is the target of our current studies, is shown (Figure 1, panel b). 18 DHPS catalyzes a strictly ordered reaction in which pterin-pyrophosphate is the lead substrate, and Lys220 has an important role to play in this mechanism. In the apo structure lacking any ligand, Lys220 is somewhat flexible, but its interaction with the pterin ring stretches out the side chain. Our structure with the product analog pteroic acid reveals that the now rigid side chain provides a binding platform for the second pABA substrate. In the absence of definitive structural data, it is generally assumed that loop1 and loop2 clamp down over the two substrates to complete the active site and promote catalysis.
Figure 2.

The B. anthracis DHPS enzyme shown with 2 bound. a) The full protein is shown with homology modeled loops colored. b) The pterin binding site with key binding residues and nearby loop residues. Hydrogen bonds are indicated by red dashes. Distances to nearby the loop residues in their modeled positions are shown.
Known Pterin-Based Inhibitors
The first compounds that were tested in these studies were selected from a series of DHPS-targeted inhibitors that were synthesized, analyzed and published in the 1980s but for which structural information was not generated (Figure 3, Panel a). 16, 17 These formally include 1 that we re-synthesized and structurally analyzed in an earlier study. 12 We demonstrated that this compound does engage the pterin pocket and interacts with five of the six pterin recognition elements. Asp101 is the exception because the electrostatic interaction is blocked by the N-methyl group. In the absence of the pyrophosphate moiety, the anion-binding pocket is occupied by a sulfate ion. This DHPS-inhibitor complex represents the starting point for our current studies. We selected three additional compounds, 2 – 4, for further analysis with B. anthracis DHPS based on a combination of potency, as judged by the published IC50 values against E. coli DHPS, chemical diversity, commercial availability, and ease of synthesis.
Figure 3.

DHPS hit compounds as evaluated by enzyme assay (>30% inhibition). Compounds are organized according to how they were identified. a) Compounds from previously reported studies. b) Compounds originating from preliminary screen. c) Compounds originating from large scale virtual screens. d) Compounds originating from the final knowledge-based search.
aCompounds for which co-crystal structures have been determined.
3, a close analog of 1, has a nitroso group substituted for a nitro group and an unsubstituted amine at the 6-position rather than the N-methyl substitution of 1. In B. anthracis, 3 shows improved inhibitory activity over 1 (Table 2). This improvement in activity can be rationalized by the crystal structure which reveals that the unsubstituted amine at the 6-position engages Asp101 in an electrostatic/hydrogen-bonding interaction that is blocked by the methyl substitution in 1 (not shown). 2 was shown to be an effective inhibitor of BaDHPS (IC50 value 19.8 μM), and the structure of the complex revealed the basis of this potency (Figure 4, Panels a-c). Although the interaction with Asp101 is blocked by the methyl substitution at the 6-position, the remaining pterin-binding residues are engaged and the carboxyl group provides an additional interaction with the anion-binding pocket which displaces the sulfate ion. In addition, there is a van der Waals interaction between the methyl group on the linker and the ring of Phe189. Finally, 4 resembles the DHPS product in which the pterin moiety is replaced by 3 and the pABA moiety is attached via an extended linker. The molecule has an IC50 of 19.3 μM in BaDHPS (Table 2). In the structure of the complex (Figure 4, panels d-f), the pterin-like half engages the pterin pocket in a similar fashion to 3 with two differences; the interaction with Asp101 is blocked by the linker and the interaction with the side chain amine of Lys220 is via the nitrogen atom of the nitroso group rather than the oxygen atom seen with 3. The latter difference is due to a slight repositioning of the pterin-like moiety in 4 that brings it closer to Lys220 to minimize a steric clash between the linker and Asp101. The pABA moiety adopts two conformations in the two molecules of the asymmetric unit. In molecule A, the moiety points down to interact with Pro69 in a partially ordered loop2 (shown in Figure 4, panels d-f), and in molecule B, it points up to interact with Phe189 (not shown). Both orientations appear to prevent sulfate binding to the anion-binding pocket.
Table 2. DHPS hit compounds with docking scores and activities.a.
| Compound | Surflex Dock Score |
B. anthracis % inhibition |
B. anthracis IC50 (μM) |
|---|---|---|---|
| Control b | 60% (250 μM) | 58.4 | |
| 1 | 6.59 | 53% (500 μM) | N/D |
| 2 | 9.48 | 97% (500 μM) | 19.8 |
| 3 | 5.12 | 81% (250μM) | 8.0 |
| 4 | 7.47 | 79% (500 μM) | 19.3 |
| 5 | 5.78 | 0% (250 μM) | N/D |
| 6 | 6.59 | 93% (250 μM) | 32.4 |
| 7 | 5.71 | 80% (250 μM) | 108.9 |
| 8 | 6.79 | 33% (500 μM) | N/D |
| 9 | 6.75 | 37% (500 μM) | N/D |
| 10 | 6.53 | 32% (500 μM) | >500 |
| 11 | 6.51 | 44% (500 μM) | N/D |
| 12 | 6.38 | 31% (500 μM) | N/D |
| 13 | 6.36 | 32% (500 μM) | N/D |
| 14 | 7.80 | 62% (500 μM) | N/D c |
| 15 | 7.22 | 35% (500 μM) | N/D |
| 16 | 6.24 | 76% (500 μM) | 86.7 |
| 17 | 7.11 | 54% (500 μM) | 215.0 |
| 18 | 5.97 | 67% (500 μM) | 212.6 |
| 19 | 8.69 | 100% (500 μM) | 25.9 |
| 20 | 5.76 | 37% (500 μM) | N/D |
| 21 | 7.71 | 76% (500 μM) | 87.1 |
| 22 | 4.91 | 70% (500 μM) | 269.3 |
Yellow Highlight indicates compounds for which crystal structures have been determined.
6-hydroxymethyl-7,8-dihydropterin (HMDP), was used as the control compound.
IC50 determination of 14 was not possible due to a limited availability of the compound.
Figure 4.

DHPS pterin site binding interactions of compounds 2 [a-c] and 4 [d-f]. a/d) Fo-Fc electron density maps contoured at 3σ using Pymol52. Maps were calculated from models refined after removing the compounds to avoid bias. b/e) Details of the interaction using MolScript53. c/f) LigPlot18 diagram of binding interactions (arrows near benzoic group in panel f indicate positional uncertainty of this group).
Preliminary Virtual Screening Results
The first stage of virtual screening utilized a simple 2D pharmacophore search with imposed distance constraints between specified donor and acceptor groups followed by flexible docking of the hit compounds of the Maybridge and NCI libraries (see Supporting Figures S2 and S3). From the hits identified we obtained three co-crystal structures (Compounds 5, 6, 7; Figure 3, Panel b). Compounds 6 and 7 were potent inhibitors of DHPS. 5, which has a pterin-like A-ring did not show inhibition in our DHPS assay. It is unclear why 5 does not inhibit the enzyme in our assay even though it was shown to bind in our co-crystal trials. Potent inhibitor 6 is similar to pterin but has a methyl substitution at position 8 that prevents interaction with Asp101, and a carboxyl group at position 6 that forms a novel salt bridge interaction with the terminal amine of Lys220 (Figure 5, Panels a-c). 7 is structurally very similar to 3, with a nitro in place of a nitroso group at the 5 position, and the co-crystal structure shows interactions that are virtually identical (Figure 5, Panels d-f).
Figure 5.

DHPS pterin site binding interactions of compounds 6 [a-c] and 7 [d-f]. a/d) Fo-Fc electron density maps contoured at 3σ using Pymol 52. Maps were calculated from models refined after removing the compounds to avoid bias. b/e) Details of the interaction using MolScript 53. c/f) LigPlot18 diagram of binding interactions.
Large scale Virtual Screening Results
To expand on the previous studies, a high-throughput virtual screen of the ZINC databases was performed. This used the crystal structure of 2 bound within the pterin pocket receptor for the docking model. We chose this structure because 2 is a potent inhibitor that accesses many of the key pterin-pyrophosphate binding residues in the pocket, and the crystal structure is well determined. Loops 1 and 2 in our BaDHPS structure are either disordered or involved in crystal packing interactions, and although the loops are not believed to play a major role in binding the pterin substrate, we built an homology model of their conformations using the E. coli and M. tuberculosis structures (Figure 2) 9, 11 and performed a 100 ps molecular dynamics simulation to refine their positions.
The virtual screening was performed using the UNITY and Surflex programs available in the Sybyl 7.3 molecular modeling suite of Tripos, Inc. 19-22 We first prepared the UNITY databases for screening from the ZINC libraries that, at the time, contained nearly 5 million compounds and included protonation variants and tautomers for the medium pH range of 5.75 to 8.25. We then prepared the pharmacophore filter from the 2 complex structure. The filter contained three elements. The first was a surface volume constraint created by including all residues surrounding the pterin pocket within 8 Å of the bound 2 with a van der Waals tolerance of 1 Å (Supporting Figure 1a). Part of this volume included residues from the modeled loops 1 and 2 which represented their primary contribution to the overall pharmacophore filter. The second element was the ligand-based hydrogen-bonding constellation of 2 (Supporting Figure 1b). These parameters were derived from test runs to derive a hit-to-failure ratio that generated a reasonable number of candidate compounds for the next stage of molecular docking. The final element of the screen was a molecular weight cutoff of 350 D and a maximum of five rotatable bonds. We applied this filter to generate lower molecular weight ‘fragment-like’ molecules that have been shown to represent better lead compounds, with more scope for elaboration and optimization. Although the lower molecular weight and complexity of the fragment compounds generally results in lower binding affinity (often high micromolar to low millimolar), they are often on par with or exceed drug like compounds in terms of ligand efficiency (binding affinity normalized by molecular weight or heavy atom count). 23-25 A further benefit of these selection criteria is that it increases the likelihood for selecting compounds with reasonable water solubility, as poor solubility was noted for some of the analogs identified in the first compound series.
5,093 compounds from the ZINC screening libraries matched the pharmacophore requirements, and when the UNITY hit lists were merged, the total number of unique compounds was 3104 indicating some redundancy in the ZINC databases. All 3104 compounds were then docked and scored by the Surflex docking tool within the Sybyl 7.3 molecular modeling suite. 21, 22 We previously reported a docking validation study of the DHPS pterin site which concluded that Surflex-Dock performs well in this particular active site. 26 The top 2% of the ranked compounds (62 compounds) were eventually selected for testing in the DHPS enzyme assay. Of this number, 17 compounds were no longer available from suppliers and the remaining 45 compounds were procured and tested. The compounds were tested at 500 μM concentration (250 μM if very poorly soluble) and a percentage inhibition was obtained. Eight compounds showing greater than 30% inhibition, an acceptable standard when dealing with fragment-like compounds, and suitable solubility were taken into crystallography trials (Compounds 8–15, Figure 3, Panel c). 27, 28
Scaffold Search Results
To maximize the return of our studies, a simple and rapid 2D scaffold search of all commercially available compounds in the CAS registry was performed using the key pharmacophoric elements discovered in our previous studies. The key elements of the scaffold search are shown in Figure 6. On the A-ring, the C2 nitrogen and the nitrogens at the 1 and 3 positions were required to be unsubstituted, and a carbonyl or the tautomeric phenol was required at the 4 position. The B-ring allowed more flexibility in the search; double or single bonds were permitted at the 5, 6 and 7, 8 positions and the 6 position substituent had no restrictions imposed. Finally, the substituent at the 8 position was restricted to an N-methyl group or unsubstituted nitrogen.
Figure 6.

Key pharmacophore elements and scaffold search criteria for pterin-like compounds that access the pterin pocket of DHPS.
43 compounds were identified using this scaffold search, of which 19 were marked as interesting and selected for procurement and testing. However, only 10 of these were commercially available for immediate testing. Seven compounds had activities above our 30% threshold and were advanced into crystallography trials (Compounds 16–22, Figure 3, Panel d), and four of these generated co-crystal structures. All four compounds have the same A-ring structure seen in the natural pterin substrate plus a nitrogen atom at the 5 position, and they engage the pocket in the expected fashion. 16 and 17 both have methyl substitutions at the N8 position which prevent interaction with Asp101, but this interaction is possible in 18 and 19 where the N8 is unsubstituted. 17 and 19 each have a side chain at the 6 position of the B-ring and both interact with the active site locale. In 17, the OH group interacts with a sulfate in the anion binding pocket. 19 is very similar to the product analog pteroic acid that we have already visualized in the active site, and the binding is virtually identical (Figure 7, Panels a-c) .12 The side chain engages the acyl chain of Lys220, and the terminal carboxyl group interacts with the OH of Ser221. As shown in Table 2, 16, 17 and 18 have relatively weak and equivalent potencies as inhibitors, but 19 is exceptional which probably reflects its close similarity to the product.
Figure 7.

DHPS pterin site binding interactions of compound 19. a) Fo-Fc electron density map contoured at 3σ using Pymol 52. Map was calculated from model refined after removing the compound to avoid bias. b) Details of the interaction using MolScript 53. c) LigPlot 18 diagram of binding interactions.
DHPS Binding Order Studies
Previous kinetic analyses of S. pneumoniae DHPS have shown that the enzyme catalyzes a strictly ordered reaction in which DHPP is the lead substrate followed by pABA.29 Our inhibitors all engage the pterin-binding pocket. Thus it is probable that pterin based inhibitors can bind in the absence of other ligands. However, in this study it was noticed that a sulfate ion in the anion-binding pocket is present in all our DHPS-inhibitor complexes, apart from compound 2 where the sulfate is displaced by the anionic carboxylate group. This raises the question if the binding of this inhibitor class may require that the anion binding pocket also be occupied. To investigate this possibility, we used an isothermal titration calorimetry (ITC) approach to confirm binding order and study the requirements for inhibitor binding. First it was verified that the B. anthracis DHPS catalyzes an ordered reaction. Co-incubation experiments show that pABA does not bind to the enzyme in the absence of pyrophosphate, which was used to mimic the presence of DHPP (Figure 8A), while pABA does bind tightly to DHPS that has been pre-incubated with pyrophosphate (Figure 8B). Thus, the B. anthracis DHPS catalytic mechanism is indeed ordered. Then the requirement for phosphate or sulfate anions to be present for the binding of pterin based inhibitors was examined using representative inhibitor 6 for which a sulfate had been clearly resolved in its complex structure (Figure 5B). Sulfate and phosphate ions were carefully removed from the enzyme and inhibitor samples prior to addition of the inhibitor to DHPS sample in the ITC cell. The ITC still showed a positive isotherm (Figure 8C) clearly demonstrating that occupancy of the anion binding pocket is not required for binding of pterin targeted inhibitors.
Figure 8.

Isothermal titration calorimetry. a) Titration of 500 μM pABA into a protein solution of 20 μM B. anthracis DHPS. No heats of binding could be detected. b) Titration of 500 μM pABA into a 20 μM protein solution containing 10 mM sodium pyrophosphate. Dissociation constant, Kd, is 5.62 × 10−6 M. c) Titration of 500 μM compound 6 into a 20 μM protein solution. Kd is 5.26 × 10−6 M.
Developed Pharmacophore Model
Using the activity and structural data obtained from pterin pocket inhibitors identified in our studies, we have derived an initial SAR (or pharmacophore) map based on the pterin two-ring structure of the natural substrate (Figure 6). The A ring, particularly the N1, C2, N3 and C4 positions that access the conserved residues deep in the pterin pocket, is least tolerant to modification. A number of compounds with A ring substitutions were tested, but only seven showed sufficient activity for structural studies. 8, 9, 13, 14 and 15 failed to produce co-structures, and 11 did not engage the pterin pocket and instead formed a stacking/covalent interaction at a remote surface location. We conclude that all six compounds have little or no affinity for the pterin pocket, which is consistent with the observed low activity of these compounds in our assay (Table 2). Although 10 also has low activity and shares minimal structural features with pterin, we were successful in visualizing it within the pterin pocket. Its interactions with the pocket residues are quite unique and it appears to represent a novel low molecular weight scaffold that we intend to pursue.
In contrast, the B ring that binds closer to the opening of the pterin pocket is far more tolerant of modifications and provides more opportunities for optimizing the potency of pterin-based inhibitors. Compounds with both six- and five-membered B rings and open B rings were visualized in our structural studies. 5 was the only five-membered ring compound identified in our screens, and this showed little or no inhibition of the enzyme. We also tested a 5 homolog in which the SH group is replaced with an OH group, and this was also shown to have minimal activity (data not shown). We therefore concentrated on the six-membered and open B ring compounds and identified three features that improve potency. First, an acceptor at the 5 position is required to form a second hydrogen-bonding interaction with conserved Lys220. An Sp2 nitrogen performs this task in the natural pterin substrate, but carbonyl, nitro or nitroso groups appear to be superior based on our structures and assay data. The second favorable feature is a carboxyl group attached to the C6 position. This feature is present in 2 and 6 which are both potent inhibitors. In 6, the carboxyl group is directly attached to C6 and forms a salt bridge with Lys220, and a sulfate ion is present in the anion-binding pocket. However, in 2 the carboxyl group is attached via a short linker which allows it to engage Arg254 in a salt bridge/hydrogen bonding interaction, and it displaces the sulfate from the anion-binding pocket. In addition, the methyl group on the linker makes van der Waal interactions with the conserved Phe189. The potencies of 2 and 6 are equivalent, and it is unclear which of the two carboxyl interactions is superior. However, the 2 co-structure suggests that extension of the linker by one or two carbon atoms would enable the carboxyl to more fully engage the anion-binding pocket. The final favorable feature is a hydrogen-bonding interaction between the conserved Asp101 and a donor at the N8 position. This interaction is possible in 3, 5, 7, 18 and 19, as well as the natural pterin substrate, but not possible in 2, 6, 16, 17 and 4 (the latter for steric reasons). We have direct evidence that this feature increases potency; 1 and 7 are identical compounds apart from a methyl substitution at the N8 position in 1, and 7 is the more potent compound. In addition, 19 is our most potent compound but introducing a double bond at N8 which removes the donor hydrogen, as in 21, significantly reduces potency (Table 2).
Crystal versus Docked Structures
The 12 crystal structures presented here, together with the compound 1 structure reported earlier 12 provide an alternative test of the docking procedure, namely, calculating the heavy atom rmsd values between the crystal structures and their corresponding docked poses. As can be seen in Table 3, the docked poses generally correspond very closely to the crystal structure positions, and we attribute this success to the prior validation that was performed to select the optimal docking software, in this case, SURFLEX-Dock. 26 In these docking validation studies, we used an rmsd value of 1.5 Å as the cutoff for success and, applying the same criteria here, it can be seen that eight of the 11 docking poses accurately predict the crystal structures. Can we rationalize the three docking failures? The reason why 3 failed is not clear, especially because the predicted pose of the similar 1 closely matches the crystal structure. One possible explanation is that the position of the compound in the docked pose is influenced by an electrostatic interaction between the 6-amino group and Asp61 which results in the loss of the electrostatic interaction with Lys220. 3 is also the smallest hit compound studied and it can sterically adopt poses that are not accessible to the larger compounds. Regarding 4 and 19, the docked positions of the pterin ring substructure were essentially correct (see numbers in parentheses in Table 3), and the deviations occurred in the pABA-like moieties that are predicted to engage the flexible loops with associated docking uncertainty.
Table 3. Calculated RMSD values for docked and crystal structure hit compounds.
| Compound | RMSD Value (Å) |
|---|---|
| 1 | 1.100 |
| 2 | 0.906 |
| 3 | 2.134 |
| 4 | 4.73 (1.148)a |
| 5 | 1.003 |
| 6 | 0.837 |
| 7 | 0.585 |
| 10 | 0.651 |
| 16 | 0.496 |
| 17 | 0.658 |
| 18 | 1.412 |
| 19 | 5.121 (0.509)a |
Values in parentheses reflect calculated rmsd values for only the pterin ring substructure of these two compounds
Limitations of pterin-based inhibitors
With one exception, all of the hit compounds are similar to the natural pterin substrate. Considering the high specificity and conserved nature of the pterin-binding pocket, this selectivity is not surprising, and it has been noted that the pocket does not easily accommodate compounds with alternate scaffolds. 16 However, from a drug discovery perspective, there are two drawbacks to pterin-like compounds. First, pterin-like compounds tend to be poorly soluble due to their planar character which results in high crystal lattice energy .30 This has led to some degree of experimental difficulty and, in some cases, necessitated activity testing at a lower concentration than our standard concentration (250 μM rather than 500 μM). It is well documented that poor solubility has negative ramifications in terms of drug discovery and clinical candidacy. 30, 31 We plan to address this problem by adding anionic functional groups that can interact with the anion binding pocket, and the addition of a carboxylate group at the 6 position is a first step in this process. Second, we would prefer our hit compounds to have more diversity in chemical structure and to include novel scaffolds. It is now recognized that ‘scaffold hopping’ is an important part of the drug discovery process .32 The use of a ligand-based filter may explain why so many pterin-like compounds were detected by virtual screening, and a receptor-based filter is planned for future studies in an attempt to identify alternate scaffolds.
One compound with a novel scaffold, 10, did result from the virtual screening studies. Although non pterin-like, the compound is able to make many of the key binding interactions defined in the pharmacophore model. Figure 9 (Panels a-c) shows how 10 accesses the pocket, and it is satisfying in terms of our general approach that docking successfully recapitulated the crystal structure. The N1 nitrogen forms an H-bonding interaction with Lys220 and a key nitrogen triad interacts with the conserved and crucial Asn120 and Asp184. Finally, due to its planar nature, 10 is also able to engage Arg254 in the characteristic stacking interaction seen with all of the pterin-like hit compounds. The positions of the carboxylate group of Asp101 and the N6 ring nitrogen of 10 indicate the presence of an electrostatic interaction at this position which facilitates binding. Microspecies and pKa calculations performed with 10 reveal that the nitrogen is weakly basic (pKa 6.13) and has a predicted microspecies population of only 2.62% at pH 7.4. 33 However, the charge on the adjacent Asp101 is likely to raise the pKa and the bound species of 10 may actually carry a significant positive charge centered at the N6 position. One key binding feature that is absent in 10 is a negatively charged group that can engage the anionic pocket. 10 has a molecular weight of 150.1 and is a bona fide ‘fragment’ molecule with many opportunities for elaboration. Future studies are planned with 10 analogues to explore the SAR of substitutions or modifications at the 7 position to take advantage of these opportunities to improve binding affinity.
Figure 9.

DHPS pterin site binding interactions of compound 10. a) Fo-Fc electron density map contoured at 3σ using Pymol 52. Map was calculated from model refined after removing the compound to avoid bias. b) Details of the interaction using MolScript 53. c) LigPlot 18 diagram of binding interactions.
CONCLUSIONS
In these studies, we have thoroughly characterized the pterin binding pocket of DHPS and generated a detailed structure-activity based pharmacophore map that will facilitate the development of novel DHPS inhibitors that specifically target the pterin site. We have identified the optimal binding features of the pterin scaffold and will apply these insights to the production of pterin-based libraries for further screening efforts. We have also identified a non-pterin scaffold that engages the pocket and we will create a second library of compounds to include in our future screening efforts based upon this scaffold. Our current compounds and future libraries all target key active site residues and, unlike the sulfonamide drugs, avoid the flexible loops that typically accrue resistance mutations.
METHODS
Compound procurement
The majority of compounds used in this study were procured from the following commercial vendors and compound repositories: 3, Toronto Research Chemicals, Inc.; 5, Ryan Scientific, Inc.; 6, 7, 10, 12, 16, 17, 19, 21, 22, National Cancer Institute’s Drug Testing Program34; 8, 11, Specs, Inc.; 9, 13, 18, 20, Sigma Aldrich, Inc.; 14, ChemDiv, Inc.; 15, ChemBridge, Inc. The remaining compounds that were not commercially available were synthesized according to the following published procedures: 1, 12, 16; 2, 35; 4. 17 Details of the preparation of 2 & 4 are provided in the supplementary data section (Scheme S1 and synthesis methods).
Compound Purity Testing
Proof of purity for all compounds purchased or synthesized was determined by analytical reverse -HPLC was conducted on a Shimadzu HPLC system using a Phenomenex Luna C18 column (100Å, 3 μm, 4.6 × 50 mm), flow rate 1.0 mL/min and a gradient of solvent A (water with 0.1% TFA) and solvent B (acetonitrile): 0–2.00 min 100% A; 2.00–8.00 min 0–100% B (linear gradient) and UV detection at 254 nm / 215 nm. Compounds were determined to be of ≥95% purity.
Enzyme Assay
Enzyme Preparation
The E. coli HPPK-GST fusion gene was provided by Dr. Honggao Yan and transformed into competent BL21 (DE3) E. coli cells (Novagen Cat. N69450). 36 The initial culture was grown overnight at 26 °C in 100 mL LB containing 100 mg/L ampicillin, and 13 mL of this culture was used to inoculate 1L of the same medium. This was grown at 37 °C until the OD reached 0.8, at which point isopropyl-B-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.4 mM. Cells were then grown overnight in IPTG at 28 °C and harvested by centrifugation at 4 °C, 4000 rpm for 15 minutes. The cell pellet was suspended in 100 mL PBS with 10 mg lysozyme and lysed by a microfluidizer (Microfluidics M-110), and the cell debris was cleared by centrifugation at 30,000 rpm for 30 min. The supernatant was filtered through a 0.45 mm syringe filter and applied to a 5 mL GSTrap FF column (Amersham Biosciences), and the column was washed with 70 mL PBS binding buffer and then step-eluted using 50 mM Tris-HCl, 10 mM reduced glutathione, pH 8.0. The eluted protein was essentially pure and adjusted to a concentration of 5 mg/mL, dialysed in 100 mM NaCl, 20 mM Tris pH 8.0, and finally stored at −80 °C. The B. anthracis DHPS enzyme was expressed in E. coli and purified as previously described 12. Protein concentrations were measured by the Bradford protein assay (Bio-Rad Laboratories) using bovine serum albumin as standard.
DHPS Substrates
6-Hydroxymethyl-7,8-dihydropterin hydrochloride was purchased from Schircks Laboratories, Switzerland. [ring--14C]para-aminobenzoic acid (14C pABA, 55 mCi/mmol) was obtained from Moravek Biochemicals, USA. 6-Hydroxymethyl-7,8-dihydropterin diphosphate is unstable and was prepared enzymatically using HPPK-GST 37, 38 6-Hydroxymethyl-7,8-dihydropterin was incubated at 37 °C for 30 minutes in 5 mM ATP, 10 mM magnesium chloride, 3% dimethyl sulfoxide, 20 μg/mL GST-HPPK, and 50 mM HEPES pH 7.6. The reaction solution was passed through the GST·Bind™ Resin (Novagen) to remove the GST-HPPK enzyme and stored in aliquots at −80 °C.
Enzyme Assay
DHPS activity was measured in a 30 μL reaction containing 5 μM 14C pABA, 10 μM 6-Hydroxymethyl-7,8-dihydropterin diphosphate, 10 mM magnesium chloride, 2% DMSO, 50 mM HEPES pH 7.6, and 10 ng DHPS. 29, 39 After 30 minutes incubation at 37 °C, the reactions were stopped by addition of 1 μL of 50% acetic acid in an ice bath. The labeled product of the reaction, 14C dihydropteroate, was separated from 14C pABA by thin layer chromatography. 15 μL aliquots of the reaction mixture were spotted onto Polygram TLC plates (CEL 300 PEI) purchased from Macherey-Nagel and developed with ascending chromatography in 100 mM phosphate buffer pH 7.0. The plates were scanned using a Typhoon (GE Healthcare) and analyzed with ImageQuant TL. Inhibitor compounds were dissolved in DMSO, and inhibition was tested at 500 μM or 250 μM depending on solubility. The final concentration of DMSO in the reaction mixture was 2%. To determine the 50% inhibitory concentration (IC50) values, DHPS activities were measured in the presence of various concentrations of the compounds using the conditions described above but with 5ng DHPS. Data were analyzed by using Prism GraphPad software.40
Crystallography
The structures of all DHPS-inhibitor complexes were obtained by soaking the small molecules into the P6222 B. anthracis DHPS crystals described earlier. 12 The small molecules were first dissolved in crystal mother liquor (1.3 M Li2SO4, 0.1 M Bis-Tris propane, pH 9.0) to the saturation allowed by their typically limited solubility (approximately 1mM), and the crystals were transferred into these solutions for 24–48 hour soaking periods. Crystals were then cryoprotected by brief immersion in 50% Paratone-N and 50% mineral oil, and flash frozen in liquid nitrogen. All data were collected at the SER-CAT beamlines 22-ID and 22-BM at the Advanced Photon Source and processed using HKL2000.41 Structures were directly refined using REFMAC 42, 43 and the deposited coordinates of B. anthracis DHPS bound with Pteroic acid (PDB code 1TX0). Model building was performed using the COOT program.44 Relevant data collection and refinement statistics are presented in Supplementary Table S1 and Table S2.
Isothermal Titration Calorimetry
The purified B. anthracis DHPS protein was dialyzed against 50 mM HEPES, 5mM MgCl2, pH 7.6. ITC titrations were performed in 40 mM HEPES, 4 mM MgCl2 at pH 7.6 and 25 °C. 5% DMSO was added to the ITC buffer for the titration experiment of Compound 6. Nineteen injections of 2 μL each (except 0.5 μL for the first injection) of 500 μM ligand solution were added to 200 μL of 20 μM protein solution. ITC titrations were performed on an ITC200 Microcalorimeter (MicroCal), and data was analyzed using MicroCal Origin 7.0 software using a one-site binding model.
Computational and Experimental Methods
The first stage of preliminary virtual screening utilized a simple 2D pharmacophore search with imposed distance constraints between specified donor and acceptor groups followed by flexible docking of the hit compounds (Supplementary Figures S2 and S3). The UNITY program implemented in the Sybyl v6.9 molecular modeling package was used to perform the 2D search and the FlexX implementation in Sybyl v6.9 was used to perform the docking. 19, 20, 45 The Maybridge and NCI databases supplied with the Sybyl package were screened. Compounds were scored using F-score, PMF-score, and ChemScore and selected on the basis of their consensus score as well as visual inspection and comparison with the DHPP substrate.
For the second stage of virtual screening (large scale screening), the screening compounds used were downloaded in sdf format from the vendors subsets of ZINC database (version 6). 46 The sdf files were converted to UNITY databases for pharmacophore screening using the UNITY tool available with the Sybyl 7.3 molecular modeling suite of Tripos, Inc. 19, 20, 45 2D and Macro fingerprints were created using default settings. Concord was used to generate 3D coordinates, when necessary. 47 Default values were accepted for all other UNITY database preparation settings. The UNITY pharmacophore filter consisted of 1 donor and 4 acceptor positions based upon the H-bonding patterns observed in the crystal structure of 2 (Figure 4) as discussed in the results and discussion section. A spatial tolerance of 0.3 Å was used for each macro and 2 partial match constraints were applied. The UNITY databases were screened using a 3D Flex search with modified “Rule of Three” search options as discussed in the results and discussion section.48 The flex ring search option was also enabled. All other settings retained their default values.
Hitlists from the pharmacophore filtering were merged to eliminate duplicate compounds and then the converted to a multi-mol2 file for docking. Charges were loaded to the compounds using the Gasteiger-Huckel method.49 Surflex docking utilized the multi-mol2 file and a protomol generated using a threshold of 0.50 and bloat of zero (default values).22 These settings are the same as those used in our previously reported docking validation study.26 An active site water was retained for all docking runs. The ring flexibility function was enabled; all other docking settings retained their default values. Compounds docked with Surflex were scored with the native Surflex scoring function; the Cscore option was disabled. The top 2% of the Surflex scored compounds were selected for procurement and testing in the enzyme assay described above.
The final stage of compound selection was used to maximize our emerging structure activity relationship results obtained in the previous stages. This screen was performed using simple 2D scaffold similarity search using SciFinder® against all commercially available compounds in the Chemical Abstracts Services (CAS) registry.50, 51 The scaffold search criteria are shown in Figure 6. The search criteria were identified by visual SAR analysis of compounds 1 through 15, which were identified in the earlier stages of screening or were known pterin site binders from previous studies. Key binding features were identified in these earlier compounds using their measured % inhibition of enzyme activity. Compounds matching the scaffold search constraints were procured and tested as described above.
Supplementary Material
ACKNOWLEDGEMENTS
Funding for this research was provided by National Institutes of Health grants AI060953 (to SWW and REL) and AI070721 (to SWW and REL), Cancer Center core grant CA21765, and the American Lebanese Syrian Associated Charities (ALSAC). SER-CAT supporting institutions may be found at www.ser.anl.gov/new/index.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. We acknowledge the technical assistance of D. Ball and J. Scarborough at the University of Tennessee, Health Science Center and of M. Frank in the Department of Infectious Diseases at St. Jude Children’s Research Hospital. Support of this research by the American Foundation for Pharmaceutical Education is also gratefully acknowledged.
Accession Codes. The atomic coordinates of the B. anthracis DHPS co-crystal complex structures have been made publicly available through the Protein Data Bank (www.rcsb.org/pdb) with the following PDB id’s: compound 2, 3H21; compound 3, 3H22; compound 4, 3H23; compound 5, 3H24; compound 6, 3H26; compound 7, 3H2A; compound 10, 3H2C; compound 11, 3H2E; compound 16, 3H2F; compound 17, 3H2M; compound 18, 3H2N; compound 19, 3H2O.
Abbreviations
- DHPS
dihydropteroate synthase
- pABA
para-aminobenzoic acid
- DHPP
dihydropterin pyrophosphate
- DHFR
dihydrofolate reductase
- PCP
Pneumocystis carinii pneumonia
- MANIC
2-amino-6-(methylamino)-5-nitropyrimidin-4(3H)-one
- CAS
Chemical Abstracts Service
- HMDP
6-hydroxymethyl-7,8-dihydropterin
Footnotes
Supporting Information Available: Pharmacophore screening methods, organic synthesis scheme and methods, statistics of x-ray crystallography data collection and refinement. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 3.Sum PE, Lee VJ, Testa RT, Hlavka JJ, Ellestad GA, Bloom JD, Gluzman Y, Tally FP. Glycylcyclines. 1 A new generation of potent antibacterial agents through modification of 9-aminotetracyclines. J Med Chem. 1994;37:184–188. doi: 10.1021/jm00027a023. [DOI] [PubMed] [Google Scholar]
- 4.Bryskier A. Ketolides-telithromycin, an example of a new class of antibacterial agents. Clin Microbiol Infect. 2000;6:661–669. doi: 10.1046/j.1469-0691.2000.00185.x. [DOI] [PubMed] [Google Scholar]
- 5.Domagk G. Ein Beitrag zur Chemotherapie der bakteriellen Infektionen. Dtsch Med Wochenschr. 1935;61:250–253. [Google Scholar]
- 6.Sköld O. Sulfonamide resistance: mechanisms and trends. Drug Resist Updat. 2000;3:155–160. doi: 10.1054/drup.2000.0146. [DOI] [PubMed] [Google Scholar]
- 7.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan JE, Benson C, Holmes KK, Brooks JT, Pau A, Masur H. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents--2009. Recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR. 2009;58(ER):1–198. [PubMed] [Google Scholar]
- 9.Achari A, Somers DO, Champness JN, Bryant PK, Rosemond J, Stammers DK. Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nat Struct Biol. 1997;4:490–497. doi: 10.1038/nsb0697-490. [DOI] [PubMed] [Google Scholar]
- 10.Hampele IC, D’Arcy A, Dale GE, Kostrewa D, Nielsen J, Oefner C, Page MG, Schonfeld HJ, Stuber D, Then RL. Structure and function of the dihydropteroate synthase from Staphylococcus aureus. J Mol Biol. 1997;268:21–30. doi: 10.1006/jmbi.1997.0944. [DOI] [PubMed] [Google Scholar]
- 11.Baca AM, Sirawaraporn R, Turley S, Sirawaraporn W, Hol WG. Crystal structure of Mycobacterium tuberculosis 7,8-dihydropteroate synthase in complex with pterin monophosphate: new insight into the enzymatic mechanism and sulfa-drug action. J Mol Biol. 2000;302:1193–1212. doi: 10.1006/jmbi.2000.4094. [DOI] [PubMed] [Google Scholar]
- 12.Babaoglu K, Qi J, Lee RE, White SW. Crystal structure of 7,8-dihydropteroate synthase from Bacillus anthracis: mechanism and novel inhibitor design. Structure. 2004;12:1705–1717. doi: 10.1016/j.str.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence MC, Iliades P, Fernley RT, Berglez J, Pilling PA, Macreadie IG. The three-dimensional structure of the bifunctional 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase/dihydropteroate synthase of Saccharomyces cerevisiae. J Mol Biol. 2005;348:655–670. doi: 10.1016/j.jmb.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 14.Bagautdinov B, Kunishima N. Crystal Structure of Dihydropteroate Synthase (FolP) from Thermus thermophilus HB8. RIKEN Structural Genomics/Proteomics Initiative (RSGI); 2006. [Google Scholar]
- 15.Levy C, Minnis D, Derrick JP. Dihydropteroate synthase from Streptococcus pneumoniae: structure, ligand recognition and mechanism of sulfonamide resistance. Biochem J. 2008 doi: 10.1042/BJ20071598. [DOI] [PubMed] [Google Scholar]
- 16.Lever OW, Jr., Bell LN, McGuire HM, Ferone R. Monocyclic pteridine analogues. Inhibition of Escherichia coli dihydropteroate synthase by 6-amino-5-nitrosoisocytosines. J Med Chem. 1985;28:1870–1874. doi: 10.1021/jm00150a019. [DOI] [PubMed] [Google Scholar]
- 17.Lever OW, Jr., Bell LN, Hyman C, McGuire HM, Ferone R. Inhibitors of dihydropteroate synthase: substituent effects in the side-chain aromatic ring of 6-[[3-(aryloxy)propyl]amino]-5-nitrosoisocytosines and synthesis and inhibitory potency of bridged 5-nitrosoisocytosine-p-aminobenzoic acid analogues. J Med Chem. 1986;29:665–670. doi: 10.1021/jm00155a014. [DOI] [PubMed] [Google Scholar]
- 18.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 19.Martin YC. 3D database searching in drug design. J Med Chem. 1992;35:2145–2154. doi: 10.1021/jm00090a001. [DOI] [PubMed] [Google Scholar]
- 20.Hurst T. Flexible 3D searching: The directed tweak technique. J Chem Inf Comput Sci. 1994;34:190–196. [Google Scholar]
- 21.Jain AN. Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J Comput Aided Mol Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 22.Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 23.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 24.Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nat Rev Drug Discov. 2004;3:660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 25.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 26.Hevener KE, Zhao W, Ball DM, Babaoglu K, Qi J, White SW, Lee RE. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J Chem Inf Model. 2009;49:444–460. doi: 10.1021/ci800293n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murray CW, Callaghan O, Chessari G, Cleasby A, Congreve M, Frederickson M, Hartshorn MJ, McMenamin R, Patel S, Wallis N. Application of fragment screening by X-ray crystallography to beta-secretase. J Med Chem. 2007;50:1116–1123. doi: 10.1021/jm0611962. [DOI] [PubMed] [Google Scholar]
- 28.Edwards PD, Albert JS, Sylvester M, Aharony D, Andisik D, Callaghan O, Campbell JB, Carr RA, Chessari G, Congreve M, Frederickson M, Folmer RH, Geschwindner S, Koether G, Kolmodin K, Krumrine J, Mauger RC, Murray CW, Olsson LL, Patel S, Spear N, Tian G. Application of fragment-based lead generation to the discovery of novel, cyclic amidine beta-secretase inhibitors with nanomolar potency, cellular activity, and high ligand efficiency. J Med Chem. 2007;50:5912–5925. doi: 10.1021/jm070829p. [DOI] [PubMed] [Google Scholar]
- 29.Vinnicombe HG, Derrick JP. Dihydropteroate synthase from Streptococcus pneumoniae: characterization of substrate binding order and sulfonamide inhibition. Biochem Biophys Res Commun. 1999;258:752–757. doi: 10.1006/bbrc.1999.0695. [DOI] [PubMed] [Google Scholar]
- 30.Huang LF, Tong WQ. Impact of solid state properties on developability assessment of drug candidates. Adv Drug Deliv Rev. 2004;56:321–334. doi: 10.1016/j.addr.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 32.Zhao H. Scaffold selection and scaffold hopping in lead generation: a medicinal chemistry perspective. Drug Discov Today. 2007;12:149–155. doi: 10.1016/j.drudis.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 33.ChemAxon Calculator Plugins were used for structure property prediction and calculation, Marvin 5.2.0, 2009. http://www.chemaxon.com.
- 34.Hergenrother PJ. Obtaining and screening compound collections: a user’s guide and a call to chemists. Curr Opin Chem Biol. 2006;10:213–218. doi: 10.1016/j.cbpa.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Morrison RW, Mallory WR, Styles VL. Pyrimido[4,5-c]pyridazines. 1 Cyclizations with alpha-keto esters. J Org Chem. 1978;43:4844–4849. [Google Scholar]
- 36.Li Y, Wu Y, Blaszczyk J, Ji X, Yan H. Catalytic roles of arginine residues 82 and 92 of Escherichia coli 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase: site-directed mutagenesis and biochemical studies. Biochemistry. 2003;42:1581–1588. doi: 10.1021/bi026800z. [DOI] [PubMed] [Google Scholar]
- 37.Triglia T, Menting JG, Wilson C, Cowman AF. Mutations in dihydropteroate synthase are responsible for sulfone and sulfonamide resistance in Plasmodium falciparum. Proc Natl Acad Sci U S A. 1997;94:13944–13949. doi: 10.1073/pnas.94.25.13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walter RD, Konigk E. 7,8-Dihydropteroate-synthesizing enzyme from Plasmodium chabaudi. Methods Enzymol. 1980;66:564–570. doi: 10.1016/0076-6879(80)66508-2. [DOI] [PubMed] [Google Scholar]
- 39.Aspinall TV, Joynson DH, Guy E, Hyde JE, Sims PF. The molecular basis of sulfonamide resistance in Toxoplasma gondii and implications for the clinical management of toxoplasmosis. J Infect Dis. 2002;185:1637–1643. doi: 10.1086/340577. [DOI] [PubMed] [Google Scholar]
- 40.GraphPad Prism 4.03 for Windows. GraphPad Software; San Diego California USA: www.graphpad.com. [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 43.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Sybyl, 8.0. Tripos, Inc.; St. Louis, MO, USA: 2007. [Google Scholar]
- 46.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearlman RS. Concord. Tripos International; St. Louis, Missouri, 43144, USA: distributed by. [Google Scholar]
- 48.Congreve M, Carr R, Murray C, Jhoti H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov Today. 2003;8:876–877. doi: 10.1016/s1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 49.Gasteiger J, Marsili M. Iterative Partial Equalization of Orbital Electronegativity - A Rapid Access to Atomic Charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 50.Wagner AB. SciFinder Scholar 2006: an empirical analysis of research topic query processing. J Chem Inf Model. 2006;46:767–774. doi: 10.1021/ci050481b. [DOI] [PubMed] [Google Scholar]
- 51.Huffenberger MA, Wigington RL. Chemical Abstracts Service approach to management of large data bases. J Chem Inf Comput Sci. 1975;15:43–47. doi: 10.1021/ci60001a013. [DOI] [PubMed] [Google Scholar]
- 52.Delano WL. The PyMOL molecular graphics system. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- 53.Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.