

Abstract
Trypanosoma cruzi and Trypanosoma brucei are parasites that cause Chagas’ disease and African sleeping sickness, respectively. Both parasites rely on essential cysteine proteases for survival, cruzain for T. cruzi and TbCatB/rhodesain for T. brucei. A recent quantitative high-throughput screen of cruzain identified triazine nitriles, which are known inhibitors of other cysteine proteases, as reversible inhibitors of the enzyme. Structural modifications detailed herein, including core scaffold modification from triazine to purine, improved the in vitro potency against both cruzain and rhodesain by 350-fold, while also gaining activity against T. brucei parasites. Selected compounds were screened against a panel of human cysteine and serine proteases to determine selectivity, and a co-crystal was obtained of our most potent analog bound to Cruzain.
Introduction
Chagas’ disease is a neglected tropical disease, affecting approximately 18 million people, predominately in Latin America.1 Its infectious agent is the protozoan parasite Trypanosoma cruzi (T. cruzi), with symptoms progressing from mild swelling to intestinal disease and ultimately heart failure. African sleeping sickness is a related disease caused by the protozoan parasite Trypanosoma brucei (T. brucei). Current chemotherapy for each disease is insufficient, as they exhibit unacceptable side effects and often do not completely eliminate the parasite despite chronic administration. Moreover, resistance to these therapies has emerged.1 Given these deficiencies, researchers have sought novel treatments of both diseases. Several biological pathways and targets have been explored in T. brucei including the cysteine protease rhodesain (also known as brucipain) and more recently a cathepsin B-like protease, TbCatB.2, 3 Potential intervention for T. cruzi includes the inhibition of the analogous cysteine protease cruzain, which is an essential protease for the survival of the T. cruzi.4, 5 Recently, Kraus et al. also reported a strategy using a rational designed sterol 14α-demethylase inhibitor which displays potent activity against T. cruzi.6 Our primary focus at the outset of this project was the discovery of non-peptidic, small molecule inhibitors of cruzain through the use of quantitative high-throughput screening (qHTS).7
Several groups have demonstrated that irreversible inhibition of cruzain by small molecules (examples include compounds 1 and 2) eradicates infection of the parasite in cell culture and animal models (Figure 1).4, 5, 8–10 Irreversible inhibitors 1 and 2 are peptidic and non-peptidic analogues, respectively, that contain an electrophilic functional group “warhead” (i.e. vinyl sulfone or 2,3,5,6-tetrafluorophenoxymethyl ketone) that can covalently bind to cruzain via nucleophilic attack of the active site cysteine.11 To date only irreversible inhibitors of cruzain have successfully cured parasitic infection5 implying that tight binding to the enzyme may be essential, though the relationship between reversible and irreversible inhibition has not been fully explored. We sought to identify and develop a small molecule reversible covalent inhibitor of cruzain, which may offer the potential of fewer off-target side-effects that are often associated with irreversible enzyme inhibitors.12, 13 The reagents developed here provide new molecular tools for the study and comparison of reversible and irreversible covalent inhibitors of cruzain.
Figure 1.

Representative covalent inhibitors of cruzain.
Design
Quantitative high-throughput screening (qHTS) was conducted at our center on ~200,000 small molecules as part of the NIH Molecular Libraries Probe Production Network (MLPCN).7, 14, 15 Several diverse chemotypes emerged as promising chemically tractable hits, including the triazine nitriles (Figure 2). In secondary assays performed at UCSF, compounds such as 3 and 4 had mid-nanomolar IC50 values with classical dose response curves. The observed inhibition was reversible by standard incubation-followed-by-dilution assays16 and was competitive with substrate, with a Ki of 180 nM obtained for compound 4 (Figure 2). Compounds in this class (triazine nitriles) have been explored for many years as potential treatments for a variety of diseases, including cancer,17 HIV,18 arthritis19, atherosclerosis and osteoporosis (Figure 3).20 Due to the strong electron-withdrawing nature of the triazine ring, the nitrile moiety in this class of compounds is particularly electrophilic21 and is thought to form a covalent but reversible bond with the active site cysteine of a respective protease.13 Though the bond is reversible, we hypothesized that the mechanism of covalent modification would prove beneficial to the inhibition of cruzain (see above). Moreover, the cysteine protease inhibitory activity of triazine nitriles (Figure 3), coupled with the privileged nature of this scaffold, should provide high hit rates of active compounds, allowing for smaller libraries to be synthesized. These factors, and the molecular simplicity of this chemotype, prompted us to pursue the triazine nitriles as inhibitors of cruzain.
Figure 2.

(a) Top actives obtained from qHTS. (b) Lineaweaver-Burk plot for compound 4, showing competitive inhibition of cruzain (Ki=180 nM)
Figure 3.

Triazine nitriles being explored for the potential treatment of cancer (5)17, HIV (6)18, arthritis (7)19, and atherosclerosis and osteoporosis (8)20.
After several rounds of optimization, and in order to expand upon our chemotype diversity, the triazine core was modified to adopt some structural rigidity. This was accomplished by incorporating one of the distal nitrogens into a heterocyclic ring, forming a purine core (Figure 4). Purine nitriles are reported to be highly potent inhibitors of the homologous cysteine proteases TbCatB3 and cathepsin K.22 It was hopeful that this trend would hold true for cruzain.
Figure 4.

Structural modification to the triazine core.
Chemistry
An initial round of analogues were pursued using compound 3 as the core structure. This led to the discovery of 4-(ethylamino)-6-(phenylamino)-1,3,5-triazine-2-carbonitrile (9) which had comparable in vitro potency yet is more amendable to parallel synthesis allowing rapid access to analogues (Scheme 1).20 Early stage structure activity relationships (SAR) were sought through common functionality scans of the aniline and alkyl-amino moieties. Equimolar amounts of either alkyl/cycloalkyl-amines or anilines (depending on modification strategy) were added to a solution of cyanuric chloride and Hunig’s base in dichloromethane at 0 °C. Depending on the starting material used in the first step, either anilines or amines were added in the second step, using the procedure described above. Finally, the nitrile was then installed using potassium cyanide (KCN) in DMSO at 120 °C for 10 minutes. Importantly, careful monitoring of the reaction was required as prolonged reaction times led to decomposition products.
Scheme 1.

Reagents: (a) (iPr)2NEt, CH2Cl2, 0 °C, various anilines or primary and secondary amines; (b) (iPr)2NEt, CH2Cl2, 0 °C, various anilines or primary and secondary amines; (c) KCN, DMSO, 120 °C.
Previously reported studies of purine nitriles against other cysteine proteases relied upon large hydrophobic groups at the 6-position and alkyl substituents at N9.3, 22 In targeting cruzain the optimal regiochemistry of the aniline and alkyl groups was not known. As such, two different synthetic strategies were employed for the preparation of isomers of Type A and Type B (Figure 4). Starting from 2,6-dichloropurine, the 9-position was alkylated using the requisite alkyl halides in the presence of K2CO3 in DMF at 60 °C (Scheme 2).22 Two regioisomers were obtained providing alkylation primarily at N9. The ratio of the product distribution was dependent on the nature of the alkyl group, and the two isomers were easily separable by column chromatography. Displacement of the 6-position chlorine was accomplished by heating various anilines in DMF at 160 °C in the presence of Hunig’s base. Finally, the nitrile was installed at the 2-position using KCN in DMSO at 120 °C. Of note, performing both of these transformations in the microwave greatly accelerated the reaction.
Scheme 2.

Reagents: (a) K2CO3, DMF, 60 °C, various primary and secondary amines; (b) (iPr)2NEt, DMF, 160 °C (μW), various anilines; (c) KCN, DMSO, 120 °C (μW).
Analogs of Type B (Figure 4) required a modified first step. The aromatic ring was first installed at the 9-position using a Buchwald-Hartwig-like copper catalyzed cross coupling between the aromatic amine and desired boronic acids (Scheme 3).23 Alkyl amines could then be installed at the 6-position in the presence of Hunig’s base in DMF at elevated temperature (120 °C). These analogues were also completed by displacing the final 2-Cl with KCN in DMSO at 120 °C in the microwave.
Scheme 3.

Reagents: (a) phenanthroline, Cu(OAc)2, 4 mol. sieves, CH2Cl2, rt, various aromatic boronic acids; (b) (iPr)2NEt, DMF, 120 °C, various primary and secondary amines; (c) KCN, DMSO, 120 °C (μW).
Results and Discussion
Our initial strategy was to modify the alkyl amine chain length and functionality of the top active from our preliminary SAR studies, compound 9. While the SAR was generally flat in this region, incorporation of the cyclopentyl group maintained potency of the lead compounds (Table 1). Also, some hydrophilicity was tolerated, which, if necessary, could be exploited at a later stage to improve pharmacokinetic properties (see below). As no appreciable improvements in potency were observed for the amine substitutions, our efforts then shifted to phenyl ring modifications. A library of compounds was synthesized to explore the effects of electron-donating/withdrawing groups in addition to incorporation of bulky hydrophobic moieties around the phenyl ring. Electron withdrawing groups at the 3-position on the phenyl ring proved to be fruitful, increasing potency 10-fold (Table 2). Placement of a nitro group at the 3-position (20) resulted in the most potent analogue. 3-fluoro and 3-chloro analogues displayed comparable potency (21 and 22, respectively), supporting the notion that electron-withdrawing groups at this position were most beneficial. Typically, electron-donating groups did not substantially improve potency, and it appeared that larger hydrophilic groups were also poorly tolerated. However, to solidify the size requirements and tolerance around the triazine scaffold, fused and pendant phenyl rings were incorporated. As anticipated, these changes resulted in decreased potency.
Table 1.
Alkyl moeity modifications
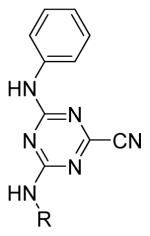 |
Compound | R | IC50 (μM)a |
| 9 | Ethyl | 0.71 | |
| 10 | Cyclopentyl | 0.71 | |
| 11 | Propyl | 0.79 | |
| 12 | Isopropyl | 0.89 | |
| 13 | Methylcyclopropyl | 0.89 | |
| 14 | Isobutyl | 0.89 | |
| 15 | Butyl | 0.89 | |
| 16 | 4-hydroxybutyl | 0.89 | |
| 17 | Methyl | 1.26 | |
| 18 | Cyclohexyl | 1.41 | |
| 19 | 2-hydroxyethyl | 1.58 | |
IC50 values were determined at NIH Chemical Genomics Center using the qHTS protocol and NCGC curve fitting software. Compounds were run in duplicate with Log IC50 standard errors ranging from 0.001 to 0.05.
Table 2.
Second round modifications
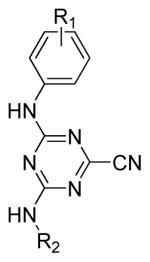 |
Compound | R1 | R2 | IC50 (μM)a |
| 20 | 3-nitro | cyclopentyl | 0.063 | |
| 21 | 3-fluoro | cyclopentyl | 0.079 | |
| 22 | 3-chloro | cyclopentyl | 0.079 | |
| 23 | 3-bromo | cyclopentyl | 0.1 | |
| 24 | 4-fluoro | cyclopentyl | 0.126 | |
| 25 | 3-methyl | cyclopentyl | 0.126 | |
| 26 | 4-bromo | cyclopentyl | 1.122 | |
| 27 | 3-phenyl | cyclopentyl | 2.239 | |
| 28 | 3-trifluoromethy1 | cyclopentyl | inactive | |
| 29 | 3,5-difluoro | cyclopentyl | 0.063 | |
| 30 | 3,5-dichloro | cyclopentyl | 0.398 | |
| 31 | 3,5-difluoro | 2,2-difluoroethyl | 0.025 | |
IC50 values were determined at NIH Chemical Genomics Center using the qHTS protocol and NCGC curve fitting software. Compounds were run in duplicate with Log IC50 standard errors ranging from 0 to 0.07.
To further explore the effect of 3-subsituted electron-withdrawing substituents, a series of compounds were synthesized including bis-meta substituted anilines (Table 2). Interestingly, 3,5-difluoro triazine nitrile 29 displayed comparable potency as the mono-m-nitro-substituted triazine 20 (both 63 nM IC50). However, 3,5-dichloro-triazine nitrile 30 was 5-fold less active than 3-chloro-triazine nitrile 22 (398 nM and 79 nM IC50, respectively) arguing again that increased bulk is poorly tolerated on this phenyl moiety. A final compound was synthesized and was designed to incorporate electron withdrawing groups on the alkyl amine. Compound 31 included the 2,2-difluoroethyl group, and this change further improved potency to 25 nM IC50 (Table 2).
The SAR generated for the triazine nitrile class of compounds greatly informed our efforts in exploring the purine nitrile chemotype. The purine core provided an additional 6-fold increase in potency for structural analogues of Type A (Figure 4). The purine analogue containing our previously most active substituents (3,5-difluoroaniline and cyclopentyl amine) improved the potency from 63 nM to 18 nM (compounds 29 and 34, respectively, Tables 2 and 3). Incorporation of the ethyl moiety (compound 32) gave a slight potency enhancement (IC50 of 10 nM) as did the 2,2-difluoroethyl moiety (compound 33) (IC50 of 13 nM). There was a noticeable decrease in potency for analogues of Type B. Incorporation of the matching substitution patterns for this chemotype (for instance 3,5-difluoroaniline at position 9 and ethyl amine substitution at position 6 in compound 40) gave a marked decrease in potency (251 nM, Table 3). Overall, the trend in potency for each substitution pattern correlated well across the two different scaffolds, with the purine core generally showing enhanced potency.
Table 3.
In vitro potency of purine nitrile analogs
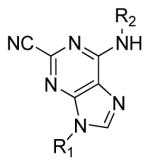 |
Compound | R1 | R2 | IC50(μM)a |
| 32 | Ethyl | 3,5-difluorophenyl | 0.010 | |
| 33 | 2,2-difluoroethyl | 3,5-difluorophenyl | 0.013 | |
| 34 | Cyclopentyl | 3,5-difluorophenyl | 0.018 | |
| 35 | Ethyl | 3-chlorophenyl | 0.018 | |
| 36 | Cyclopentyl | 3-chlorophenyl | 0.040 | |
| 37 | 3,5-difluorophenyl | 2,2-difluoroethyl | 0.050 | |
| 38 | 3-chlorophenyl | Ethyl | 0.063 | |
| 39 | 3-chlorophenyl | 2,2-difluoroethyl | 0.071 | |
| 40 | 3,5-difluorophenyl | Ethyl | 0.251 | |
| 41 | 3,5-difluorophenyl | Cyclopentyl | 0.316 | |
IC50 values were determined at NIH Chemical Genomics Center using the qHTS protocol and NCGC curve fitting software. Compounds were run in duplicate with Log IC50 standard errors ranging from 0 to 0.02.
As a result of the homologous relationship between T. cruzi and T. brucei, we tested selected compounds for inhibition of rhodesain and TbCatB (Table 4). The activity of compounds 3, 20–32, 35, and 36 against rhodesain were quite comparable to those observed for cruzain. The activities of these compounds were consistently more potent in 96-well plate cruzain assays than the corresponding 1536-well format, varying from 5–50 fold. Although the precise underlying mechanism for these differences is presently unclear, they appear to be related to the differences in enzyme availability in the two assay conditions (96-well plate with large reaction volume and low surface-to-volume ratio and 1536-format polystyrene well with low reaction volume and very high surface-to-volume ratio) and the method for compound delivery (pipetting into 96-well plate versus pintool transfer into a 1536-well plate), ultimately resulting in different detection limits in the presence of low concentration (~1 nM) of enzyme. The difference in apparent IC50s notwithstanding, there was consistency in the ranking of compound activities between this assay platform and the original screening technique throughout all analogue production cycles. The similar inhibitory potential for these agents against cruzain and rhodesain is not surprising given the structural homology of these two enzymes. As we expected based on lower homology to cruzain, activity against TbCatB was more modest. We were however encouraged by the observation that some compounds showed low micromolar IC50 values against this enzyme, which has been shown to be essential for T. brucei survival based on RNA interference studies in vitro2 and in mice.24 In contrast, knocking-down rhodesain produced a phenotype only in vivo, when it prolonged life of infected mice, but didn’t cure parasitemia as observed for TbCatB knock-down. These studies suggested that the relevant cysteine protease target for cure of sleeping sickness is probably TbCatB, while rhodesain seems to be a virulence factor, facilitating crossing the blood brain barrier24 of the host. While encouraged by the sub-nanomolar in vitro potency of our lead compounds against cruzain, and in some cases the low micromolar levels of activity against TbCatB, we were eager to investigate the activity against parasites in vivo.
Table 4.
In vitro and in vivo potency of active compounds against cysteine protease homologs
| Compound | cruzain IC50 (μM)a | rhodesain IC50 (μM)a | TbCatB IC50 (μM)a | Tbb MIC50 (μM) |
|---|---|---|---|---|
| 3 | 0.09 | 0.3 | >1 | >100 |
| 4 | 0.07 | >10 | >1 | >100 |
| 20 | 0.001 | 0.008 | >1 | 100 |
| 21 | 0.01 | 0.02 | >1 | 100 |
| 22 | 0.002 | 0.006 | >100 | 6 |
| 23 | 0.002 | 0.006 | >100 | 6 |
| 24 | 0.03 | 0.04 | >1 | >100 |
| 25 | 0.03 | 0.05 | 6 | 25 |
| 26 | 0.06 | 0.1 | >10 | >100 |
| 27 | 0.05 | 0.03 | >10 | 25 |
| 28 | 0.04 | 0.07 | >10 | 25 |
| 29 | 0.004 | 0.005 | 4 | 6 |
| 30 | 0.01 | 0.03 | >10 | 25 |
| 31 | 0.001 | <0.006 | 1 | 6 |
| 32 | 0.0002 | <0.006 | 6 | 25 |
| 35 | 0.0004 | 0.01 | >100 | 25 |
| 36 | 0.0003 | <0.006 | >10 | 100 |
IC50 values were determined at UCSF. Curve fitting errors ranged between 7% an 32%.
To assess the in vivo activity of these agents, a subset of optimized compounds and top actives from the primary screen were tested against the T. brucei parasite (Tbb, Table 4). The screening leads 3 and 4 showed no trypanocidal activity up to 100 μM. Gratifyingly, several of the lead triazines (22–23, 29) showed activity against T. brucei with MIC50 values of 6 μM. Furthermore, similar purine nitrile analogs 32 and 35 were also active (MIC50 = 25 μM). Among these compounds 29, 31 and 32 showed inhibition of TbCatB at low micromolar levels (IC50 of 4, 1 and 6 μM respectively), at concentrations similar to the necessary for anti-parasitic activity. However, for some compounds such as 22, 23 and 35 IC50 values against this enzyme are over 100 μM, despite trypanocidal activity.
Triazines 22, 23, 25, 27–30 and purine nitriles 32, 35, and 36 had no effect against the T. cruzi parasite in cell culture despite the potent in vitro activity. The T. cruzi assay is substantially more stringent than the T. brucei assay, as the former measures parasite infection of human macrophages over several weeks, demanding penetration of the host cell membrane and stability over a long period of time10, whereas the T. brucei assay is conducted on free living parasites over a few days. Given the disparity in activity of these compounds in the in vitro and cell-based assays, it is likely that cell permeability and solubility is lacking. Guy and co-workers reported that incorporation of hydrophilic alkyl moieties at N9 greatly improved activity against Tbb.3 As such, we are hopeful that future studies directed towards the improvement of the pharmacokinetic properties of these molecules could provide a platform for the treatment of T. cruzi utilizing reversible covalent inhibitors.
To better understand the mechanism of action of these covalent but reversible inhibitors of cruzain, we determined an x-ray crystal structure of the cruzain/32 complex, to 1.1 Å resolution (Table 5). The ultra-high resolution of this structure allowed us to analyze the enzyme-inhibitor interactions in detail. Even before fitting and refining compound 32, the unbiased Fo-Fc electron density was unambiguous for the inhibitor (Figure 5). The covalent adduct with the catalytic Cys25 is clear, with the formally linear nitrile becoming a planar imino-moiety, with a Cysteine-sulfur to inhibitor carbon distance of 1.75 Å and a sulfur-carbon-nitrogen angle of 125°, in agreement with standard values. Several polar interactions are observed between nitrogens in the purine ring in 32 and waters or cruzain residues. The nitrogen in the newly formed iminothioether hydrogen-bonds to water 281 (3.17 Å) and to the Nε in Gln19 (2.96 Å), which is part of the oxyanion hole in cysteine proteases of the papain family, such as cruzain26. The anilinic nitrogen and N7 are solvent exposed and form hydrogen bonds to water 294 (distances 2.92 Å and 3.15 Å, respectively). N7 is also involved in dipole-dipole interactions with waters 292 (3.14 Å) and 368 (3.36 Å). The inhibitor purine packs against the surface between S1 and S2 pockets of the cruzain active site, while the aromatic ring off the 6-position extends into the S2 pocket of the enzyme. Good van der Waals complementarity is observed in this mostly hydrophobic pocket, which is completely filled by the 3,5-difluorophenyl ring from 32. This supported our observation that larger, more hydrophobic groups (i.e. 3,5-dichloro) in this region result in a decrease in potency. Double conformation is observed for the terminal carbon atom in the ethyl substituent at N9. This alkyl moiety reaches toward the surface of the enzyme with no direct interactions with the binding site being observed (Figure 5). As such, a wide variety of substituents could be tolerated at this position and ultimately exploited for the improvement of pharmacokinetic properties (above).
Table 5.
Data collection and refinement statistics (molecular replacement) for the cruzain/32 complex structure
| Data collection | |
|---|---|
| Space group | P6522 |
| Cell dimensions | |
| a, b, c (Å) | 82.97, 82.97, 101.90 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 41.56 (1.10) * |
| Rsym or Rmerge | 0.052 (0.407) |
| I/σI | 33.6 (2.8) |
| Completeness (%) | 92.7 (65.1) |
| Redundancy | 11.6 (4.3) |
| Refinement | |
| Resolution (Å) | 1.10 |
| No. reflections | 73808 |
| Rwork/Rfree | 0.141/0.117 |
| No. atoms | 2075 |
| Protein | 1705 |
| Ligand/ion | 23 |
| Water | 351 |
| B-factors | |
| Protein | 8.29 |
| Ligand/ion | 10.43 |
| Water | 22.34 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.020 |
| Bond angles (°) | 2.006 |
Values in parentheses are for highest-resolution shell.
Figure 5.

(a) Unbiased Fo-Fc 3σ electron density from the complex between molecule 32 and cruzain, determined to 1.1 Å resolution, stereo view. (b) Key interactions observed in the refined cruzain/32 x-ray structure, stereo view. Dashed lines represent hydrogen bonds (green) and dipole-dipole interactions (salmon), waters are shown as red spheres. Protein carbon atoms colored gray, inhibitor carbon atoms colored orange, oxygen, nitrogen, sulfur and fluorine atoms colored red, blue, yellow and cyan respectively. Second conformation of terminal ethyl carbon in 32 colored magenta. PDB entry 3I06. Images prepared with Pymol.25
Protease Activity Profiling
The lead compounds 29, 22, 32, and 40 were also screened against a human protease panel (Figure 6) to identify possible off-target activity. As expected, these analogues were active against a few cathepsins that are highly homologous with human papain cysteine proteases.27 These results, however, are not necessarily adverse. For example, previously reported vinyl sulfone inhibitors of cruzain which were efficacious in mice models of Chagas’ disease were potent inhibitors of cathepsins B, L, S. However, no toxicity was observed in mice, rats or dogs. 28 Cathepsins are located in the lysosymes of cells, whereas the parasites are located in the more accessible cytoplasm. Potent compounds are likely to preferentially inhibit the target parasite as a result. Aside from the cathepsins, the lead compounds gave little to no inhibition against a panel of caspases, peptidases, matrix metalloproteinases (MMPs), and serine proteases. This protease profile demonstrates the specificity and broad utility of these triazine and purine nitriles.
Figure 6.

Protease profiling of compounds 29, 22,32, and 40. IC50s were determined for all four compounds against a panel of cysteine porteases, aspartyl proteases, peptidases, MMPs, and serine proteases. Black corresponds to inactives, in yellow are IC50s > 1uM, and in red are IC50s < 1uM. Data was determined by Reaction Biology, Inc., using a 10-dose, 3-fold serial dilution of compounds. The protease activities were monitored as a time-course measurement of the increase in fluorescence signal from fluorescently labeled peptide substrate, and initial linear portion slope (signal/min) was analyzed.
Conclusion
In summary, our initial screen focused on identification of reversible, non-peptidic small molecule cruzain inhibitors. The screen identified the covalent reversible triazine nitriles, among other classes of molecules (unpublished results), which are known inhibitors of several human cysteine proteases. The original hits, while modestly potent in biochemical assays (down to mid-nanomolar IC50 values), were inactive against T. brucei parasites. Modifications to the extended structure both improved potency and achieved activity in vivo. The determination of the crystal structure of the cruzain/32 complex, at ultra-high resolution, may help guide additional medicinal chemistry efforts against this important parasite target.
Experimental Methods
Chemistry
Unless otherwise stated, all reactions were carried out under an atmosphere of dry argon or nitrogen in dried glassware. Indicated reaction temperatures refer to those of the reaction bath, while room temperature (rt) is noted as 25 °C. All solvents were of anhydrous quality purchased from Aldrich Chemical Co. and used as received. Commercially available starting materials and reagents were purchased from Aldrich, Alfa Aesar, Acros, and Synquest and were used as received.
Analytical thin layer chromatography (TLC) was performed with Sigma Aldrich TLC plates (5 × 20 cm, 60 Å, 250 μm). Visualization was accomplished by irradiation under a 254 nm UV lamp. Chromatography on silica gel was performed using forced flow (liquid) of the indicated solvent system on Biotage KP-Sil pre-packed cartridges and using the Biotage SP-1 automated chromatography system. 1H- and 13C NMR spectra were recorded on a Varian Inova 400 MHz spectrometer. Chemical shifts are reported in ppm with the solvent resonance as the internal standard (CDCl3 7.26 ppm, 77.00 ppm, DMSO-d6 2.49 ppm, 39.51 ppm for 1H, 13C respectively). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constants, and number of protons. Low resolution mass spectra (electrospray ionization) were acquired on an Agilent Technologies 6130 quadrupole spectrometer coupled to the HPLC system. High resolution mass spectral data was collected in-house using and Agilent 6210 time-of-flight mass spectrometer, also coupled to an Agilent Technologies 1200 series HPLC system. If needed, products were purified via a Waters semi-preparative HPLC equipped with a Phenomenex Luna® C18 reverse phase (5 micron, 30 × 75 mm) column having a flow rate of 45 mL/min. The mobile phase was a mixture of acetonitrile and H2O each containing 0.1% trifluoroacetic acid. Samples were analyzed for purity on an Agilent 1200 series LC/MS equipped with a Luna® C18 reverse phase (3 micron, 3 × 75 mm) column having a flow rate of 0.8–1.0 mL/min over a 3-minute gradient and a 4.5 minute run time. The mobile phase was a mixture of acetonitrile (0.025% TFA) and H2O (0.05% TFA), and a temperature was maintained at 50 °C.. Purity of final compounds was determined to be >95%, using a 3 μL injection with quantitation by AUC at 220 and 254 nM (Agilent Diode Array Detector).
Experimental Procedures
Example procedure for the formation of 4,6-dichloro-N-alkyl-1,3,5-triazine-2-amines (Step 1)
4,6-dichloro-N-cylopentyl-1,3,5-triazin-2-amine
To a solution of cyanuric chloride (1.25 g, 6.78 mmol) in CH2Cl2 (50 mL) at 0 °C was added Hünig’s base (1.12 mL, 6.78 mmol). After 5 minutes, cyclopentylamine (0.67 mL, 6.78 mmol) was added and the reaction mixture was allowed to stir for 15 minutes at 0 °C, upon which time the ice bath was removed. The reaction mixture was stirred at rt for 30 min, then concentrated under reduced pressure and directly purified on silica column. Gradient elution with ethyl acetate (2→40%) in hexanes provided 4,6-dichloro-N-cylopentyl-1,3,5-triazin-2-amine as a colorless solid: yield (1.62 g, 5.85 mmol, 86 %).
General procedure for the formation of 6-chloro- N2-alkyl-N4-phenyl-1,3-5-triazine-2,4-diamines (Step 2)
6-chloro-N2-cyclopentyl-N4-(3,5-difluorophenyl)-1,3,5-triazine-2,4-diamine
To a solution of 4,6-dichloro-N-cyclopentyl-1,3,5-triazin-2-amine (1.5 g, 6.44 mmol) in CH2Cl2 (50 mL) at 0 °C was added Hünig’s base (1.12 mL, 6.44 mmol). After 5 minutes, 3,5-difluoroaniline (0.83 g, 6.44 mmol) was added and the reaction mixture was allowed to stir for 15 minutes at 0 °C, upon which time the ice bath was removed. The reaction mixture was stirred at rt for 30 min, then concentrated under reduced pressure and directly purified on silica column. Gradient elution with ethyl acetate (2→40%) in hexanes provided 6-chloro-N2-cyclopentyl-N4-(3,5-difluorophenyl)-1,3,5-triazine-2,4-diamine as a colorless solid: yield (1.9 g, 5.83 mmol, 91%).
General procedure for the formation of 4-alkylamino-6-phenylamino-1,3,5-triazine-2-carbonitriles (Step 3)
4-(cyclopentylamino)-6-(3,5-difluorophenylamino)-1,3,5-triazine-2-carbonitrile (29)
To a solution of 6-chloro-N2-cyclopentyl-N4-(3,5-difluorophenyl)-1,3,5-triazine-2,4-diamine (1.0 g, 3.07 mmol) in DMSO (25 mL) was added KCN (0.220 g, 3.38 mmol). The reaction mixture was sealed and heated to 120 °C for 10 minutes. Upon completion, the reaction mixture was diluted with ethyl acetate and washed several times with saturated sodium chloride solution. The organic layer was dried on magnesium sulfate, filtered, and concentrated. The resulting residue was purified on silica column. Gradient elution with ethyl acetate (2→40%) in hexanes provided 4-(cyclopentylamino)-6-(3,5-difluorophenylamino)-1,3,5-triazine-2-carbonitrile (29) as a colorless solid: yield (863 mg, 2.73 mmol, 89%); 1H (DMSO-d6) δ 1.45–1.63 (m, 4H), 1.63–1.79 (m, 2H), 1.84–2.02 (m, 2H), 4.06–4.27 (m, 1H), 6.82–6.92 (m, 1H), 7.49 (d, J = 8.7 Hz, 1H), 7.56 (d, J = 8.1 Hz, 1H), 8.48 (d, J = 6.3 Hz, 1H), 8.61 (brs, 1H), 10.39 (brs, 1H) and 10.52 (brs, 1H); 13C NMR (DMSO-d6) δ 23.82, 23.87, 32.15, 32.41, 52.57, 98.39, 103.43, 115.67, 141.85, 151.50, 151.93, 161.63, and 164.10; HRMS (ESI) m/z = 317.1319 (M+H)+ (C15H15F2N6 requires 317.1316); LC-MS: rt (min) = 3.94; LRMS (ESI) m/z = 317.1.
Example procedure for the formation of 2,6-dichloro-9-alkyl-9H-purines(Step 1)
2,6-dichloro-9-ethyl-9H-purine
To a solution of 2,6-dichloro-9H-purine (2.0 g, 10.6 mmol) in acetone (45 mL) was added sodium carbonate (2.25 g, 21.2 mmol). The reaction vessel was equipped with a reflux condenser, and the mixture was heated under reflux conditions for 20 minutes. After that time, iodoethane (0.86 mL, 10.6 mmol) was added in one portion, and the reaction mixture was allowed to stir for 5 h. Upon completion, the reaction mixture was concentrated under reduced pressure and directly purified on silica column. Gradient elution with ethyl acetate (2→40%) in hexanes provided regioisomers 2,6-dichloro-9-ethyl-9H-purine and 2,6-dichloro-7-ethyl-7H-purine as pale yellow solids: yield (1.5 g, 6.91 mmol, 82 %; 0.4 g, 1.84 mmol, respectively).
Example procedure for the formation of 2-chloro-N-phenyl-9-ethyl-9H-purin-6-amines (Step 2)
2-chloro-N-(3-chlorophenyl)-9-ethyl-9H-purin-6-amine
To a solution of 2,6-dichloro-9-ethyl-9H-purine (0.5 g, 2.30 mmol) in DMF (2 mL) was added 3-chloroaniline (0.29 g, 2.30 mmol) and Hunig’s Base (0.40 mL, 2.30 mmol). The reaction mixture was sealed in a microwave tube (2-5 mL) and heated to 110 °C for 30 min at 90 W in a Biotage Initiator® microwave. Upon completion, the reaction mixture was diluted with ethyl acetate and washed several times with 3 N lithium chloride solution. The organic layer was separated, dried on magnesium sulfate, filtered, concentrated under reduced pressure and directly purified on silica column. Gradient elution with ethyl acetate (5→50%) in hexanes provided 2-chloro-N-(3-chlorophenyl)-9-ethyl-9H-purin-6-amine as a pale yellow solid: yield (0.64 g, 2.08 mmol, 90 %).
Example procedure for the formation of 6-phenylamino-9-ethyl-9H-purine-2-carbonitriles (Step 3)
6-(3-chlorophenylamino)-9-ethyl-9H-purine-2-carbonitrile (35)
To a solution of 2-chloro-N-(3-chlorophenyl)-9-ethyl-9H-purin-6-amine (0.05 g, 0.16 mmol) in DMSO (1 mL) was added KCN (10.5 mg, 0.16 mmol). The reaction mixture was sealed in a microwave tube (0.5–2 mL) and heated in a microwave to 140 °C for 1 h at 90 W. Upon completion, the reaction mixture was diluted with ethyl acetate and washed several times with saturated sodium chloride solution. The organic layer was separated, dried on magnesium sulfate, filtered, concentrated under reduced pressure and directly purified on silica column. Gradient elution with ethyl acetate (10→70%) in hexanes provided 35 as a colorless solid: yield (45 mg, 0.15 mmol, 93 %); 1H (CDCl3) δ 1.60 (t, J = 7.3 Hz, 3H), 4.34 (q, J = 7.4 Hz, 2H), 7.15 (ddd, J = 8.1, 2.0 and 0.9 Hz, 1H), 7.35 (t, J = 8.1 Hz, 1H), 7.75 (ddd, J = 8.2, 2.2 and 0.9 Hz, 1H), 7.87 (t, J = 2.1 Hz, 1H), 7.89 (s, 1H) and 8.03 (s, 1H); LC-MS: rt (min) = 3.67; LRMS (ESI) m/z = 299.0.
6-(3,5-difluorophenylamino)-9-ethyl-9H-purine-2-carbonitrile (32)
The title compound was prepared using a procedure similar to that detailed for 35, substituting 3,5-difluoroaniline in Step 2, providing 32 as a colorless solid: yield (35 mg, 90%); 1H NMR (DMSO-d6) δ 1.46 (t, J = 7.3 Hz, 3H), 4.30 (q, J = 7.3 Hz, 2H), 6.94 (tt, J = 9.3 and 2.4 Hz, 1H), 7.63–7.83 (m, 2H), 8.67 (s, 1H), and 10.79 (s, 1H); HRMS (ESI) m/z = 301.1008 (M+H)+ (C14H11F2N6 requires 301.1006); LC-MS: rt (min) = 3.70; LRMS (ESI) m/z = 301.0.
Example procedure for the formation of 2,6-dichloro-9-phenyl-9H-purines (Step 5)
2,6-dichloro-9-(3,5-difluorophenyl)-9H-purine
To a solution of 2,6-dichloro-9H-purine (0.6 g, 3.17 mmol) in CH2Cl2 (15 mL) was added 3,5-difluorophenylboronic acid (1.0 g, 6.35 mmol), copper (II) acetate (1.15 g, 6.35 mmol), 4 Å molecular sieves (~250 mg), and NEt3 (1.3 mL, 9.5 mmol). The reaction mixture was stirred at 100 °C for 16 h. Upon completion, the reaction mixture was diluted with ethyl acetate, filtered through a pad of celite, washed with water and brine (3 × 30 mL), and the solvent was removed under reduced pressure. The remaining residue was directly purified on silica column. Gradient elution with ethyl acetate (5→65%) in hexanes provided 2,6-dichloro-9-(3,5-difluorophenyl)-9H-purine as a pale yellow solid: (200 mg, 0.66 mmol, 21 %).
2-chloro-9-(3,5-difluorophenyl)-N-ethyl-9H-purin-6-amine
The title compound was prepared using a procedure similar to that detailed in Step 2 above, substituting ethylamine, providing the product as a pale yellow solid: yield (60 mg, 83%).
9-(3,5-difluorophenyl)-6-(ethylamino)-9H-purine-2-carbonitrile (40)
The title compound was prepared using a procedure similar to that detailed in Step 3 above, providing 40 as a colorless solid: yield (23 mg, 48%). 1H NMR (CDCl3) δ 1.34–1.40 (m, 3H), 3.73 (brs, 2H), 6.02 (brs, 1H), 6.94 (tt, J = 8.7 and 2.3 Hz, 1 H), 7.39 (dd, J = 7.2, 1.9 Hz, 2H) and 8.16 (s, 1H); 13C NMR (CDCl3) δ 14.70, 35.94, 103.72, 103.97, 104.21, 106.34, 106.62, 116.59, 136.23, 138.93, 139.77, 155.00, 162.14, and 164.77; HRMS (ESI) m/z = 301.1008 (M+H)+ (C14H11F2N6 requires 301.1003).
Methods
Cruzain enzymatic assays in 1536-well format
The compounds were initially prepared as 10 mM DMSO stock solutions and were arrayed for testing as serial twofold dilutions at 5 μL per well in 1,536-well Greiner polypropylene compound plates following previously described protocols.29 Cruzain activity was assayed in freshly prepared 100 mM acetate buffer pH 5.5, containing 5 mM dithiothreitol (DTT) and 0.01 % Triton X-100. Three μl of reagents (buffer as negative control and cruzain at 1.5 nM final concentration in the remainder of the plate) were dispensed by Flying Reagent Dispenser™ (FRD) (Beckman Coulter.Inc., Fullerton, CA) into a black solid-bottom 1,536-well plate (Greiner Bio-One, Monroe, NC). Inhibitors were delivered as 23 nL of DMSO solutions via pintool transfer; vehicle-only control consisted of 23 nL DMSO. The plate was incubated for 15 min at room temperature, and then 1 μL of cruzain fluorogenic substrate (Z-FR-AMC, Bachem, 2 uM final concentration) was added to start the reaction. Following substrate dispense, the plate was immediately transferred into ViewLux High-throughput CCD imager (Perkin-Elmer, Waltham, MA) for kinetic fluorescence data collection utilizing standard 340 nm excitation and 450 nm emission filter set. Percent inhibition was calculated relative to the no-enzyme and uninhibited controls (48 wells averaged per condition) by using the fluorescence intensity change recorded during the first 60 seconds of reaction.
Cruzain, rhodesain and tbcatB enzymatic assays in 96-well plate format
100 μL per well of recombinant enzyme cruzain, rhodesain or tbcatB (in a buffer solution consists of 100 mM sodium acetate pH 5.5, 5 mM DTT and 0.001% Triton X-100) was added to a 96-well black plate that contained 1 μL of test compound (in DMSO). The enzyme-compound mixture was incubated for 5 min at room temperature. Then 100 μL per well of substrate Z-FR-AMC (in the same buffer solution as above) was added to the enzyme-compound mixture to initiate the reaction. The rate of increase in fluorescence (units/sec), resulting from the proteolytic cleavage of the substrate leading to the release of fluorogenic AMC was monitored as with an automated microtiter plate spectrofluorimeter (SpectraMax M5, Molecular Devices) with fluorescence readout setting of excitation at 355 nm and emission at 460 nm. The assay concentrations of enzyme and substrate are 4 nM (cruzain), 4 nM (rhodesain), 258 nM (tbcatB) and 10 μM Z-Phe-Arg-AMC, respectively. Positive control wells contained 1 μL of DMSO. IC50 curve fitting was performed with Prism 4 software (GraphPad, San Diego, CA).
Competition assays and Ki determination
Cruzain activity was conducted in 96-well plates as described above, for varying concentrations of compound 5 and Z-FR-AMC, in presence of 0.01% Triton. Each concentration of 5 (0, 25, 50, 100, 200 and 500 nM), was tested in seven concentrations of substrate (0.31 to 20 uM, in 2-fold increments). Compound was incubated with cruzain for 15 minutes prior to addition of substrate and enzyme activity was then monitored for five minutes. Given the covalent reversible mechanism of this compound, initially an increase in rates of activity was observed, until equilibrium was reached. Calculations of rates of cruzain activity were based on time points after equilibrium, when a uniform rate was observed with time. All assays were performed in duplicate. Lineaweaver-Burk plot was built in Prism 4.
Trypanosoma brucei brucei assay
Trypanosoma brucei brucei strain 221 was grown in complete HMI-9 medium containing 10% FBS, 10% Serum Plus medium (Sigma Inc. St. Louis Mo. USA) and 1X penicillin/streptomycin. The trypanosomes were diluted to 1×105 per mL in complete HMI-9 medium. 95 μL per well of the diluted trypanosomes was added to sterile Greiner 96-well flat white opaque culture plates that contained 5 μL of test samples (in 10% DMSO). Control wells contained 95 μL of the diluted trypanosomes and 5 μL of 10% DMSO while control wells for 100% inhibition contained 95 μL of the diluted trypanosomes and 5 μL of 1mM Thimerosal (in 10% DMSO). Trypanosomes were incubated with test samples for 48 h at 37°C with 5% CO2 before monitoring viability. Trypanosomes were then lysed in the wells by adding 50 μl of CellTiter-GloTM (Promega Inc., Madison, WI, USA). Lysed trypanosomes were placed on an orbital shaker at room temperature for 2 min. The resulting ATP-bioluminescence of the trypanosomes in the 96-well plates was measured at room temperature using an Analyst HT plate reader (Molecular Devices, Sunnyvale, CA, USA). Each compound was evaluated in eight concentrations, in four-fold dilutions starting at 100 μM. The reported MIC50 is the lowest tested compound concentration which inhibited parasite growth by at least 50%.
Trypanosoma cruzi assay
Assays were conducted as previously described.10 Briefly, irradiated (1000 rad) J774 macrophages were plated overnight onto sterile tissue culture plates prior to infection with Y strain T. cruzi trypomastigotes for 2 h. Parasites were then removed and cells cultured with RPMI-1640 medium with the addition of compounds at the appropriate concentration (n=3 per compound). Untreated infected controls and cultures treated with 10 μM K777 were included in each independent experiment. Treatment was continued for 27 days and medium replaced every 48 h. Cultures were observed daily by contrast phase microscopy (400×) for the appearance of free trypomastigotes in the medium and or compound toxicity (i.e. granulation, detachment and death) for host macrophages. After completion of the treatment period, cultures were maintained for an additional 14 days to ensure ‘cure’ of the host cells. Under these experimental conditions, T. cruzi completed the intracellular cycle in 5 days in untreated controls, while K777 cured cell infection and no free parasites were observed. Seven triazine nitriles (compounds 22, 23, 25, 27–30) were tested at 1, 5 and 10 uM. All triazines were toxic against macrophages at 10 μM, inactive against T. cruzi at 1 μM and either toxic (23 and 29) or inactive (22, 25, 27, 28, 30) at 5 μM. Purines 32, 35, and 36 were tested at 10 μM and were inactive.
Cruzain expression and purification
Procruzain truncated at the C-terminal30 was expressed and purified using a modified protocol (Lee, Balouch and Craik, unpublished results). A 0.5 mg/mL solution of procruzain (in 100 mM sodium acetate pH 5.5, 10 mM EDTA, 5 mM DTT and 1 M NaCl, pH adjusted to 5.3) was activated at 37 °C for three hours. After activation, cruzain was dialyzed in 10 mM Tris buffer pH 7.5, inhibited with the covalent reversible inhibitor methyl methanethiosulfonate (MMTS) and dialyzed again in the same buffer. Finally, the protein was purified in a MONO-Q anion exchange column, using a 0 to 500 mM NaCl gradient in 10 mM Tris buffer (pH 7.5).
Crystallography
MMTS inhibited cruzain was concentrated in to 1 mg/mL in 2 mM bis tris pH 5.8. In order to reverse MMTS inhibition, 5 mM DTT was added, followed by addition of 400 uM of compound 32. The solution was stirred for a few hours at 4 °C, until cruzain was completely inhibited, and the protein was concentrated down to 7.5 mg/mL. Hanging drops for 384 crystallography conditions (Joint Center Structure Genomics screens I–IV, Qiagen) were set up using a Mosquito® (TTP Labtech). Each condition was screened in 1:1 and 2:1 ratio between protein solution and mother liquor. After two weeks incubation at 18 °C a 200 um crystal was obtained in 0.1 M Tris pH 8.5, 2.0 M NH4H2PO4, in the (200 nL protein solution): (100 nL mother liquor) drop. Crystals were then reproduced in the same conditions in 2–4 μL hanging drops, were transferred to a solution of 25% glycerol in mother liquor and cryocooled in liquid nitrogen.
Data collection was performed on frozen crystals in beamline 7-1 in the Stanford Synchrotron Radiation Lightsource at the SLAC National Accelerator Laboratory. Reflections were indexed and integrated using Ipmosflm. PHASER31 was used for the initially phasing, based on a previously reported structure of cruzain bound to a non-covalent inhibitor (PDB entry 1ME3)32, with the ligand, water molecules and ions removed. The space group was P6522. Data refinement was performed using REFMAC533 package and models were built and waters placed using Coot34.
Supplementary Material
Acknowledgments
The authors thank Jeremy Smith, Paul Shinn, and Danielle van Leer for assistance with compound management. We also thank Eaman Balouch for providing the cruzain construct and information on cruzain expression protocol, and Dr. Linda Brinen for helpful discussions and suggestions. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research and by NIH Grant GM59957 (to BKS) and by the Sandler Center for Basic Research in Parasitic Diseases (to JHM).
Abbreviations
- qHTS
quantitative High-Throughput Screening
- MLPCN
NIH Molecular Libraries Probe Production Network
Footnotes
Supporting Information Available: Experimental procedure and spectroscopic data (1H, 13C, LC/MS and HRMS) are listed for representative compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References and notes
- 1.http://www.who.int/tdr/diseases/chagas/direction.htm
- 2.Mackey ZB, OBrien TC, Greenbaum DC, Blank RB, McKerrow JH. A cathepsin B-like protease Is required for host protein degradation in Trypanosoma brucei. J Bio Chem. 2004;279:48426–48433. doi: 10.1074/jbc.M402470200. [DOI] [PubMed] [Google Scholar]
- 3.Mallari JP, Shelat AA, Obrien T, Caffrey CR, Kosinksi A, Connelly M, Harbut M, Greenbaum D, McKerrow JH, Guy RK. Development of potent purine-derived nitrile inhibitors of the trypanosomal protease TbcatB. J Med Chem. 2008;51:545–552. doi: 10.1021/jm070760l. [DOI] [PubMed] [Google Scholar]
- 4.Barr SC, Warner KL, Kornreic BC, Pisctelli J, Wolfe A, Benet L, McKerrow JH. A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob Agents Chemother. 2005;49:5160–5161. doi: 10.1128/AAC.49.12.5160-5161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brak K, Doyle PS, McKerrow JH, Ellman JA. Identification of a new class of nonpeptidic inhibitors of cruzain. J Am Chem Soc. 2008;130:6404–6410. doi: 10.1021/ja710254m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kraus JM, Verlinde CLJ, Karimi M, Lepesheva GI, Gelb MH, Buckner FS. Rational modification of a candidate cancer drug for use against Chagas disease. J Med Chem. 2009;52:1639–1647. doi: 10.1021/jm801313t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: A titration based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci USA. 2006:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doyle PS, Zhou YM, Engel JC, McKerrow JH. A cysteine protease inhibitor cures Chagas disease in an immunodeficient-mouse model of infection. Antimicrob Agents Chemother. 2007;51:3932–3939. doi: 10.1128/AAC.00436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobsen W, Christians U, Benet LZ. In vitro evaluation of the disposition of a novel cysteine protease inhibitor. Drug Metab Dispos. 2000;28:1343–1351. [PubMed] [Google Scholar]
- 10.Engel JC, Doyle PS, Hsieh I, McKerrow JH. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J Exp Med. 1998;188:725–734. doi: 10.1084/jem.188.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Roush WR, Gwaltney SL, II, Cheng J, Scheidt KA, McKerrow JH, Hansell E. Vinyl sulfonate esters and vinyl sulfonamides: potent, irreversible inhibitors of cysteine proteases. J Am Chem Soc. 1998;120:10994–10995. [Google Scholar]; b) Roush WR, Cheng J, Knapp-Reed B, Alvarez-Hernandez A, McKerrow JH, Hansell E, Engel JC. Potent second generation vinyl sulfonamide inhibitors of the typanosomal cysteine protease cruzain. Bioorg Med Chem Lett. 2001;11:2759–2762. doi: 10.1016/s0960-894x(01)00566-2. [DOI] [PubMed] [Google Scholar]
- 12.K11777 -. This is despite the fact that compound 1 has not shown significant off-target side effects to date.
- 13.Hickey ER, Bekkali Y, Patel UR, Spero DM, Thomson DS, Young ERR. Nitriles useful as reversible inhibitors of cysteine proteases. 2006;6:263. [Google Scholar]
- 14.Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 15.Jadhav A, Ferreira RS, Klumpp C, Mott BT, Austin CP, Inglese J, Thomas CJ, Maloney DJ, Shoichet BK, Simeonov A. Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Arifacts in a Screen for Inhibitors of a Thiol Protease. J Med Chem. doi: 10.1021/jm901070c. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 17.Bhatt R, Gong B, Hong F, Jenkins SA, Klein PJ, Kumar AM, Tulinsky J. 6-phenyl-N-phenyl-(1,3,5)-triazine-2,4-diamine derivatives and related compounds with lysophphosphatidic acid acyltransferase beta (LPAAT-BETA) inhibitory activity for use in the treatment of cancer. 2003 WO 03037346/2003. [Google Scholar]
- 18.Kukla MJ, Ludovici DW, Kavash RW, De Corte BLD, Heeres J, Janssen PAJ, Koymans LMH, de Jonge MR, Van Aken A, Krief A, Gerardus R, Leenders G. HIV replication inhibitors. 2003;0:171–374. [Google Scholar]
- 19.Bailey A, Pairaudeau G, Patel A, Thom S. Use of pyrimidine - or triazine - 2 carbonitriles for treating diseases associated with cysteine protease activity and novel pyrimidine-2-carbonitrile derivatives. 2006;7:125–881. [Google Scholar]
- 20.Rankovic Z, Cai J, Cumming I. 2-cyano-1,3,5-triazine-4,6-diamine derivatives. 2005 WO 2005/011703. [Google Scholar]
- 21.Oballa RM, Truchon JF, Bayly CI, Chauret N, Day S, Crane S, Berthelette C. A generally applicable method for assesing the electrophilicity and reactivity of diverse nitrile-containing compounds. Bioorg Med Chem Lett. 2007;17:998–1002. doi: 10.1016/j.bmcl.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 22.Altmann E, Cowan-Jacob SW, Missbach M. Novel purine nitrile derived inhibitors of the cysteine protease cathepsin K. J Med Chem. 2004;47:5833–5836. doi: 10.1021/jm0493111. [DOI] [PubMed] [Google Scholar]
- 23.Bakkestuen AK, Gundersen LL. Regioselective N-9 Arylation of purines employing arylboronic acids in the presence of Cu(II) Tetrahedron Lett. 2003;44:3359–3362. [Google Scholar]
- 24.Abdulla MH, O’Brien T, Mackey ZB, Sajid M, Grab DJ, McKerrow JH. RNA Interference of Trypanosoma brucei Cathepsin B and L Affects Disease Progression in a Mouse Model. PLoS Negl Trop Dis. 2008;2:e298. doi: 10.1371/journal.pntd.0000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLano WL. The Pymol Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 26.Ménard R, Carriere J, Laflamme P, Plouffe C, Khouri HE, Vernet T, Tessier DC, Thomas DY, Storer AC. Contribution of the glutamine 19 side chain to transition-state stabilization in the oxyanion hole of papain. Biochemistry. 1991;30:8924–8928. doi: 10.1021/bi00101a002. [DOI] [PubMed] [Google Scholar]
- 27.Despite the potent inhibition of several cathepsins (notably, Cat C, L, and K), these analogues were essentially inactive across 60 relevant human cysteine proteases.
- 28.McKerrow JH. Development of cysteine protease inhibitors as chemotherapy for parasitic diseases: insights on safety, target validation, and mechanism of action. Int J Parasitol. 1999;29:833–837. doi: 10.1016/s0020-7519(99)00044-2. [DOI] [PubMed] [Google Scholar]
- 29.Yasgar A, Shinn P, Michael S, Zheng W, Jadhav A, Auld DS, Austin CP, Inglese J, Simeonov A. Compound management for quantitative high-throughput screening. J Assoc Lab Automat. 2008;13:79–89. doi: 10.1016/j.jala.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eakin AE, McGrath ME, McKerrow JH, Fletterick RJ, Craik CS. Production of crystallizable cruzain, the major cysteine protease from Trypanosoma cruzi. J Biol Chem. 1993;268:6115–6118. [PubMed] [Google Scholar]
- 31.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr, Sect D: Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 32.Huang L, Brinen LS, Ellman JA. Crystal structures of reversible ketone-based inhibitors of the cysteine protease cruzain. Bioorg Med Chem. 2003;11:21–29. doi: 10.1016/s0968-0896(02)00427-3. [DOI] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.