

Abstract
TGFβ1 is a polypeptide growth modulatory and differentiation factor involved in many biological processes including immune homeostasis and self-tolerance. Tgfb1 knockout mice die around weaning age due to severe inflammation in most major organ systems, but the mechanism underlying this disease is not understood. In this study we demonstrate that Tgfb1−/− CD4+CD8+ and CD4+CD8− thymocytes are hyperresponsive to receptor-mediated and receptor-independent mitogenic stimulation. A suboptimal concentration of ionomycin in the presence of PMA fully activates Tgfb1−/− thymocytes, whereas the inhibitors of Ca2+ influx and calcineurin, EGTA and FK506, eliminate the hyperresponsiveness. Hence, the hypersensitivity of Tgfb1−/− thymocytes is due to a lowered threshold for Ca2+-dependent activation. Further, we demonstrate that the hypersensitivity of thymocytes results from the absence of TGFβ1 and not from the inflammatory environment because the thymocytes are hyperresponsive in preinflammatory-stage Tgfb1−/− mice. Our results suggest for the first time that TGFβ1 functions to inhibit aberrant T cell expansion by maintaining intracellular calcium concentration levels low enough to prevent a mitogenic response by Ca2+-independent stimulatory pathways alone. Consequently, TGFβ1 prevents autoimmune disease through a Ca2+ regulatory pathway that maintains the activation threshold above that inducible by self-MHC-TCR interactions.
Transforming growth factor β1 is a polypeptide growth regulatory factor that functions both during development and in the adult by affecting cell differentiation, growth, apoptosis, adhesion, and immune response (1–5). Tgfb1 knockout mice have defects in preimplantation development (6), yolk sac development (7), tooth development (8), genetic stability (9, 10), platelet activation (11), and cancer (9, 12–14). In the immune system, TGFβ1 plays an important role in regulating inflammation (15, 16), induction of self- and oral-tolerance (17–19), and auto-immunity (20). TGFβ1 has been shown to induce Ag-specific unresponsiveness in naive T cells (21), and TGFβ1-secreting Th3 type regulatory T cells and CD8+ suppressor T cells maintain immune homeostasis and inhibition of autoimmune disorders (22–24). TGFβ1 is secreted by T cells that are activated by IL-2 or PHA-P/PMA (25), by cross-linking of CTLA-4, and by activation-induced cell death, suggesting an involvement in lymphocyte homeostasis (26, 27). Fetal thymic organ culture studies have demonstrated that TGFβ1 and TGFβ2 inhibit growth and induce differentiation of thymocytes (28). In vitro studies also have shown that TGFβ1 can act as a positive as well as a negative regulator of lymphocyte proliferation and apoptosis (29, 30).
Tgfb1 knockout mice develop severe inflammatory lesions in multiple organs and die within 3 wk after birth (15, 16). Anti-LFA-1 Ab (31) and fibronectin peptides (32) rescue these mice from inflammatory disorders, suggesting that either extravasation or T cell activation is affected. T lymphocytes are the primary effectors in the pathological phenomenon in these mice as Tgfb1−/− Scid (31), Tgfb1−/− Rag2−/− (14), Tgfb1−/− Rag1−/− (33), and Tgfb1−/− athymic nude mice (T. Doetschman, unpublished observations) survive months longer than do immunocompetent Tgfb1−/− mice. A study using mice expressing dominant-negative TGFβ receptor type II transgenes under T cell-specific promoters revealed that T cells are hyperresponsive, acquire a memory phenotype, generate autoantibodies, and produce inflammatory lesions in lung and colon tissues (34). However, another study found that only CD8+ T cells are hyperresponsive in dominant-negative TGFβ receptor type II transgenic mice (35). Also, TGFβ1 can inhibit differentiation of T cells into the Th2 phenotype by inhibiting IL-4 induction (36). Breeding of Tgfb1−/− mice onto an Ifnγ−/− background but not onto an Il4−/− background enhances the life span by 3–4 wk and eliminates necroinflammatory hepatitis (37). Finally, Tgfb1−/− mice on a MHC class I-deficient background (β2-microglobulin null mice) live much longer than on a class II-deficient background, suggesting that abnormal expression and presentation of self-Ags by class I molecules to CD8+ T cells may also contribute to the inflammatory phenotype (38).
Complicating these studies is the possibility that the inflammatory disorder may have secondary effects that are independent of or synergistic with the absence of TGFβ1. For example, in Tgfb1 knockout mice inflammatory cytokine levels are increased (15), the percentage of CD4+ single-positive (SP)3 thymocytes increases 7-fold, and hyperproliferation leads to enlarged spleens and lymph nodes (39). However, these are common inflammatory stress responses. Consequently, it is not clear how TGFβ1-deficient lymphocytes would behave in the absence of inflammatory stress. To avoid these complications we have investigated thymocyte function in very young Tgfb1−/− mice. We have utilized PMA and ionomycin to stimulate thymocytes in a TCR- and APC-independent manner (40, 41). Our results indicate that thymocytes from mice with no inflammatory load are hyperresponsive to calcium-calcineurin-mediated mitogenic stimuli.
Materials and Methods
Mice
Tgfb1−/− mice (129 × CF-1) were generated as described (15). These mice were bred onto BALB/c, C3H, and 129 backgrounds. Tgfb1 heterozygous male and female mice were housed in a specific pathogen-free mouse facility at the University of Cincinnati Medical Center.
Reagents
RPMI 1640 medium, AIM-V medium, Dulbecco’s PBS, and HBSS were purchased from Life Technologies (Rockville, MD). RBC lysing buffer, trypan blue, Con A, ionomycin, PMA, rapamycin, EGTA, and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). FK506 was purchased from Biomol (Plymouth Meeting, PA). Indo-1 acetoxmethyl ester (Indo-1-AM) and NF-κB SN50 (cell-permeable inhibitor peptide) were purchased from Calbiochem (La Jolla, CA). Pluronic F-127 was purchased from Molecular Probes (Eugene, OR). Paraformaldehyde was purchased from Electron Microscopy Sciences (Fort Washington, PA). [methyl-3H]Thymidine (sp. act., 6.7 Ci/mmol) was purchased from NEN (Boston, MA). FALCON flat-bottom 96-well Microtest Primaria tissue culture plates and 24-well Multiwell tissue culture plates were purchased from BD Biosciences (Franklin Lakes, NJ). FK506 (200 nM), rapamycin (100 nM), PMA (10 μM), and ionomycin (100 μM) stocks were prepared in DMSO, aliquoted, and stored at −80°C in a freezer.
Abs and staining kits
Purified anti-mouse CD3ε, anti-mouse CD28, anti-mouse CD16/CD32 (FcγRIII/II), FITC anti-mouse CD3ε, FITC anti-mouse CD4 (L3T4), Cy-Chrome anti-mouse CD4, R-PE anti-mouse CD8α (Ly-2), fluorochrome-conjugated isotype control Abs, Annexin V FITC apoptosis kit, Annexin V PE apoptosis kit, and 5-bromo-2′-deoxyuridine (BrdU) Flow kit were purchased from BD PharMingen (San Diego, CA).
PCR genotyping
Genotype of the newborn pups from heterozygous matings was determined by PCR amplification of tail DNA and size fractionation on agarose gels (14).
Inflammation index
After the thymus was removed for in vitro studies, Tgfb1 null mice were fixed in 10% neutral-buffered formalin. Tissue sections were scored for inflammatory lesions with a severity scale from 0 to 4. Although inflammation in 25–30 organs was measured, only 8–12 organs were found to be the most severely affected (15, 39). The sum of scores for all organs was divided by the number of organs analyzed to generate an inflammation index. Mice severely affected by multifocal inflammation had inflammation indices ranging from 0.5 to 1.0 (3 wk old), whereas wild-type mice had no inflammation. Tgfb1−/− mice up to 1 wk of age had inflammation indices ranging from 0 to <0.1. Inflammation indices for older mice are given in the figures.
Preparation of thymocytes
Newborn pups and young mice (<3 wk old) were sacrificed by isoflurane overdose and thymii were dissected out aseptically into a petri dish containing RPMI 1640 medium. Tissue was mechanically separated into individual cells with a syringe and 22-gauge needle. Cells were centrifuged at 1000 rpm for 5 min and RBCs were depleted with RBC lysing buffer. The cells were washed once with RPMI 1640 medium and were suspended in Aim-V serum-free medium. Viable cells were counted using trypan blue dye exclusion as an indicator. Cells were resuspended at 2 × 106/ml in Aim-V serum-free medium for all in vitro culture studies. Thymocytes were prepared in Dulbecco’s PBS or HBSS for staining and flow cytometry.
Phenotype analysis of thymocytes
One million thymocytes from each mouse were stained on ice for 30 min for CD3, CD4, and CD8 expression using fluorochrome-conjugated mAbs in the presence of 10% FBS and Fc blocking Ab. After the cells were washed once they were resuspended in HBSS containing 0.09% NaN3 and analyzed in Beckman Coulter Epics XL flow cytometer (Fullerton, CA), FACSCaliber, or BD-LSR flow cytometers (BD Biosciences). Side and forward angle light scattering was used to electronically gate the cells of choice and exclude debris. Some 10,000 events within the gate region were collected for each sample. Fluorochrome-conjugated isotype control Abs were used to control for the nonspecific binding. Cells stained with each Ab individually were used to set compensation networks for each fluorochrome. In all experiments, control cells were taken from either Tgfb1+/+ littermates or Tgfb1+/− littermates when a wild-type littermate was not available. Data were analyzed by System II or CellQuest software (BD Biosciences). Cells were stained for apoptosis using annexin V according to the manufacturer’s protocol.
Assessment of thymocyte proliferation
A total of 2 × 105 thymocytes were cultured in 200 μl Aim-V serum-free medium in the presence of various concentrations of mitogens in triplicate in FALCON flat-bottom 96-well Microtest Primaria tissue culture plates (BD Biosciences). After 3 days of in vitro culture at 37°C and 5% CO2, cultures were pulsed with 0.5 μCi of tritiated thymidine (NEN) for a period of 14–16 h, cells were harvested onto glass fiber filters, and radioactivity was counted in a Beckman Coulter scintillation counter. Data are represented as mean dpm from triplicate cultures ±SD. Background incorporation of unstimulated cultures was always <600 dpm. Optimization of cell number, mitogen concentration, and serum replacements were conducted in the preliminary studies. The optimum concentration of Con A was 0.5 μg/ml; anti-CD3 Ab, 0.5 μg/ml; PMA, 1 nM; and ionomycin, 250 nM in Aim-V serum-free medium supplemented with L-glutamine, streptomycin sulfate, gentamicin sulfate, and HSA. For phenotype analysis of hyperresponsive thymocytes, 2 × 106 cells were cultured in 2 ml Aim-V serum-free medium in the presence of mitogens in 24-well Multiwell tissue culture plates at 37°C and 5% CO2 and were pulsed with 10 μM BrdU for the last 12–14 h. Cultures were harvested after 2 days of culture and were stained for surface Ags and incorporated BrdU according to the manufacturer’s protocol.
Analysis of intracellular calcium concentration [Ca2+]i levels by flow cytometry
A total of 1 × 107/ml day 3–11 thymocytes in cell-loading medium (CLM; HBSS-containing Ca2+ and Mg2+, 1% FBS) were loaded with 10 μM Indo-1-AM (stock solution; 2 mg/ml dissolved in Pluronic F-127) for 40 min at 30°C (42). Cells were washed three times with CLM and resuspended in CLM, surface stained with azide-free fluorochrome-conjugated anti-CD4 and anti-CD8 Abs for 20 min on ice, washed once with CLM, and left on ice until analyzed. Cells were prewarmed at 37°C for 10 min before acquiring for Ca2+ measurements. Cells were acquired for 30 s before adding the agonist. For each analysis, cells were acquired for 512 s at a flow rate of 300–400 events per second in a BD-LSR FACS machine equipped with UV lasers. Ratio metric analyses of Ca2+-bound Indo-1 (FL5)/Ca2+-free Indo-1 (FL4) were done by CellQuest (BD Biosciences) and Flow Jo (Tree Star, San Carlos, CA) software programs. Basal [Ca2+]i levels and changes in [Ca2+]i levels in CD4+CD8−, CD4−CD8+ and CD4+CD8+ subpopulations were analyzed after gating on these cells. Based on the concentration of [Ca2+]i levels after acquiring each sample for 512 s, cells were further grouped into low (M1), intermediate (M2), medium high (M3), and high (M4) [Ca2+]i cells (43).
Statistical analysis
Significance of the differences between wild-type and mutant mice responses was calculated using Student’s t test.
Results
Normal thymic phenotype in young Tgfb1−/− mice
The total number and percent of SP and double-positive (DP) cell populations in the thymus of young Tgfb1−/− mice (postnatal day 3–7) is not different from those of Tgfb1−/− mice (Fig. 1, A and B). This suggests that there is no obvious defect in the selection and maturation of thymocytes in Tgfb1−/− mice. However, in older mice (2–3 wk old) we have also noticed a decrease in the DP population and an increase in both CD4−CD8− double-negative and CD4+ SP cells (39). Older mice also show increased apoptosis similar to a corticosteroid-induced stress response (data not shown).
FIGURE 1.

Normal thymic phenotype in young Tgfb1−/− mice. A, Total number of cells from thymii of three to five mice for each age (days) represented with error bars as mean ± SD. B, Phenotypic analysis of thymocytes. Thymocytes from day 7 mice were stained with fluorochrome-labeled anti-CD4 and anti-CD8 Abs as described in Materials and Methods. Numbers in each quadrant represent the percentage of cells positive for the indicated surface Ag(s).
Thymocytes are hyperresponsive to Ca2+-mediated mitogenic stimulus
Engagement of TCR by anti-CD3 or Con A results in activation of T cells and mitogenesis. However, immature T cells undergo apoptosis instead of mitogenesis upon TCR stimulation. Consequently, the mitogenic response of APC-dependent, receptor-mediated activation of postnatal day 7–17 Tgfb1−/− thymocytes by Con A suggests that they are immature. However, Tgfb1−/− thymocytes are relatively hyperresponsive (Fig. 2A). Stimulation of thymocytes from day 5 pups by optimal concentrations of PMA plus ionomycin results in activation and proliferation of both wild-type and mutant thymocytes, but at suboptimal concentrations only mutant cells are responsive (Fig. 2B). Either PMA or ionomycin is not mitogenic on its own but both of them act synergistically and induce mitogenic response in lymphocytes (data not shown and Fig. 2B).
FIGURE 2.

Tgfb1−/− thymocytes are hyperresponsive to mitogenic stimulation. Thymocytes from day 3–7 mice were cultured either in the presence of Con A (A), PMA + ionomycin(B), anti-CD3 + anti-CD28(C), anti-CD28 + PMA (D), or anti-CD3 + PMA (E) and pulsed with [3H]thymidine as described in Materials and Methods. Legend and ordinate within D and E are the same as that in C. Data are presented as mean value of dpm from triplicate wells and error bars represent ±SD. Similar responses are observed in all day 3–21 mice tested, independent of severity of inflammation (p for all mice from day 3–21 is <0.0005 for PMA + 125 nM ionomycin stimulation, <0.02 for anti-CD3 + PMA stimulation, and <0.05 for anti-CD3 + anti-CD28 and anti-CD28 + PMA stimulations).
Hyperresponsiveness of thymocytes to anti-CD3 plus anti-CD28 stimulation
To address whether dysregulation of [Ca2+]i mobilization causes the hyperresponsiveness of Tgfb1−/− thymocytes, cells were treated with anti-CD3 plus anti-CD28 Abs. Treatment with anti-CD3 causes elevation of [Ca2+]i and activation of protein kinase pathways resulting in activation and nuclear translocation of NF-ATc, NF-κB, AP-1, and other transcription factors involved in lymphokine gene transcription. Treatment with anti-CD28 provides a costimulatory signal resulting in activation of phosphatidylinositol 3 kinase and protein kinase pathways such as mitogen-activated protein kinase, c-Jun N-terminal kinase, and protein kinase C (PKC). Thymocytes from day 3 Tgfb1+/+ mice do not respond to either anti-CD3 alone or in combination with anti-CD28 because they are immature cells. However, thymocytes from age-matched Tgfb1−/− mice respond to anti-CD3 plus anti-CD28 stimulation, suggesting that these cells are inherently hyperresponsive (Fig. 2C).
To address whether altered [Ca2+]i levels cause the hyperresponsiveness in Tgfb1−/− thymocytes, cells were next treated with PMA and anti-CD28. Treatment with PMA results in activation of the PKC pathway, which in turn activates NF-κB but does not elevate [Ca2+]i. Hence, PMA plus anti-CD28 stimulate lymphocytes in a Ca2+-calmodulin-independent manner. Without additional stimulation through the Ca2+-calmodulin pathway, normal lymphocytes will not proliferate in response to either PMA, anti-CD28, or both (44, 45). As expected, treatment of Tgfb1+/+ thymocytes with PMA plus anti-CD28 does not result in a mitogenic response, whereas Tgfb1−/− thymocytes do respond to the same mitogenic stimulation (Fig. 2D). The data also indicate that the Ca2+-calmodulin-calcineurin signaling pathway may be up-regulated and in synergy with PMA and anti-CD28 signaling. Similar responses are also observed with anti-CD3 plus PMA treatments (Fig. 2E), although it must be noted that the magnitude of mitogenesis in response to PMA plus ionomycin is much larger than that achievable by any other mitogenic regimen tested. Together, these results suggest that Tgfb1−/− thymocytes are inherently hyperresponsive to mitogenic stimulation.
Our results on preinflammatory-stage Tgfb1−/− thymocytes demonstrate that mitogenic responses to Con A, anti-CD3 plus anti-CD28, and PMA plus ionomycin are clearly altered. Consequently, the hypersensitivity of Tgfb1−/− thymocytes to the suboptimal concentration of ionomycin (125 nM) in the presence of the optimal concentration of PMA (1 nM) is due to the absence of TGFβ1 and not due to the inflammatory environment.
TGFβ1 inhibits Ca2+-calcineurin-mediated thymocyte activation
The mitogenic response of thymocytes is dependent, in part, on the mobilization and elevation of [Ca2+]i, which leads to calcineurin activation, nuclear translocation of dephosphorylated NF-ATc, and transcriptional activation of IL-2. Our PMA plus ionomycin experiments suggest that the Tgfb1−/− thymocyte response could be due to elevated [Ca2+]i levels. To determine whether TGFβ1 alters the [Ca2+]i levels and/or calcineurin activation, we have tested the effects of EGTA and FK506, which block Ca2+ mobilization and NF-ATc activation, respectively. If the absence of TGFβ1 leads to high [Ca2+]i levels, then Tgfb1−/− thymocytes should require more EGTA or FK506 to reach the same level of inhibition as in wild-type cells.
Thymocytes from Tgfb1−/− mice are less sensitive to FK506, indicating increased NF-ATc activation. The proliferation of wild-type cells stimulated with 125 nM or 250 nM ionomycin, plus 1 nM PMA, is completely blocked by 0.1 nM FK506. Mutant thy-mocyte proliferation, on the other hand, is not completely blocked by 0.1 nM FK506 when stimulated by 250 nM ionomycin plus 1 nM PMA (Fig. 3A). FK506 (1.0 nM) completely blocks 250 nM ionomycin-stimulated mitogenesis of both cell types in the presence of 1 nM PMA. Hyperresponsiveness of Tgfb1−/− thymocytes to 125 nM ionomycin plus 1 nM PMA is also inhibited by EGTA (Fig. 3B), indicating increased Ca2+ mobilization. However, 0.4 mM EGTA does not completely abolish the mitogenic response of 125 nM ionomycin plus 1 nM PMA-stimulated mutant thymocytes because it is still ~60-fold higher than a background at ~140 dpm. On the other hand, 0.4 mM EGTA completely inhibits the mitogenic response of 125 nM ionomycin plus 1 nM PMA-stimulated wild-type cells because the response is not above background levels. At 0.8 mM EGTA, the mitogenic response is not above background levels either in mutant or wild-type mice. Likewise, 1.0 nM FK506 reverses the hyperresponsiveness of Tgfb1−/− thymocytes to stimulation by PMA plus anti-CD28 (Fig. 3C). Together, these results suggest that the Ca2+-calcineurin pathway could be one of the primary targets of TGFβ1 function in thymocytes.
FIGURE 3.

Thymocytes from Tgfb1−/− mice are less sensitive to FK506 and EGTA, and equally sensitive to rapamycin and SN50. Thymocytes from day 5 mice (A and C), day 13 mice (B), or day 10 mice (D) were cultured in the presence of indicated concentrations of PMA + ionomycin with or without FK506 (A), PMA + ionomycin with or without EGTA (B), anti-CD28 + PMA with or without FK506 (C), or PMA + ionomycin with or without rapamycin or NF-κB inhibitor SN50 (D) and pulsed with [3H]thymidine as described in Materials and Methods. Inflammation indices: 0 for the day 5 Tgfb1−/− mouse (A and C), and 0.18 for the day 10 Tgfb1−/− mouse (D). Inflammation index for the day 13 mouse used in B was not determined, but average index for four other day 13 mice was 0.18 ± 0.15. For comparison, inflammation index for the 50% of Tgfb1−/− mice that have not yet died by day 21 from severe inflammation ranges from 0.5 to 1.0. Data are presented as mean value of dpm from triplicate wells and error bars represent ±SD. Control cultures were included and either received one of the mitogens or medium only. A value of p < 0.01 for 0.1 nM FK506 mediated inhibition of 1 nM PMA + 250 nM ionomycin stimulation of thymocytes from six mutant mice compared with age-matched littermate control mice.
To determine whether TGFβ1 specifically regulates calcineurin rather than p70 S6 kinase- or PKC-mediated pathways, we treated thymocytes with rapamycin or NF-κB SN50 and stimulated with 1 nM PMA and 250 nM ionomycin. Rapamycin and NF-κB SN50 inhibit p70S6 kinase- and PKC-mediated signaling, respectively. Thymocytes from both Tgfb1+/− and Tgfb1−/− mice are equally sensitive to rapamycin and NF-κB SN50 (Fig. 3D), demonstrating that TGFβ1 provides a check on thymocyte activation by inhibiting Ca2+-calcineurin signaling rather than p70 S6 kinase or PKC signaling.
Thymocyte [Ca2+]i levels altered in Tgfb1−/− mice
To confirm that TGFβ1 regulates calcium-calcineurin signaling, we directly measured thymocyte [Ca2+]i levels and changes in [Ca2+]i levels upon stimulation. Basal [Ca2+]i levels are higher in mutant thymocytes, and the [Ca2+]i levels increase with age of the mice (Fig. 4A, compare top panels with bottom panels) and as the cells mature from DP stage to SP stage (Fig. 4A, compare right panels (CD4+CD8+DP thymocytes) with left (CD4+CD8−SP) and middle (CD4−CD8+SP) panels in the same row). Percentages of cells in the M2 region of Tgfb1+/+ and Tgfb1−/− mice also suggest that there is a 2-fold increase in the number of cells with elevated [Ca2+]i in Tgfb1−/− mice. Numbers in the histograms of Fig. 4A represent the percentage of cells in the M2 region. Stimulation of thymocytes with anti-CD3 results in elevation of [Ca2+]i, which is more evident in CD4+CD8− SP than in CD4+CD8+ DP thymocytes. There are noticeable differences in percentages of cells in gates M1 and M2 between Tgfb1+/+ and Tgfb1−/− CD4+CD8− SP thymocytes. However, there are no differences in DP thymocytes (Fig. 4B, compare top left panel with bottom left panel). Stimulation with a suboptimal concentration of ionomycin (100 nM) leads to elevation of [Ca2+]i to a greater extent as against anti-CD3 stimulation and is more elevated in mutant than wild-type thymocytes (note the rightward shift of the red tracing relative to the green in Fig. 4B, right panels).
FIGURE 4.

[Ca2+]i levels are higher in Tgfb1−/− thymocytes. Fluorescence ratio of Ca2+-bound Indo-1 (FL5)/Ca2+-free Indo-1 (FL4) was measured as described in Materials and Methods. A, Overlays of FL5/FL4 vs cell-count histograms of Tgfb1+/+ (filled) and Tgfb1−/− (open) CD4+ SP thymocytes (left panels), CD8+SP thymocytes (middle panels), CD4+CD8+DP thymocytes (right panels), from day 3 (upper panels) and day 8 (lower panels) mice. Percentages of cells in M2 gate are shown. B, Overlays of FL5/FL4 vs cell-count histograms of day 7 Tgfb1+/+ (filled) and Tgfb1−/− (open) CD4+ SP thymocytes (upper panels) and CD4+CD8+DP thymocytes (lower panels) of anti-CD3-stimulated, 20 μg/ml (left panels) and ionomycin-stimulated, 100 nM (right panels). C, Overlays of mean FL5/FL4 ([Ca2+]i levels) vs time of Tgfb1+/+ (red) and Tgfb1−/− (blue) CD4+CD8+DP thymocytes stimulated with ionomycin 100 nM (upper panel) or anti-CD3 (lower panel). (D) Overlays of mean FL5/FL4 ratio ([Ca2+]i levels) vs time of Tgfb1+/+ (red) and Tgfb1−/− (blue) CD4+CD8− SP thymocytes stimulated with anti-CD3 (upper panel) or anti-CD28, 2 μg/ml (lower panel). Note that upon stimulation with anti-CD3, the magnitude of increase in [Ca2+]i levels in DP thymocytes (C, lower panel) is lower than the CD4+ SP thymocytes (D, upper panel). Arrow indicates the time of addition of agonist in C and D. Genotype of Tgfb1+/+ (+/+) and Tgfb1−/− (−/−) mice are shown.
Upon anti-CD3-stimulation, increase in [Ca2+]i levels in DP thymocytes of both wild-type and mutant mice is not significantly different, and the magnitude of [Ca2+]i levels is much lower (mean ratio of 180, Fig. 4C, bottom panel) than in mature CD4+CD8− thymocytes (mean ratio of 260, Fig. 4D, upper panel). As expected, anti-CD3-induced elevation in [Ca2+]i levels in mature CD4+CD8− thymocytes is higher in mutant mice (mean ratio of Tgfb1−/− SP thymocytes is 20% more than Tgfb1+/+ SP thymocytes upon stimulation with anti-CD3, Fig. 4D, upper panel), and anti-CD28 and PMA do not alter [Ca2+]i levels in mutant or wild-type thymocytes (Fig. 4D, PMA data not shown). Hence, lack of sufficient elevation in [Ca2+]i levels explains why the immature thymocytes do not respond to receptor-mediated mitogenic stimuli in wild-type mice.
Phenotype of hyperresponsive thymocytes in Tgfb1−/− mice
The thymus is composed of mainly DP cells (85–90%) and a small proportion of mature SP cells and double-negative T cell precursors. Based on the thymidine incorporation data previously presented, it is reasonable to conclude that DP cells are the primary effectors in the hyperresponsive cultures because there would not be enough SP cells to yield such a massive proliferation seen at suboptimal concentrations of ionomycin in the presence of PMA. Upon stimulation, thymocytes down-regulate one of their coreceptors and mature into either CD4+ SP or CD8+ SP cells (46). Mutant, but not wild-type cells, undergo mitogenesis in response to a suboptimal concentration of ionomycin in the presence of PMA in day 9 mice (Fig. 5A). This again confirms that DP thymocytes respond to suboptimal mitogenic stimulus and differentiate to CD4+ SP cells in Tgfb1−/− mice (Fig. 5B). A similar response is observed in day 16 Tgfb1−/− mice, except that DP thymocytes differentiate to both CD4+ SP and CD8+ SP cells (data not shown). This suggests that CD4+ SP cells are the initiators of inflammation and assist in the generation of effector CD8+ SP cells in Tgfb1−/− mice. These data also suggest that only a portion (10–20%) of thymocytes proliferate in response to mitogenic stimulus even at optimal concentrations of PMA and ionomycin. The majority of thymocytes undergo apoptosis in both wild-type and mutant thymocyte cultures upon stimulation with PMA plus ionomycin (data not shown). Hence, the differences in mitogenic responses observed between wild-type and mutant mice is actually due to the presence of a small percentage of hyperresponsive thymocytes in mutant mice.
FIGURE 5.
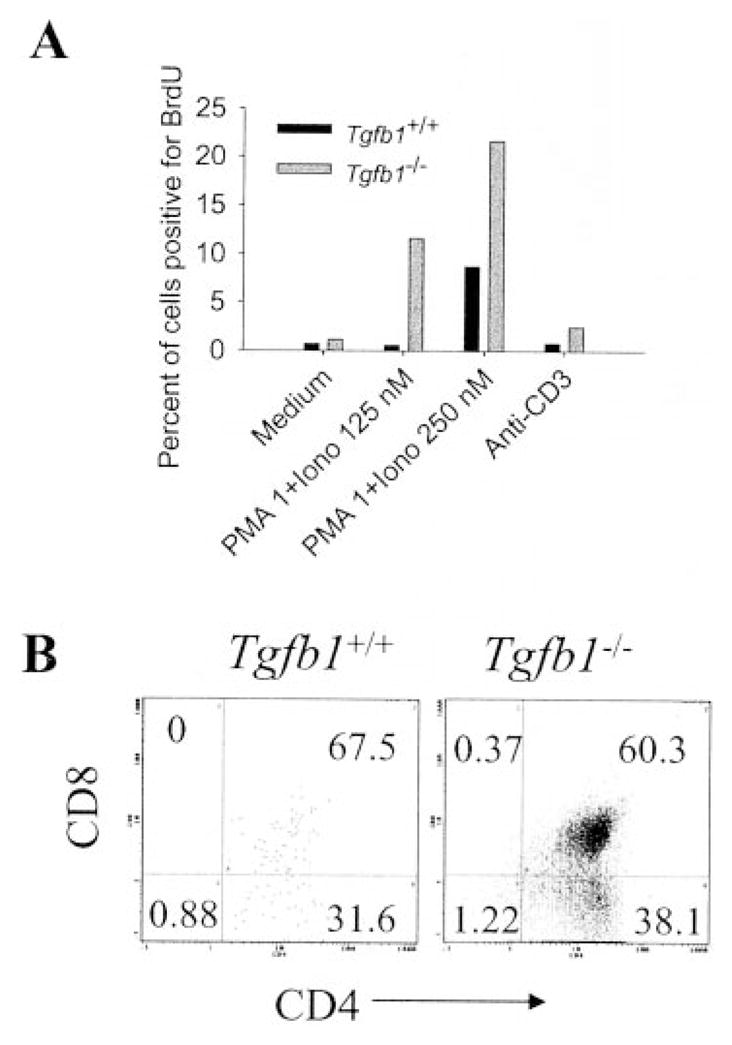
CD4+ SP Tgfb1−/− thymocytes initiate hyperresponsive phenotype. Thymocytes from day 9 mice were cultured in the presence of PMA plus ionomycin, anti-CD3 or medium, pulsed with the thymidine analog BrdU, and stained for surface Ags and BrdU. Percentage of cells positive for BrdU (A) and phenotype of 1 nM PMA plus 125 nM ionomycin-stimulated, BrdU-positive cells (B) was determined by FACS analysis as described in Materials and Methods. Note that both the immature DP and mature CD4+ SP thymocytes are hyperresponsive.
Discussion
Tgfb1−/− mice develop severe inflammatory lesions in multiple organs and die around 3 wk of age. Backcrossing these mice onto immunodeficient backgrounds has revealed that T cells are the primary effectors in the inflammatory phenotype of Tgfb1−/− mice. It is clear that there is a positive relationship between age and the degree of inflammation in Tgfb1−/− mice (39), and that many phenotypes, such as epithelial cell proliferation and ICAM and MHC class II up-regulation, are eliminated if the inflammation is removed by genetically combining Tgfb1−/− and Scid mice (31). The present study demonstrates that the distribution of thymocyte subsets is not affected up to 1 wk after birth. Our proliferation studies unequivocally demonstrate for the first time that Tgfb1−/− thymocytes are inherently hyperresponsive to a suboptimal concentration of ionomycin plus PMA, irrespective of the age or inflammation index of the mice. They are also hyperresponsive to other forms of mitogenic stimulation such as Con A and anti-CD3 plus anti-CD28. However at ages ≥2 wk, the inflammation-induced stress response makes it difficult to interpret whether the observed decrease in thymic cellularity results from the presence of inflammation or the absence of TGFβ1. Tgfb1−/− thymocytes are also hyperresponsive to Ca2+-independent (PMA plus anti-CD28) mitogenic stimulus. Because Tgfb1+/+ thymocytes do not respond well to stimulation by PMA plus anti-CD28 or to suboptimal concentrations of ionomycin plus PMA, TGFβ1 could be acting on the [Ca2+]i channels through the FK506 binding protein, FKBP12, and calcineurin.
FKBP12 is known to be released through association with the TGFβ type I receptor upon ligand binding (47) and to recruit calcineurin to the inositol 1,4,5-triphosphate (IP3) receptor complex (48), where together they are required for fully functional IP3 receptor activity (49, 50). That FKBP12 can function in TGFβ signaling is strongly suggested by the fact that Fkbp12 knockout mice have cardiomyocyte defects that involve abnormal Ca2+ channel function (51), and Fkbp12 and Tgfb2 knockout mice have similar morphological defects in the developing heart (51, 52). However, it is still unclear which isoform of FKBP is involved in TGFβ signaling in T cells (53–56). In the absence of TGFβ1, there could be disequilibria of TGFβ type I receptor-associated FKBPs, resulting in leaky Ca2+ release channels. In this study we have shown that Tgfb1−/− thymocytes are less sensitive than wild-type thymocytes to Ca2+ion chelation by EGTA and to calcineurin inhibition by FK506. This suggests that TGFβ1 inhibits activation of thymocytes in vivo by blocking [Ca2+]i mobilization and uptake by activated cells. The observation that both Tgfb1+/+ and Tgfb1−/− thymocytes are equally sensitive to rapamycin and NF-κB SN50, while Tgfb1+/+ cells are more sensitive to FK506, suggests that TGFβ1 does not affect PKC and p70S6 kinase signaling. Finally, BrdU incorporation studies demonstrate that DP thymocytes are hyperresponsive and that CD4+ SP thymocytes develop at a higher rate than CD8+ SP thymocytes. [Ca2+]i measurements of unstimulated and stimulated thymocytes confirm that the basal [Ca2+]i levels in Tgfb1−/− thymocytes are elevated, and that the increase in [Ca2+]i levels is greater upon stimulation with suboptimal concentration of ionomycin and anti-CD3. Also, the difference between Tgfb1+/+ and Tgfb1−/− is more evident in CD4+ SP thymocytes.
TGFβ1 is known to have multiple suppressive actions on immune cells. Based on the data presented in this study, we hypothesize that TGFβ1 prevents autoimmune disorders by increasing the signal threshold level of activation. It is known that alteration of signal threshold levels due to mutations affecting the TCR complex and its downstream effectors leads to altered thymic selection and hyperresponsiveness (42, 57–59). It is therefore possible that in the absence of TGFβ1, more hyperresponsive, self-reactive T cells that escape thymic negative selection are exported to the periphery. Escape to the periphery of a few autoreactive thymocytes with a lowered threshold for activation could result in enough expansion to cause autoimmune disease. It is also known that a few autoreactive cells always escape to the periphery, but their activation is inhibited by TGFβ1-secreting regulatory T cells (60). Thymocytes and mature T cells must recognize self-MHC molecules in the thymus and periphery for their survival (40, 61). Lowering of the activation threshold could therefore result in positive selection of T cells that would otherwise have been deleted. However, recognition of self-MHC molecules by TCR alone can lead to activation if there are disturbances in the signaling environment (57, 58). Activated T cells are known to exhibit enhanced adhesion to endothelial cells (62), TGFβ1 has been shown to inhibit the adhesion of neutrophils and T cells to endothelial cells (63, 64), splenocytes from Tgfb1−/− mice exhibit enhanced adhesiveness to endothelial cells (32), and TGFβ1 is known to affect integrin activity (11). Hence, it is reasonable to speculate that hyperresponsive Tgfb1−/− T cells adhere to endothelial cells and extravasate into peripheral organs more rapidly in Tgfb1−/− mice.
Thymectomy of neonatal mice in the first week of birth but not on the day of birth or 7 days after birth results in the development of autoimmune disease that can be reversed by adoptive transfer of either thymocytes from neonates or lymphocytes from spleen and lymph nodes of adult mice (60, 65). Suppressor and regulatory T cells secrete TGFβ1 (22, 23). Because TGFβ1 is produced in the thymus and secreted by both suppressor and regulatory T cells, depletion of TGFβ1 with Ab reverses the regulatory function of CD4+CD25+ T cells and makes them proliferate in response to mitogenic stimuli (66). Hence, it is reasonable to speculate that absence of TGFβ1 would lead to export of autoreactive T cells into the periphery, resulting in autoimmune disease.
Autoreactive T cells are deleted in the thymus by negative selection. In this process T cells that recognize self-Ags with a low enough affinity that they are not activated will survive negative selection and be exported to the periphery. Autoimmune disease can occur if there is up-regulation of self-MHC molecules and presentation of self-Ags, up-regulation of costimulatory molecules, or a lowering of the threshold level of T cell activation due to increased or decreased production of stimulatory or inhibitory cytokines (67). Naive T cells need to interact with self-MHC molecules for their survival in the periphery. Normally, interaction of TCR with self-MHC per se is not sufficient to activate a naive T cell in the periphery (61). However, because TGFβ1 is a negative regulator of T cells, TGFβ1-deficient T cells that exhibit a lower threshold level of activation may become activated upon engagement with self-MHC molecules.
TGFβ1 mediates its inhibitory effects on many cell types including lymphocytes through SMAD2 and SMAD3. However, there is evidence suggesting that TGFβ1 can exert its effects such as inducing fibronectin expression through SMAD-independent mechanisms. Consequently, Smad3 null thymocytes and mature T cells were found to be normal in their mitogenic response, although resistant to TGFβ1-mediated suppression of mitogenic response (68, 69). Because SMAD3-deficient mice live much longer than TGFβ1-deficient mice due to less severe forms of inflammatory disorders and because SMAD3-deficient thymocytes are not hyperresponsive to mitogenic stimulation, our data demonstrate that regulation of [Ca2+]i levels by TGFβ1 is SMAD3 independent and is critical for maintaining immune homeostasis and prevention of autoimmunity.
In summary, our studies demonstrate that Tgfb1−/− thymocytes taken from animals with no complicating effects of multifocal inflammation are hyperresponsive to mitogenic stimuli. Analysis of thymocytes from these animals has led to the conclusion that TGFβ1 functions to inhibit aberrant T cell expansion by maintaining low enough [Ca2+]i levels to prevent a mitogenic response by Ca2+-independent stimulatory pathways alone. Consequently, TGFβ1 ensures that all T cell stimulatory pathways must be extant before a mitogenic response occurs. In its absence, there is a low level of stimulation through the Ca2+-calcineurin pathway, which when combined with stimulation through other pathways becomes sufficient for T cells to proliferate rather than die, thereby leading to autoimmune disease. Because mature SP thymocytes have more free [Ca2+]i levels than their precursor DP cells, they may be more prone to autoreactivity when they emigrate to the periphery.
Acknowledgments
We thank Sandi Engle for suggesting the use of an inflammation index. We thank Wen Yun Sun for expert assistance in PCR genotyping and James Cornelius for expert assistance in flow cytometry. The assistance of Sandy Schwemberger in the flow cytometry and analysis of [Ca2+]i measurements is greatly appreciated.
Footnotes
Abbreviations used in this paper: SP, single-positive; BrdU, 5-bromo-2′-deoxyuridine; CLM, cell loading medium; DP, double-positive; PKC, protein kinase C; [Ca2+]i, intracellular calcium concentration.
This work was supported by National Institutes of Health Grants HD26471, ES05652, ES06096, and CA84291 (to T.D.) and by a grant from Shriners of North America (to G.F.B.).
References
- 1.Massague J. How cells read TGF-β signals. Nat Rev Mol Cell Biol. 2000;1:169. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 2.Barnard JA, Lyons RM, Moses HL. The cell biology of transforming growth factor β. Biochim Biophys Acta. 1990;1032:79. doi: 10.1016/0304-419x(90)90013-q. [DOI] [PubMed] [Google Scholar]
- 3.Roberts AB, Sporn MB. The transforming growth factor βs. In: Sporn MB, Roberts AB, editors. Peptide Growth Factors and Their Receptors: Handbook of Experimental Pathology. Springer-Verlag; Heidelberg: 1990. p. 419. [Google Scholar]
- 4.Wahl SM, Orenstein JM, Chen W. TGF-β influences the life and death decisions of T lymphocytes. Cytokine Growth Factor Rev. 2000;11:71. doi: 10.1016/s1359-6101(99)00030-1. [DOI] [PubMed] [Google Scholar]
- 5.Letterio JJ. Murine models define the role of TGF-β as a master regulator of immune cell function. Cytokine Growth Factor Rev. 2000;11:81. doi: 10.1016/s1359-6101(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 6.Kallapur S, Ormsby I, Doetschman T. Strain dependency of TGFβ1 function during embryogenesis. Mol Reprod Dev. 1999;52:341. doi: 10.1002/(SICI)1098-2795(199904)52:4<341::AID-MRD2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 7.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-β1 knock out mice. Development. 1995;121:1845. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 8.D’Souza RN, Cavender A, Dickinson D, Roberts A, Letterio J. TGF-β1 is essential for the homeostasis of the dentin-pulp complex. Eur J Oral Sci. 1998;106(Suppl 1):185. doi: 10.1111/j.1600-0722.1998.tb02174.x. [DOI] [PubMed] [Google Scholar]
- 9.Glick AB, Lee MM, Darwiche N, Kulkarni AB, Karlsson S, Yuspa SH. Targeted deletion of the TGF-β1 gene causes rapid progression to squamous cell carcinoma. Genes Dev. 1994;8:2429. doi: 10.1101/gad.8.20.2429. [DOI] [PubMed] [Google Scholar]
- 10.Glick AB, Weinberg WC, Wu IH, Quan W, Yuspa SH. Transforming growth factor β1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 1996;56:3645. [PubMed] [Google Scholar]
- 11.Hoying JB, Yin M, Diebold R, Ormsby I, Becker A, Doetschman T. Transforming growth factor β1 enhances platelet aggregation through a non-transcriptional effect on the fibrinogen receptor. J Biol Chem. 1999;274:31008. doi: 10.1074/jbc.274.43.31008. [DOI] [PubMed] [Google Scholar]
- 12.Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, Akhurst RJ. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- 13.Tang B, Bottinger EP, Jakowlew SB, Bagnall KM, Mariano J, Anver MR, Letterio JJ, Wakefield LM. Transforming growth factor-β1 is a new form of tumor suppressor with true haploid insufficiency. Nat Med. 1998;4:802. doi: 10.1038/nm0798-802. [DOI] [PubMed] [Google Scholar]
- 14.Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor β1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379. [PubMed] [Google Scholar]
- 15.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Doetschman T. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teng YT, Gorczynski RM, Hozumi N. The function of TGF-β-mediated innocent bystander suppression associated with physiological self-tolerance in vivo. Cell Immunol. 1998;190:51. doi: 10.1006/cimm.1998.1389. [DOI] [PubMed] [Google Scholar]
- 18.Khoury SJ, Hancock WW, Weiner HL. Oral tolerance to myelin basic protein and natural recovery from experimental autoimmune encephalomyelitis are associated with downregulation of inflammatory cytokines and differential upregulation of transforming growth factor β, interleukin 4, and prostaglandin E expression in the brain. J Exp Med. 1992;176:1355. doi: 10.1084/jem.176.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barone KS, Tolarova DD, Ormsby I, Doetschman T, Michael JG. Induction of oral tolerance in TGF-β1 null mice. J Immunol. 1998;161:154. [PubMed] [Google Scholar]
- 20.Thorbecke GJ, Umetsu DT, deKruyff RH, Hansen G, Chen LZ, Hochwald GM. When engineered to produce latent TGF-β1, antigen specific T cells down regulate Th1 cell-mediated autoimmune and Th2 cell-mediated allergic inflammatory processes. Cytokine Growth Factor Rev. 2000;11:89. doi: 10.1016/s1359-6101(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 21.Gilbert KM, Thoman M, Bauche K, Pham T, Weigle WO. Transforming growth factor-β1 induces antigen-specific unresponsiveness in naive T cells. Immunol Invest. 1997;26:459. doi: 10.3109/08820139709022702. [DOI] [PubMed] [Google Scholar]
- 22.Kitani A, Chua K, Nakamura K, Strober W. Activated self-MHC-reactive T cells have the cytokine phenotype of Th3/T regulatory cell 1 T cells. J Immunol. 2000;165:691. doi: 10.4049/jimmunol.165.2.691. [DOI] [PubMed] [Google Scholar]
- 23.Prud’homme GJ, Piccirillo CA. The inhibitory effects of transforming growth factor-β-1 (TGF-β1) in autoimmune diseases. J Autoimmun. 2000;14:23. doi: 10.1006/jaut.1999.0339. [DOI] [PubMed] [Google Scholar]
- 24.Miller A, Lider O, Roberts AB, Sporn MB, Weiner HL. Suppressor T cells generated by oral tolerization to myelin basic protein suppress both in vitro and in vivo immune responses by the release of transforming growth factor β after antigen-specific triggering. Proc Natl Acad Sci USA. 1992;89:421. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lucas C, Bald LN, Fendly BM, Mora-Worms M, Figari IS, Patzer EJ, Palladino MA. The autocrine production of transforming growth factor-β1 during lymphocyte activation: a study with a monoclonal antibody-based ELISA. J Immunol. 1990;145:1415. [PubMed] [Google Scholar]
- 26.Chen W, Jin W, Wahl SM. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor β (TGF-β) production by murine CD4+ T cells. J Exp Med. 1998;188:1849. doi: 10.1084/jem.188.10.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen W, Jin W, Tian H, Sicurello P, Frank M, Orenstein JM, Wahl SM. Requirement for transforming growth factor β1 in controlling T cell apoptosis. J Exp Med. 2001;194:439. doi: 10.1084/jem.194.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plum J, De Smedt M, Leclercq G, Vandekerckhove B. Influence of TGF-β on murine thymocyte development in fetal thymus organ culture. J Immunol. 1995;154:5789. [PubMed] [Google Scholar]
- 29.Schiott A, Sjogren HO, Lindvall M. The three isoforms of transforming growth factor-β co-stimulate rat T cells and inhibit lymphocyte apoptosis. Scand J Immunol. 1998;48:371. doi: 10.1046/j.1365-3083.1998.00405.x. [DOI] [PubMed] [Google Scholar]
- 30.Chung EJ, Choi SH, Shim YH, Bang YJ, Hur KC, Kim CW. Transforming growth factor-β induces apoptosis in activated murine T cells through the activation of caspase 1-like protease. Cell Immunol. 2000;204:46. doi: 10.1006/cimm.2000.1694. [DOI] [PubMed] [Google Scholar]
- 31.Diebold RJ, Eis MJ, Yin M, Ormsby I, Boivin GP, Darrow BJ, Saffitz JE, Doetschman T. Early-onset multifocal inflammation in the transforming growth factor β1-null mouse is lymphocyte mediated. Proc Natl Acad Sci USA. 1995;92:12215. doi: 10.1073/pnas.92.26.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hines KL, Kulkarni AB, McCarthy JB, Tian H, Ward JM, Christ M, McCartney-Francis NL, Furcht LT, Karlsson S, Wahl SM. Synthetic fibronectin peptides interrupt inflammatory cell infiltration in transforming growth factor β1 knockout mice. Proc Natl Acad Sci USA. 1994;91:5187. doi: 10.1073/pnas.91.11.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultz JJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T. TGFβ1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorelik L, Flavell RA. Abrogation of TGFβ signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 35.Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor β II receptor. J Exp Med. 2000;191:1187. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-β inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. 2000;165:4773. doi: 10.4049/jimmunol.165.9.4773. [DOI] [PubMed] [Google Scholar]
- 37.Gorham JD, Lin JT, Sung JL, Rudner LA, French MA. Genetic regulation of autoimmune disease: BALB/c background TGF-β1-deficient mice develop necroinflammatory IFN-γ-dependent hepatitis. J Immunol. 2001;166:6413. doi: 10.4049/jimmunol.166.10.6413. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi S, Yoshida K, Ward JM, Letterio JJ, Longenecker G, Yaswen L, Mittleman B, Mozes E, Roberts AB, Karlsson S, Kulkarni AB. β2-microglobulin-deficient background ameliorates lethal phenotype of the TGF-β1 null mouse. J Immunol. 1999;163:4013. [PubMed] [Google Scholar]
- 39.Boivin GP, O’Toole BA, Orsmby IE, Diebold RJ, Eis MJ, Doetschman T, Kier AB. Onset and progression of pathological lesions in transforming growth factor-β1-deficient mice. Am J Pathol. 1995;146:276. [PMC free article] [PubMed] [Google Scholar]
- 40.Takahama Y, Nakauchi H. Phorbol ester and calcium ionophore can replace TCR signals that induce positive selection of CD4 T cells. J Immunol. 1996;157:1508. [PubMed] [Google Scholar]
- 41.Truneh A, Albert F, Golstein P, Schmitt-Verhulst AM. Early steps of lymphocyte activation bypassed by synergy between calcium ionophores and phorbol ester. Nature. 1985;313:318. doi: 10.1038/313318a0. [DOI] [PubMed] [Google Scholar]
- 42.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 43.Davey GM, Schober SL, Endrizzi BT, Dutcher AK, Jameson SC, Hogquist KA. Preselection thymocytes are more sensitive to T cell receptor stimulation than mature T cells. J Exp Med. 1998;188:1867. doi: 10.1084/jem.188.10.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin CS, Boltz RC, Siekierka JJ, Sigal NH. FK-506 and cyclosporin A inhibit highly similar signal transduction pathways in human T lymphocytes. Cell Immunol. 1991;133:269. doi: 10.1016/0008-8749(91)90103-i. [DOI] [PubMed] [Google Scholar]
- 45.Boulougouris G, McLeod JD, Patel YI, Ellwood CN, Walker LS, Sansom DM. IL-2-independent activation and proliferation in human T cells induced by CD28. J Immunol. 1999;163:1809. [PubMed] [Google Scholar]
- 46.Anderson SJ, Coleclough C. Regulation of CD4 and CD8 expression on mouse T cells: active removal from the cell surface by two mechanisms. J Immunol. 1993;151:5123. [PubMed] [Google Scholar]
- 47.Wang T, Li BY, Danielson PD, Shah PC, Rockwell S, Lechleider RJ, Martin J, Manganaro T, Donahoe PK. The immunophilin FKBP12 functions as a common inhibitor of the TGF β family type I receptors. Cell. 1996;86:435. doi: 10.1016/s0092-8674(00)80116-6. [DOI] [PubMed] [Google Scholar]
- 48.Cameron AM, Nucifora FCJ, Fung ET, Livingston DJ, Aldape RA, Ross CA, Snyder SH. FKBP12 binds the inositol 1,4,5-trisphosphate receptor at leucine-proline (1400–1401) and anchors calcineurin to this FK506-like domain. J Biol Chem. 1997;272:27582. doi: 10.1074/jbc.272.44.27582. [DOI] [PubMed] [Google Scholar]
- 49.Cameron AM, Steiner JP, Sabatini DM, Kaplin AI, Walensky LD, Snyder SH. Immunophilin FK506 binding protein associated with inositol 1,4,5-trisphosphate receptor modulates calcium flux. Proc Natl Acad Sci USA. 1995;92:1784. doi: 10.1073/pnas.92.5.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83:463. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 51.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 52.Sanford LP, Ormsby I, Gittenbergerde GA, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development. 1997;124:2659. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aghdasi B, Ye K, Resnick A, Huang A, Ha HC, Guo X, Dawson TM, Dawson VL, Snyder SH. FKBP12, the 12-kDa FK506-binding protein, is a physiologic regulator of the cell cycle. Proc Natl Acad Sci USA. 2001;98:2425. doi: 10.1073/pnas.041614198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bassing CH, Shou W, Muir S, Heitman J, Matzuk MM, Wang XF. FKBP12 is not required for the modulation of transforming growth factor β receptor I signaling activity in embryonic fibroblasts and thymocytes. Cell Growth Differ. 1998;9:223. [PubMed] [Google Scholar]
- 55.Xu X, Su B, Barndt RJ, Chen H, Xin H, Yan G, Chen L, Cheng D, Heitman J, Zhuang Y, Fleischer S, Shou W. FKBP12 is the only FK506 binding protein mediating T-cell inhibition by the immunosuppressant FK506. Transplantation. 2002;73:1835. doi: 10.1097/00007890-200206150-00023. [DOI] [PubMed] [Google Scholar]
- 56.Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, Collier ML, Deng KY, Jeyakumar LH, Magnuson MA, et al. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature. 2002;416:334. doi: 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- 57.Yamazaki T, Arase H, Ono S, Ohno H, Watanabe H, Saito T. A shift from negative to positive selection of autoreactive T cells by the reduced level of TCR signal in TCR-transgenic CD3 ζ-deficient mice. J Immunol. 1997;158:1634. [PubMed] [Google Scholar]
- 58.Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- 59.Gong Q, Cheng AM, Akk AM, Alberola-Ila J, Gong G, Pawson T, Chan AC. Disruption of T cell signaling networks and development by Grb2 haploid insufficiency. Nat Immun. 2001;2:29. doi: 10.1038/83134. [DOI] [PubMed] [Google Scholar]
- 60.Bonomo A, Kehn PJ, Shevach EM. Premature escape of double-positive thymocytes to the periphery of young mice: possible role in autoimmunity. J Immunol. 1994;152:1509. [PubMed] [Google Scholar]
- 61.Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 62.Haskard D, Cavender D, Ziff M. Phorbol ester-stimulated T lymphocytes show enhanced adhesion to human endothelial cell monolayers. J Immunol. 1986;137:1429. [PubMed] [Google Scholar]
- 63.Gamble JR, Vadas MA. Endothelial adhesiveness for blood neutrophils is inhibited by transforming growth factor-β. Science. 1988;242:97. doi: 10.1126/science.3175638. [DOI] [PubMed] [Google Scholar]
- 64.Gamble JR, Vadas MA. Endothelial cell adhesiveness for human T lymphocytes is inhibited by transforming growth factor-β1. J Immunol. 1991;146:1149. [PubMed] [Google Scholar]
- 65.Shevach EM. Regulatory T cells in autoimmmunity. Annu Rev Immunol. 2000;18:423. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 66.Yamashiro H, Hozumi N, Nakano N. Development of CD25+ T cells secreting transforming growth factor-β1 by altered peptide ligands expressed as self-antigens. Int Immunol. 2002;14:857. doi: 10.1093/intimm/dxf061. [DOI] [PubMed] [Google Scholar]
- 67.Rose NR, Herskowitz A, Neumann DA, Neu N. Autoimmune myocarditis: a paradigm of post-infection autoimmune disease. Immunol Today. 1988;9:117. doi: 10.1016/0167-5699(88)91282-0. [DOI] [PubMed] [Google Scholar]
- 68.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18:1280. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dong Y, Tang L, Letterio JJ, Benveniste EN. The Smad3 protein is involved in TGF-β inhibition of class II transactivator and class II MHC expression. J Immunol. 2001;167:311. doi: 10.4049/jimmunol.167.1.311. [DOI] [PubMed] [Google Scholar]