

When invited to contribute to the Reflections series, I pondered what aspects of my career I should focus on. Hoping to lay down a strong narrative arc, I have chosen to link my earliest beginnings with RNA, as a graduate student studying the ribosome, to my later adventures in what hence became known as “the RNA World,” as a tenured professor studying the spliceosome. I believe that some of my most unusual scientific insights have come from this particular path, and it is those examples I have highlighted here. Given that these articles are intentionally highly personal narratives, I want to ask at the outset for forbearance from the many members of the RNA World community whose invaluable contributions I have not been able to cite, given the length constraints, but without which my own progress would not have been possible.
Prologue: The Ribosome
When I went to the University of Michigan in Ann Arbor as a freshman in 1962, I wanted to major in biochemistry. When I found out there was no such major, I toyed with English. (My mother was a writer.) I enjoyed the reading, but I resented being evaluated for my personal, obviously subjective views in writing. When my essay on Moby Dick came back covered with red ink and a large angry “C,” I decided I had had it. I would pursue science, unassailable in its objectivity! (Or so I thought.) I chose a major in zoology rather than biology, which spared me from having to memorize long lists of plant names in Latin. However, in my senior year, I was transfixed by a lecture in which it was revealed that someone had succeeded in splitting a bacterial ribosome into several pieces and putting it back together again, a biochemical tour de force. Yes, this is what I liked best of all.
In a remarkable coincidence, the next year found me sitting in the office of the very man who had accomplished this feat, Masayasu Nomura. I had recently married my high school sweetheart, and he was accepted only at a single medical school, the University of Wisconsin, where I dutifully followed him. I had never thought seriously about getting a Ph.D. Only three other girls in my high school class had even gone to college. Along the way, however, my husband-to-be persuaded me to consider trying for a Master's, so I had applied to graduate school in Madison and been accepted. “I want to work on the ribosome,” I told Professor Nomura. “What grades did you get in organic chemistry?” he parried. Hearing of my 10 h of Cs (and ignoring my straight As in biology), he brusquely told me to look elsewhere.
When I persevered, he told me, “Girls can't do biochemistry. They can't lift heavy rotors or spend long hours in the cold room.” “What can they do?” I asked. “Genetics,” he said.
As it turned out, I would have to wait several years before a suitable ribosome genetics project was conceived. In the interim, Nomura allowed me to do some experiments analyzing how ribosomes initiated protein synthesis in vitro. This indeed entailed biochemistry. To establish that the initiator tRNA bound first to the 30S not the 70S ribosome required a complex labeling strategy utilizing radioactively (14C and tritium) and density (15N and deuterium) labeled components. The experiments were technically difficult and emotionally stressful. For example, because of the high cost of “heavy water,” every drop of D2O had to be redistilled from the growth medium for reuse. Eventually, I was able to show that the initiator tRNA was found exclusively on hybrid (heavy/light) 70S ribosomes, i.e. which had to have undergone prior initiation on 30S ribosomes. This work was published in Nature in 1968 (1). After that, I was definitely ready for some genetics.
During that time, the main focus of the rest of the lab had remained the elusive search for ribosomal protein structure: function relationships. Nomura believed that, when each protein was eventually purified, it could be assigned a specific function by asking what happened to ribosome performance when this single protein was omitted. Numerous male postdoctoral fellows put in their long hours in the cold room, trying to achieve “total reconstitution” of the ribosome. (The work I had heard about in Ann Arbor was, by comparison, only “partial,” i.e. a soluble protein fraction could be split off from the ribonucleoprotein (RNP) core particles in high salt and, after dialysis, etc., added back to the cores to regain activity.) For total reconstitution, a mixture of 21 proteins of the 30S ribosomal subunit was combined with purified 16S rRNA, yet despite repeated tries, no active ribosomes were produced. In what I consider to this day a grand cosmic joke, the breakthrough came when Peter Traub, a tenaciously driven German postdoctoral student, reasoned that, because cells grow at physiological temperatures, ribosome reconstitution might in fact require a temperature-dependent step. Out from the cold room he came. Indeed, when heated to 40 °C, active 30S ribosomes were reconstituted! It was a great day in the laboratory, but as more and more elegant experiments were carried out to describe the in vitro biochemical pathway, the question loomed whether this pathway had anything to do with biology.
Thus was my genetics project finally born. The reasoning was that if the process of ribosomal subunit assembly in vivo was also temperature-dependent, mutants should manifest themselves as cold-sensitive (cs). Thus, I mutagenized bacteria and replica-plated them to low and high temperatures. Indeed, I found that some 30% of cs mutants in Escherichia coli were ribosomal subunit assembly-defective (sad) at low temperatures. This required me to perform double-labeling experiments (wild-type versus mutant) of cells shifted to the cold; extracts were then analyzed by sucrose gradients. In contrast to wild-type profiles containing only 30S and 50S peaks, sad mutant cells accumulated species with unusual sedimentation values. Most interestingly, in one case, the size approximated that found for in vitro reconstitution intermediates (reconstitution intermediate particles) formed without the heat incubation step (and subsequent analysis revealed a significant overlap in protein composition). Although the specific molecular blocks in my sad mutants were never determined, these experiments were taken to confirm the biological relevance of the in vitro pathway, consistent with the hypothesis that ribosome assembly requires a temperature-dependent RNA conformational rearrangement (2).
The timing could not have been better. The 1969 Cold Spring Harbor Symposium was entitled “The Mechanism of Protein Synthesis,” and the lab's combined stories were granted three back-to-back presentations in the opening session. I was completely overwhelmed to be given this rare opportunity to talk as only a third-year graduate student, and I barely slept the night before my talk. (Probably, Joan Steitz had the same problem; we shared a bathroom in the Page Motel, and I threw up repeatedly.) Nonetheless, the talk was enthusiastically received, and I was invited to give a seminar the next fall at Harvard University (to Nomura's chagrin).
The ribosome was a magnet for driven scientists at the time, and competition between laboratories in the United States and Europe was fierce. Unfortunately for me, two of the fiercest competitors were Nomura and Chuck Kurland, another Madison biochemist. I had greatly enjoyed taking courses from Chuck in the newly emerging field of molecular genetics, and he had become an invaluable mentor to me as I struggled to stay free of the fallout between them, but it was not to be. Ultimately refusing to tolerate any connection between Kurland and myself, Nomura ordered me to leave the laboratory. In the early spring of 1970, my thesis committee was informed that I should receive my Ph.D. forthwith, and that was that. Adrift, I was overwhelmed by doubts that it would ever be possible to do science in a nurturing environment (Fig. 1). Indeed, this became my major goal when I finally established my own laboratory at the University of California, San Francisco, in 1973. Little did I realize at the time how profound were my “lessons from the ribosome,” both personally and scientifically.
FIGURE 1.

Adrift in 1970.
The Spliceosome
The discovery of “split genes” in 1977 was absolutely breathtaking. How could it be? Why? For those of us who had been working in the new field of “RNA processing” (first so dubbed in 1974 at a symposium in Brookhaven), this “amazing sequence arrangement” (3) spurred a thrilling and highly competitive search for mechanism. In 1980, Joan Steitz published the provocative idea that small nuclear ribonucleoprotein particles (snRNPs) might mediate the specificity of the reaction by virtue of complementarity between sequences at the 5′-ends of U1 small nuclear RNA (snRNA) to short sequences at the splice sites (4). Although this was a highly appealing paradigm, the data were circumstantial at best. At the time, I had been trying to understand the mechanism by which introns were removed from certain yeast tRNAs using a system of suppressor genetics derived from the pioneering studies of bacterial tRNAs by the group led by John Smith and Sydney Brenner at the Medical Research Council in Cambridge, England. Although still very much a novice at yeast genetics, I was captivated by the possibility of using this type of approach to understand the removal of introns from yeast mRNAs. Specifically, if Joan's hypothesis were correct, it should be possible to make splicing suppressors by introducing complementary base pair changes between a 5′-splice site and U1 and thereby prove the model genetically.
I was convinced that this approach would work, but I found little support from my colleagues in the processing community. It repeatedly was pointed out to me that only a handful of yeast genes even had introns, somehow implying to them that the mechanism would be “special” and mechanistically unrelated to that in higher eukaryotes. Even more problematical was the fact that Joan had failed to detect snRNPs in Saccharomyces cerevisiae when she performed immunoprecipitations with “anti-Sm” antibodies derived from patients with the autoimmune disease systemic lupus erythematosus. The very fact that these antibodies could bring down snRNPs from all other species tested (including something called the fall armyworm) had been used to argue the high conservation of snRNP-based splicing across evolution. Ipso facto, yeast must use a different mechanism. I had a different conviction, that the failure of the antibody experiment rather reflected the divergence of the so-called Sm antigen; thus, the best approach should be to look directly for the snRNAs themselves.
I based this argument on observations gleaned from ongoing analyses of bacterial ribosomes. As I described above, the efforts to get at the mechanism of translation had to date been focused almost exclusively on the roles of the ribosomal proteins. This came from a strong conceptual bias that only proteins could perform functionally sophisticated roles. Although many ribosomal proteins could be omitted in ribosome reconstitution experiments, inactivation of rRNA followed single-hit kinetics. By far the most persuasive argument for the functional importance of the RNA came from phylogenetic sequence comparisons, where it emerged that the secondary structure of bacterial 16S rRNAs was remarkably conserved. The beautiful exposition of these data by Noller and Woese in 1981 (5) persuaded me beyond any doubt that phylogenetics must similarly underlie the most important functional conservation among snRNAs, from yeast to mammals, yet when I announced my intentions to use phylogenetics to get at the mechanism of splicing, I was generally laughed at.
Indeed, there was little to encourage me as I began my hunt for snRNAs in budding yeast. Southern analyses using cloned probes from mammalian U1, U2, and U4 RNAs were uniformly negative, forcing me to look directly for the RNAs themselves. In consultation with Joan Steitz and Alan Weiner, I chose to focus on three criteria for RNAs of interest: small size, possession of a unique 5′-cap (trimethylguanosine), and metabolic stability. From years of analyzing metabolically labeled yeast tRNAs on polyacrylamide gels, I knew that other RNAs in that size range would likely be present in very low abundance, but even when I scaled up to 50-mCi 32P labelings, there were only faint candidates, which themselves required a second dimension to be resolved from the more abundant breakdown products of rRNA. The key diagnostic was to determine which, if any, of these RNAs possessed 5′-caps. This entailed a series of enzymatic digests and subsequent analyses by thin-layer chromatography, requiring long exposures of the autoradiographs. Of course, there had been the real possibility that yeast caps might be different from the 2,2,7-trimethyl caps of mammals, but I managed to identify spots that contained at least dimethyl caps. Curiously, the number of these species was larger than I had anticipated, but that seemed the least of my problems. Cloning proved to be the most difficult step of all, and I was fortunate to attract an ambitious and talented postdoctoral student, David Tollervey, who finally succeeded. In 1983, the lab (Fig. 2) was able to publish two back-to-back papers in Cell demonstrating that yeast did indeed have snRNAs and, most importantly for subsequent genetic analysis, that these were encoded by single-copy genes, in striking contrast to the large multigene families in mammals (6, 7).
FIGURE 2.

My lab, circa 1980.
Although I was feeling triumphant at this proof-of-principle stage in my program, I was increasingly troubled by the fact that sequence analysis of the first six of the cloned genes failed to reveal any obvious homology to mammalian U1–U6 snRNAs. Even harder to explain was the finding that deletion of these genes (an experiment that could be done only in yeast) produced no phenotype whatsoever. In 1985, we finally found that cells deleted for snR10 were cold-sensitive. To test the hypothesis that we were dealing with functional redundancy, we constructed multiple mutants, an increasingly tedious process given the small number of selectable markers available to track the deleted gene copies. Ultimately, we made the sextuple mutant only to find that this strain was not any more cold-sensitive than the single deletion of snR10. My talks at meetings were greeted by increasingly bored audiences, and even good friends seemed embarrassed for me, suggesting that I reconsider my research goals, but I could not let go. Where were the expected essential snRNAs, and what in the world were the functions of all these dispensable genes?
While awaiting the identification of the longed-for spliceosomal snRNAs, I was fortunate to attract a graduate student who had been well schooled in yeast genetics by Beth Jones. Roy Parker brought joy to my heart by totally buying into the splicing suppressor project, so while others toiled away, screening new snRNA genes, Roy embarked on the suppressor end of things. Because the majority of known intron-containing genes were essential, he designed a reporter gene in which the intron (and short 5′-exon) was derived from actin, and the 3′-exon encoded a gene allowing cells to grow on histidinol (Hol), a histidine precursor. Thus, cells that were specifically unable to splice the reporter gene could be identified as Hol−. Because little was known about the cis-acting requirements for intron removal, Roy proceeded to mutagenize the reporter and sequence mutants. On his first try, he identified a Hol− mutant that accumulated unspliced precursor due to a mutation (G → A) at position 5 of the 5′-splice site. This work was published in Cell in 1985 (8). Now, if we only had a U1 snRNA…
In an important breakthrough, John Abelson's lab had recently developed an in vitro splicing system and subsequently, by running the extracts over a sucrose gradient, identified an ∼40S particle containing 32P-labeled splicing intermediates, which they dubbed the “spliceosome” (9). This provided an opportunity to see if any of the cloned RNAs co-migrated with this peak, experiments we carried out with Ed Brody on his sabbatical in John's lab at the California Institute of Technology, but the resolution was poor and signals low. Joan Steitz also came to do a sabbatical there, hoping to identify yeast U1 using an RNase H strategy, but this approach was equally unsuccessful. In the meantime, the competition was finally starting to heat up, and my frustration was at an all-time high. Having finally identified an essential small capped RNA, snR7, I essentially ordered the graduate student working on it to find some homology before his group meeting. Remarkably, the next day, Bruce Patterson came in smiling with the news that snR7 was none other than yeast U5: only 9 nucleotides of sequence identity, but tellingly located in a region of highly conserved secondary structure! To prove his case, he went on to clone the gene under the inducible GAL promoter, which allowed him to delete the chromosomal copy when cells were growing in galactose; in glucose, these cells accumulated unspliced actin mRNA, as predicted for an essential spliceosomal snRNA (10). I was jubilant, thinking it would be just a short time before we could establish a 1:1 relationship with all the mammalian RNAs.
My hopes were buoyed by experiments we had been performing in collaboration with Sandra Wolin, who had perfected the technique of microinjection into Xenopus oocytes. The idea was to introduce a collection of yeast RNAs into amphibian nuclei, which stockpile the Sm antigen prior to the mid-blastula transition; the in vivo assembled snRNPs should then become immunoprecipitable with mammalian anti-Sm antibodies. Gratifyingly, one of the RNAs we identified was snR7, which indeed contains a putative Sm-binding site (AAUUUUUG in a single-stranded region) (11). A second immunoprecipitable RNA was snR14, which we then predicted to also be an essential gene. When this expectation was fulfilled, we were highly optimistic that the approach was robust, although puzzled that several other immunoprecipitated RNAs were far from “small.”
At about the same time, Manny Ares had made the remarkable discovery that an ∼1000-nucleotide yeast RNA contained a region with high primary sequence identity to the 5′-end of mammalian U2 snRNA. Curiously, as he continued to examine the RNA, he identified small patches of potential homology to other mammalian snRNAs, including U5. He published the sequence of what he dubbed to be a “poly-snRNA” in Cell in 1986 (12), a highly satisfying idea to those who had always maintained the conviction that yeast were “different,” yet seemingly in conflict with our identification of snR7 as U5, which was not published until 1987. Clearly, resolution of this apparent paradox would ultimately require identification of each yeast homolog as well as determination of the function of the remainder of the poly-snRNA sequences.
In the meantime, I had been fascinated by the U2-like structure of the 5′-end of Manny's large spliceosomal snRNA (Lsr1). Despite some initial confusion, we soon established that this RNA was indeed represented among our cloned capped species (snR20), but which we had paid little attention to because of its suspiciously large size. It had been previously established that yeast introns shared a unique consensus, UACUAAC, upstream of the 3′-splice site. Initially, this was taken as firm evidence that yeast was “different,” as no such consensus could be found in mammalian introns. Importantly, however, it was subsequently shown that this sequence contained the site of lariat formation in the first chemical step of splicing. I was struck by the realization that Lsr1/snR20 contained a sequence with almost perfect complementarity to the so-called “TACTAAC box,” almost because it required a single nucleotide to be bulged out of an otherwise perfectly base-paired helix. I recently had been devouring papers about the self-splicing introns of the so-called Group II class because it had been shown that splicing of this class used an identical two-step mechanism involving lariat formation. That breathtaking discovery had riveted the field, as it strongly implied that, as with the ribosome, the spliceosomal snRNAs might themselves be catalytic. An elegant experiment recently had been published in which it was shown that the lariat formed on a conserved adenosine bulged out of an intramolecular helix and that this bulge was essential for catalysis. With that, I had what seemed to me the perfect rationalization for the pairing of the intronic UACUAAC with GUAGUA near the 5′-end of the yeast U2-like snRNA.
The beauty of the hypothesis was that we finally had in hand the tools to perform the long-dreamed of suppressor experiment, but with U2 instead of U1. Roy Parker was just about out the door to do a postdoctoral program, but I managed to persuade him to do the test: 1) mutate UACUAAC in the actin reporter and look for cells that could no longer grow on histidinol; 2) mutate GUAGUA in a plasmid-borne copy of snR20, leaving the chromosomal copy intact; and 3) make all pairwise combinations. If the model were correct, suppression (growth on histidinol) should be observed only with the complementary pair of mutations in the snRNA and the reporter pre-mRNA. Despite the conceptual simplicity of the experiment, there were many caveats. For example, compensatory base pair analysis can work only if the sequence per se is not absolutely essential, which is a counterintuitive argument when dealing with apparently invariant sequences. Thus, a likely outcome is weak partial suppression. Moreover, mutation of an important sequence in an snRNA might confer a dominant-negative phenotype, precluding any useful interpretation. Indeed, we saw examples of both situations, keeping me on pins and needles until all combinations could be carefully analyzed and all results repeated. In the end, the data clearly supported allele-specific suppression, the genetic equivalent of atomic resolution! U2 snRNA recognizes the branch point by base pairing (Fig. 3).
FIGURE 3.
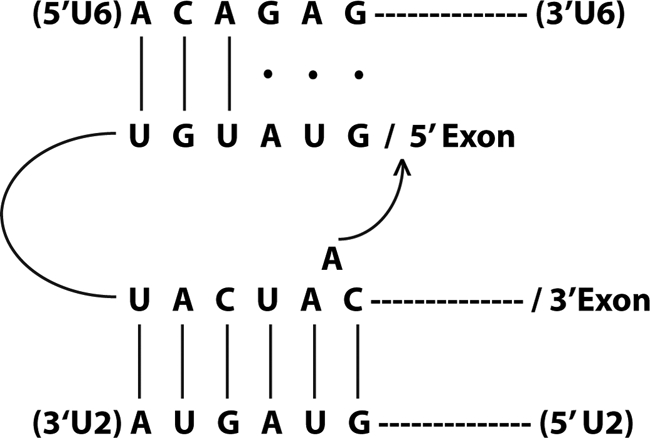
U2 snRNA recognizes the branch point by base pairing.
I was exhilarated and could not wait to see the reaction when our results were presented at the upcoming Cold Spring Harbor meeting, but the anticipated moment of triumph was dashed by the silence of the audience. Finally, a senior member of the eukaryotic splicing field sputtered, “What are we supposed to make of this? Surely this can't have anything to do with mammals!” It was emblematic of the deep suspicions held by most RNA processors, and it did not help that our primary results were genetic rather than derived from in vitro biochemistry. The paper was published in Cell in 1987 (13); we proposed that the same hydrogen-bonding interactions occurred in higher eukaryotes (the GUAGUA sequence in U2 is essentially invariant) but with shorter complementarity due to the weak adherence of the sequences surrounding the branch point to the UACUAAC consensus. Nonetheless, it would be several more years and many more experiments before the relevance of our findings was widely accepted.
Using a cis-competition experiment, Alan Weiner showed that UACUAAC was indeed preferred in favor of a natural branch point, and examples mounted in which the natural branch point sequence of efficiently spliced metazoan genes was indeed UACUAAC (14). In 1988, we showed that all but the U2-like sequences of snR20 could be deleted without consequence and, in 1990, that the human U2 gene could in fact efficiently complement an snR20 deletion (15). To this day, it is unclear what function, if any, the ∼1000 nonessential nucleotides perform. A similar quandary persists about yeast U1. In 1987, we showed that snR19, a second very large capped RNA, was the homolog of U1 both structurally and, using suppressor genetics, functionally (16). (Notably, the first U1 suppressor experiment was actually performed in mammalian tissue culture cells by Weiner's group in 1986 (17).) Similar results were reported by Michael Rosbash (18).
Having accounted for U1, U2, and U5, it was left to identify the yeast homologs of U4 and U6, specific roles for which were unknown. In fact, we had been skeptical about looking for yeast U6 from the outset because of its unusual properties in mammals. The smallest of the snRNAs, it lacks both a trimethylguanosine cap and an Sm-binding site. In 1987, a new postdoctoral fellow came to the laboratory. David Brow had studied yeast 5S ribosomal RNPs as a graduate student and was curious to use his two-dimensional gel methodology to look at yeast snRNPs, about which little to nothing was known. In preparation for analyzing yeast cell extracts, he insisted on performing a control experiment in which he analyzed phenol-extracted RNAs, subjecting them to native gel electrophoresis in the first dimension followed by a denaturing gel in the second. He then performed a Northern blot to identify the positions of several of our cloned snRNAs. Although he had only been in the lab a short time, he came to my office very excited to announce that there was something very interesting about the mobility of snR14. “What is that?” I asked, unable to see anything unusual. He insisted that it was migrating anomalously in the second dimension, slightly off the diagonal. “So what?” I asked, wondering if David was going to turn out to be tediously hung up on minor experimental details. He pointed out that it could be an effect of unusual intramolecular structure or indicate that snR14 was associated with a second RNA that dissociated in the denaturing dimension.
We had had inordinate trouble convincing ourselves of the homology between snR14 and a mammalian counterpart; it had no compelling primary sequence homology to any known snRNA. Somehow we had persuaded ourselves that it could be the U1 ortholog (prior to our analysis of snR19), and we had just submitted a manuscript to Nature to that effect. When David's subsequent control experiments convinced him that snR14 was likely associated with a second RNA, we realized that this behavior was strongly reminiscent of that of mammalian U4 and U6; Joan Steitz and Reinhard Luhrmann had previously found that these two RNAs were associated with one another. If this analogy were true, then snR14 must be U4 and the “invisible” RNA, U6. To quickly test this hypothesis, David hybridized his blot to a DNA probe from mammalian U6. Bingo! While terrifically excited by this discovery, I was simultaneously horrified that we would have to retract our manuscript from Nature. Beside myself, I somehow figured out how to contact Miranda Robertson, the Editor, at her London home in what turned out to be the middle of the night. Furious, she quickly informed me of two things: that it had not yet been sent out for review and that I better not try such a stunt again. Duly chastised but shaking with relief, I sat down to plan the next set of experiments.
As it turned out, U6 snRNA is the most highly conserved snRNA in size and in primary sequence. I have often thought how different things would have been if I had simply used U6 to probe my Southern blots in 1980. Nonetheless, I felt vindicated that my original assertion that we could gain important mechanistic insights into splicing by using phylogenetics was substantively correct. In 1988, we published our results (in Nature), wherein we presented a phylogenetically proven intermolecular structure of U4/U6 (19). In this Y-shaped structure, the central portion of U6 forms 23 contiguous base pairs with U4, consistent with our experimentally determined Tm of 53 °C for the deproteinized complex. We argued that this finding was highly significant in light of recent demonstrations by others that, during the course of splicing in vitro, U4 dissociates from U6 and the rest of the spliceosome. Moreover, we were struck by the fact that the most highly conserved sequences in U6 were those base-paired to U4, whereas the sequences of U4 were themselves variable. Taken together, these results supported what we called the “U6 ribozyme hypothesis.” Briefly, U6 is highly constrained by size and sequence, reflecting its position at the heart of the spliceosome's catalytic RNA core. To prevent premature activation, this region of U6 is sequestered by base pairing with U4, which thus functions as an antisense negative regulator. When appropriate signals have been recognized, U4 is actively displaced (presumably by an ATP-dependent mechanism), allowing U6 to refold into a catalytically active conformation.
The remaining challenge was thus to figure out the catalytically active conformation of U6. Hiten Madhani came to the lab as a highly ambitious M.D./Ph.D. student. One day in group meeting, I commented that we would never understand splicing unless we could change every single nucleotide in U6, and Hiten took me at my word. Using state-of-the-art techniques, he carried out saturation mutagenesis of U6 and assayed for effects on growth and splicing. He found a predominance of mutants in the most phylogenetically conserved sequence, the so-called “ACAGA box.” Subsequently, he proposed a model in which sequences immediately downstream of ACAGA could base pair with sequences in U2 immediately upstream of the branch point-interacting sequence GUAGUA. Although these two regions were strongly conserved, there were several potential instances of compensatory phylogenetic changes that prompted Hiten to perform a comprehensive mutagenic analysis. The important take-home of the paper published in Cell in 1992 was that the U2/U6 base pairing scheme involved mutually exclusive pairings with U4/U6 as well as with an intramolecular stem at the 5′-end of U2 (20). The next year, Cammie Lesser, another M.D./Ph.D. student in the lab, demonstrated that the ACAGA box actually pairs with the 5′-splice site, the pairing of which is itself mutually exclusive with the U1/5′-splice site helix (Fig. 3). This work was published in Science in 1993 (21) (and agreed with similar conclusions drawn by Kandels-Lewis and Séraphin (22)). Taken together, Hiten's and Cammie's results provided a dramatic description of highly dynamic changes in RNA pairing partners required for catalytic activation of the spliceosome. Although the precise order of these exchanges is still the subject of active study, it readily explains why the active spliceosome lacks U1 and U4. More broadly, it is the elucidation of this conformationally complex design principle that I think is perhaps the most important of our contributions to the understanding of the RNA components of the spliceosome (Fig. 4).
FIGURE 4.

The RNA world, circa 1988.
Although much remained to be done with the snRNAs, my attention began to shift to the factors responsible for their dynamic conformational rearrangements. Ironically, we had actually identified one of these genes in 1987 but did not realize it as such for many years to come. This story began with a mutant hunt for trans-acting suppressors of a mutation in the branch point (A → C) of our actin reporter, carried out by a highly determined team of two postdoctoral fellows, Joe Couto and Joe Tamm (23). After many false starts and much hard work, a single mutant, prp16-1, was identified that improved splicing of the mutant substrate. Because the mutant allowed the usage of a normally proscribed intron sequence, we proposed that the factor encoded by wild-type PRP16 was responsible for fidelity maintenance.
As a wide-eyed rotation student, Sean Burgess became fascinated with figuring out the role of this protein and joined the laboratory. Cloning the gene was a difficult process, but it eventually fell to Sean to make something out of the sequence. One day in group meeting, she passed around her handout, entitled “Prp16, the Muscle of the Spliceosome.” She explained that PRP16 had a bit of homology to myosin: the bit containing the Rossman fold! Deeply intrigued by this suggestive link between ATP hydrolysis and the fidelity of splicing, we published her findings in Cell in 1990 (24). Shortly thereafter, Beate Schwer, a new postdoctoral student with remarkable biochemical skills, showed that the Prp16 protein was in fact an RNA-stimulated ATPase. Using a very clever experimental strategy, she was able to demonstrate that the protein associated with the spliceosome only transiently, unless ATP hydrolysis was blocked. This work was published in Nature in 1991 (25). That same year, another enthusiastically driven graduate student, Evi Strauss, isolated a cs splicing mutant, prp28-1, and showed that it also encoded a Rossman-fold protein. As she perspicaciously pointed out in her 1991 Genes & Development paper (26), both Prp28 and Prp16 fell into a rapidly expanding family designated as “DEAD-box” ATPases, named for an eponymous tetrapeptide motif. Because members of this family had documented roles in many other nucleic acid transactions, it was clear that anything we could uncover about the spliceosomal functions would have broad ramifications. Conversely, we could exploit available data from other systems to test specific hypotheses. For example, the best studied DEAD-box protein in eukaryotic translation was eIF4A, a presumptive ATP-dependent RNA helicase. However, Prp28 failed to show unwinding activity.
Ribosome Redux
Of greatest interest to me at the time was the role of ATP hydrolysis in fidelity per se. Harkening back to the ribosome, which I had never truly left in my heart or in my mind, I was struck by the potential analogy to the role of GTP hydrolysis and fidelity in decoding at the ribosomal A site. Based on theoretical arguments, John Hopfield and Jacques Ninio had earlier developed a kinetic proofreading hypothesis, in which it was stated that to enhance the fidelity of an error-prone step, such as aminoacyl-tRNA selection, two sequential inspections would have to be separated by an irreversible step, viz. NTP hydrolysis. This strategy would create a branched pathway; “correct” (cognate) associations would proceed along the productive branch, whereas “incorrect” (non-cognate) intermediates would be discarded. Importantly, the fidelity characteristics of such a scheme would depend on the relative kinetic constants for the forward versus the discard pathways. Early experimental tests of this scheme suggested that the ribosome indeed used such a mechanism, and it was noted that the performance could be influenced by elongation factor (EF) Tu itself as well as the genetic status of several ribosomal proteins.
Sean Burgess was highly motivated to test the idea that Prp16 indeed functioned as an ATP-dependent “proofreading clock.” By saturation mutagenesis of the cloned PRP16 gene, she first showed the generality of her findings for the prp16-1 allele, namely all mutations that gave rise to the A → C suppression phenotype mapped to the ∼300-amino acid DEAD-box domain. Interestingly, the suppressor mutants all exhibited reduced ATPase activity in vitro, and the genetic and biochemical phenotypes were generally inversely correlated: the strongest suppressors were the weakest ATPases. These results suggested a conceptually simple basis for suppression. We hypothesized that splicing intermediates had to undergo a conformational rearrangement in which the branch point was inspected and that this rearrangement was in competition with ATP hydrolysis. In wild-type PRP16 cells, intermediates containing the mutant intron undergo this rearrangement slowly relative to ATP hydrolysis and are discarded. By slowing the rate of the Prp16 ATPase, the suppressors allow more time for the unfavorable rearrangement to occur, thus sparing the mutant intermediates from discard. Indeed, Sean found that the mutant molecules, i.e. lariat intermediates containing the A → C mutation, appeared to be stabilized in vivo. Her findings were published in Cell in 1993 (27), followed by a review article (in Trends in Biomedical Sciences (28)), in which we predicted that each ATP-dependent step in splicing (we currently know of eight in yeast) would similarly provide an opportunity for inspection and ATP-dependent discard. We were initially criticized for “trying to do kinetics by genetics,” but gratifyingly, recent data from several laboratories have now confirmed the generality of this notion. This paradigm linking fidelity and ATP hydrolysis remains one of the most intellectually exciting and satisfying themes of our work.
Although many extremely important questions still remain about these proofreading clocks, we have been particularly intrigued about the relationship between fidelity and the mechanics of splicing per se. For example, one might have imagined that Prp16 was a dedicated fidelity factor, yet it was clear that the protein was required for normal progression through the splicing pathway. As Beate Schwer showed in 1991, without Prp16 in vitro, spliceosomes stall prior to the second chemical step. Following Evi Strauss's interesting identification of prp28-1 as a cs mutant, Suzanne Noble, a talented M.D./Ph.D. student, decided to test the generality of this finding. She obtained a bank of cs mutants from David Botstein's lab and screened them for splicing defects in vivo. To our delight and amazement, the vast majority of mutants she identified turned out to be DEAD-box ATPases. Reminiscing about the cs phenotypes of my ribosomal sad mutants, we speculated in Suzanne's 1996 Genetics paper (29) that the rearrangements promoted by the spliceosomal proteins involve disruption of RNA secondary structures, an inherently temperature-dependent process.
The significance of this simple idea was seized upon by Jon Staley, a postdoctoral student who came to the lab from the world of protein folding. Impeccably trained as a biophysicist with Peter Kim, Staley originally thought to study yeast spliceosomal snRNAs by NMR, but while gearing up to do such experiments, he was intrigued by the genetics going on around him. I encouraged him to take a dip in these unfamiliar waters and was delighted when he came to my office several weeks later, with a Petri plate in his hand and a gleam in his eye. “This is a lot more fun than protein folding!” he announced. Shortly after that revelation, he proposed a genetic solution to a major problem in splicing. At the time, we knew the identity of most of the ATPases, but none of their targets. On the other hand, we did know some of the targeted transitions. For example, from Cammie Lesser's work, we knew that U1 must be replaced by U6 at the 5′-splice site before the first chemical step of splicing. He proposed a two-step procedure to identify the relevant DEAD-box protein: 1) hyperstabilize a U1/5′-splice site interaction, thereby creating a cs phenotype, and 2) introduce this modified U1/intron reporter system into each of the ATPase mutants and look for exacerbation of the growth phenotype.
To my amazement, Staley's scheme worked precisely as advertised. Although it took some tinkering to get the number of base pairs right, he generated the anticipated cs phenotype with an actin reporter construct and then rapidly screened our collection of splicing mutants. One and only one “winner” emerged: prp28-1. The triumph was his ability to recapitulate the cs splicing phenotype in vitro: U1-containing spliceosomes were stalled at low temperature in vitro but could be rapidly chased into productive complexes simply by raising the temperature. This reaction required wild-type Prp28 and ATP. Interestingly, Staley also showed that the defect could be suppressed by hyperstabilizing the U6/5′-splice site helix, arguing that these interactions were in equilibrium with one another. Thus, as we published in Molecular Cell in 1999 (30), he had succeeded in identifying an “RNA switch” dependent on a specific DEAD-box ATPase by an approach that should be generally applicable.
Of course the biggest switch in splicing is that from U4/U6 to U2/U6. One of the novel mutants to emerge from Suzanne Noble's cs screen was brr2-1 (named by her for bad response to refrigeration). Although clearly related to other DEAD-box spliceosomal ATPases, it had novel structural features. Moreover, whereas each of the other ATPases could be isolated as “soluble” factors, Brr2 was an integral component of the U5 and U4/U6·U5 triple snRNPs. Pratima Raghunathan, a graduate student and meticulous experimentalist, developed an elegant assay to test the hypothesis that Brr2 was required for U4 unwinding from U6; indeed, as she showed in her 1998 paper in Current Biology (31), triple snRNPs derived from brr2-1 cells failed to release U4 snRNA. This finding raised the intriguing question of how Brr2 activity would be regulated. This has turned into a fascinating, still-emerging story involving two tri-snRNP proteins: a ribosomal translocase-like GTPase, Snu114, and the largest most conserved protein in the spliceosome, Prp8.
Two students, Jim Umen and Cathy Collins, had focused their attention on the latter. Starting with intron mutations at the 3′-splice site (Jim) or 5′-splice site (Cathy) in our actin reporter, they each looked for suppressors after mutagenizing PRP8. Because this protein lacked any obvious structural motifs, I believed we could identify separable domains of structure/function by genetic dissection. After painstaking analyses, however, Jim and Cathy made the astonishing discovery that they had isolated identical alleles, arguing that this protein could interact with both splice sites simultaneously to control the fidelity of intron recognition. Because Prp8 had been previously shown to cross-link to these intron sites, in Cathy's 1999 paper in Genes & Development (32), we proposed that Prp8 played an important function at the spliceosome's catalytic core. Similar conclusions were reached independently by Magda Konarska (33). A long battle in the field then ensued as to whether Prp8 might actually contribute catalytic residues, and as of this writing, this seems a real possibility (34).
In the meantime, David Brow (by then a professor at the University of Wisconsin) gathered genetic evidence that Prp8 controlled fidelity by regulating the activities of several DEAD-box ATPases. Notably, for example, he found that several prp8 alleles could suppress the cs phenotype of brr2-1, suggesting a relief of inhibition. Other groups had used two-hybrid analysis to implicate physical interactions between Brr2 and Prp8. Of particular interest to us were interactions involving the C terminus of Prp8, in a region containing many of our splice site suppressor alleles. Moreover, Alan Kutach, a postdoctoral fellow in the lab, had carved out several C-terminal regions of the protein using protease hypersensitivity as a guide. Using these cloned fragments, he was able to verify a physical interaction between Brr2 and what we called the Prp8 C-terminal fragment (CTF). In collaboration with Corina Maeder, a new postdoctoral student in the lab, Alan developed a totally recombinant system to determine whether the ATPase or helicase activities of Brr2 were influenced by the presence of the Prp8 CTF. Remarkably, as published last year (2008) in Nature Structural & Molecular Biology (35), they showed that the CTF was necessary to activate the ability of Brr2 to unwind synthetically synthesized U4/U6 snRNAs. Moreover, this activity was lost when the CTF contained mutations known to cause retinitis pigmentosa in humans, demonstrating physiological relevance. This work stands as the first identification of a helicase regulator with a known RNA target.
As so often the case, this finding raised a series of new questions. In particular, because both Prp8 and Brr2 are integral snRNP proteins, how would the interaction with the CTF portion of the activator itself be regulated? Earlier work by Tamara Brenner when she was a graduate student in the lab predicted an enticing mechanism. Intrigued by the structural parallels between the U5 protein Snu114 and EF-2/EF-G, she carried out a comprehensive screen to identify genetic interactions between SNU114 and other spliceosomal proteins. As she described in her 2005 Genetics paper (36), the results highlighted strong functional interactions involving the C terminus of Snu114 with Prp8. Given the precedent that EF-2 undergoes a major conformational change upon GTP hydrolysis, we proposed that a similar rearrangement of Snu114 could thereby alter interactions with Prp8. This idea has recently been beautifully supported by elegant biochemical experiments by Jon Staley (now a professor at the University of Chicago), showing that the particular nucleotide state of the Snu114 GTPase is critical for controlling the activity of Brr2 (37).
We continue to be fascinated by the prospect of further analogies between the ribosome and the spliceosome (a theme also embraced, eloquently, by Konarska and Charles Query). For example, Tamara's work showed that the site of Prp8 interactions with Snu114 involves a domain in EF-2 (domain IV) hypothesized to mimic the anticodon of tRNAs, thereby mediating its function in translocation. Given the previous speculation by Jon Staley and I that the central stem/loop of U5 resembles a tRNA anticodon stem/loop (in a Cell review in 1998 (38)), this is a hypothesis we are quite eager to explore.
Finally, because the dynamic conformational changes that dominate the spliceosome's assembly and activity are so difficult to tackle by traditional experimental methods, we have recently initiated a novel approach using single-molecule fluorescence resonance energy transfer (FRET). This effort has been pioneered by John Abelson, a California Institute of Technology Professor Emeritus now working in the lab (and my husband). By placing FRET probes adjacent to the 5′- and 3′-splice sites, we have been able to monitor conformational dynamics during the course of spliceosome assembly using total internal reflection fluorescence microscopy (carried out in the laboratory of Nils Walter, our collaborator at the University of Michigan). We have found that the precursor undergoes a large number of ATP-dependent conformational rearrangements. Remarkably, the majority of these appear to be completely reversible. This finding has two important ramifications, as we point out in our upcoming publication. First, this realization prompts a provocative re-evaluation of the role of the spliceosomal ATPases, which have long been assumed to drive the pathway unidirectionally. Rather, the primary function of hydrolysis is to improve accuracy, driving the correct substrate to completion while incorrect alternatives are rejected. Second, the widespread reversibility of the spliceosomal dynamics we have observed is strikingly similar to recent single-molecule FRET observations of subunit rotation in the ribosome, which occurs reversibly in the presence or absence of GTP. Thus, we speculate that the spliceosome and the ribosome are both macromolecular RNP machines operating close to thermal equilibrium.
I will close my story here, adding a final note describing yet one more surprising connection between the two machines. Although I myself have always been most interested by mechanism, I have found increasingly of late that audiences seem to favor questions about regulation. Several years ago, I was able to entice a delightfully curious rotation student, Megan Bergkessel, to consider the hypothesis that the presence of introns in a limited subset of yeast genes (5%) would have significance for the regulation of those genes. In particular, the preponderance of the intron-containing genes encode ribosomal proteins (ribosomal protein genes). To test the hypothesis that cells would respond to amino acid starvation by down-regulating the production of ribosomal proteins at the level of splicing (in addition to transcription), Megan took advantage of a custom-designed microarray designed by Jeff Pleiss, an experimentally gifted postdoctoral fellow in the lab, which enabled us to simultaneously detect the levels of spliced and unspliced RNAs of all intron-containing genes in a single experiment. Jeff and Megan were joined in their efforts by Gregg Whitworth, another highly motivated graduate student with expertise in computational analysis. The experiment was to starve cells (by the addition of a histidine analog, 3-amino-1,2,4-triazole), extract RNA at intervals thereafter, and quantitate the ratios of spliced and unspliced RNAs by microarray analysis. The results were striking and unequivocal. As published in Molecular Cell in 2007 (39), Jeff, Gregg, and Megan showed that, within minutes after 3-amino-1,2,4-triazole addition, splicing of the majority of ribosomal protein genes is inhibited. Moreover, the effect is highly specific for this set of intron-containing genes.
Megan is now hard at work, determined to understand the molecular mechanism of this novel and fascinating example of signal transduction between the ribosome and the spliceosome. As for me? I am marveling that, in my career, I seem to have “beat on, boats against the current, borne back ceaselessly into the past” (40).
Acknowledgments
First and foremost, I express my gratitude to all of the marvelous students and postdoctoral fellows whom has been my profound privilege to mentor (many of whom did not appear in this narrowly focused narrative). It is my greatest fulfillment that, through them, I have been able to create the nurturing environment I so longed for at the start of my career. The path forward has been very difficult at times, and I owe my survival to the remarkable members of a group of female scientists who have continued to meet, enrich, and empower one another, every other Thursday, for more than 30 years (41). Finally, I am deeply grateful to members of the RNA World community, in particular John Abelson, Tom Cech, Jim Dahlberg, Elsebet Lund, Harry Noller, Joan Steitz, and Olke Uhlenbeck. Their generous encouragement and deep personal friendship have nourished me through much of my journey. I end by acknowledging the sustained support by the National Institutes of Health (Grant GM21119) and the American Cancer Society, which designated me a Research Professor of Molecular Genetics in 1992.
REFERENCES
- 1.Guthrie C., Nomura M. (1968) Initiation of protein synthesis: a critical test of the 30S subunit model. Nature 219, 232–235 [DOI] [PubMed] [Google Scholar]
- 2.Guthrie C., Nashimoto H., Nomura M. (1969) Studies on the assembly of ribosomes in vivo. Cold Spring Harbor Symp. Quant. Biol. XXXIV, 69–75 [DOI] [PubMed] [Google Scholar]
- 3.Chow L. T., Gelinas R. E., Broker T. R., Roberts R. J. (1977) An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12, 1–8 [DOI] [PubMed] [Google Scholar]
- 4.Lerner M. R., Boyle J. A., Mount S. M., Wolin S. L., Steitz J. A. (1980) Are snRNPs involved in splicing? Nature 283, 220–224 [DOI] [PubMed] [Google Scholar]
- 5.Noller H. F., Woese C. (1981) Secondary structure of 16S ribosomal RNA. Science 212, 403–411 [DOI] [PubMed] [Google Scholar]
- 6.Wise J. A., Tollervey D., Maloney D., Swerdlow H., Dunn E. J., Guthrie C. (1983) Yeast contains small nuclear RNAs encoded by single copy genes. Cell 35, 743–751 [DOI] [PubMed] [Google Scholar]
- 7.Tollervey D., Wise J. A., Guthrie C. (1983) A U4-like small nuclear RNA is dispensable in yeast. Cell 35, 753–762 [DOI] [PubMed] [Google Scholar]
- 8.Parker R., Guthrie C. (1985) A point mutation in the conserved hexanucleotide at the 5′ splice junction of the yeast actin intron uncouples recognition, cleavage, and ligation. Cell 41, 107–118 [DOI] [PubMed] [Google Scholar]
- 9.Brody E., Abelson J. (1985) The “spliceosome”: yeast pre-messenger RNA associates with a 40S complex in a splicing-dependent reaction. Science 228, 963–967 [DOI] [PubMed] [Google Scholar]
- 10.Patterson B., Guthrie C. (1987) An essential yeast snRNA with a U5-like domain is required for splicing. Cell 49, 613–624 [DOI] [PubMed] [Google Scholar]
- 11.Riedel N., Wolin S., Guthrie C. (1987) A subset of yeast snRNAs contain functional binding sites for the highly conserved Sm antigen. Science 235, 328–331 [DOI] [PubMed] [Google Scholar]
- 12.Ares M., Jr. (1986) U2 RNA from yeast is unexpectedly large and contains homology to vertebrate U4, U5, and U6 small nuclear RNAs. Cell 47, 49–59 [DOI] [PubMed] [Google Scholar]
- 13.Parker R., Siliciano P. G., Guthrie C. (1987) Recognition of the TACTAAC box during mRNA splicing in yeast involves base-pairing with the U2-like snRNA. Cell 49, 229–239 [DOI] [PubMed] [Google Scholar]
- 14.Zhuang Y. A., Goldstein A. M., Weiner A. M. (1989) UACUAAC is the preferred branch site for mammalian mRNA splicing. Proc. Natl. Acad. Sci. U.S.A. 86, 2752–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shuster E. O., Guthrie C. (1990) Human U2 snRNA can function in pre-mRNA splicing in yeast. Nature 345, 270–273 [DOI] [PubMed] [Google Scholar]
- 16.Siliciano P. G., Jones M. H., Guthrie C. (1987) S. cerevisiae has a U1-like snRNA with unexpected properties. Science 237, 1484–1487 [DOI] [PubMed] [Google Scholar]
- 17.Zhuang Y., Weiner A. M. (1986) A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell 46, 827–835 [DOI] [PubMed] [Google Scholar]
- 18.Kretzner L., Rymond B. C., Rosbash M. (1987) S. cerevisiae Ul RNA is large and has limited primary sequence homology to metazoan Ul snRNA. Cell 50, 593–602 [DOI] [PubMed] [Google Scholar]
- 19.Brow D. A., Guthrie C. (1988) Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature 334, 213–218 [DOI] [PubMed] [Google Scholar]
- 20.Madhani H. D., Guthrie C. (1992) A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell 71, 803–817 [DOI] [PubMed] [Google Scholar]
- 21.Lesser C. F., Guthrie C. (1993) Mutations in U6 snRNA that alter splice site specificity: implications for the active site. Science 262, 1982–1988 [DOI] [PubMed] [Google Scholar]
- 22.Kandels-Lewis S., Séraphin B. (1993) Involvement of U6 snRNA in 5′ splice site selection. Science 262, 2035–2039 [DOI] [PubMed] [Google Scholar]
- 23.Couto J. R., Tamm J., Parker R., Guthrie C. (1987) A trans-acting suppressor restores splicing of a yeast intron with a branch point mutation. Genes Dev. 1, 445–455 [DOI] [PubMed] [Google Scholar]
- 24.Burgess S., Couto J. R., Guthrie C. (1990) A putative ATP-binding protein influences the fidelity of branch point recognition in yeast splicing. Cell 60, 705–717 [DOI] [PubMed] [Google Scholar]
- 25.Schwer B., Guthrie C. (1991) PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature 349, 494–499 [DOI] [PubMed] [Google Scholar]
- 26.Strauss E. J., Guthrie C. (1991) A cold-sensitive mRNA splicing mutant is a member of the RNA helicase gene family. Genes Dev. 5, 629–641 [DOI] [PubMed] [Google Scholar]
- 27.Burgess S. M., Guthrie C. (1993) A mechanism for enhancing mRNA splicing fidelity by ATP hydrolysis: the RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell 73, 1377–1391 [DOI] [PubMed] [Google Scholar]
- 28.Burgess S. M., Guthrie C. (1993) Beat the clock: paradigms for NTPases in the maintenance of biological fidelity. Trends Biochem. Sci. 18, 381–384 [DOI] [PubMed] [Google Scholar]
- 29.Noble S. M., Guthrie C. (1996) Identification of novel components of the yeast pre-mRNA splicing machinery by means of cold-sensitive mutants. Genetics 143, 67–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staley J. P., Guthrie C. (1999) An RNA switch at the 5′ splice site requires ATP and the DEAD-box protein Prp28p. Mol. Cell 3, 55–64 [DOI] [PubMed] [Google Scholar]
- 31.Raghunathan P. L., Guthrie C. (1998) RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 8, 847–855 [DOI] [PubMed] [Google Scholar]
- 32.Collins C. A., Guthrie C. (1999) Allele-specific genetic interactions between Prp8 and RNA active site residues suggest a function for Prp8 at the catalytic core of the spliceosome. Genes Dev. 13, 1970–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siatecka M., Reyes J. L., Konarska M. M. (1999) Functional interactions of Prp8 with both splice sites at the spliceosomal catalytic center. Genes Dev. 13, 1983–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abelson J. (2008) Is the spliceosome a ribonucleoprotein enzyme? Nat. Struct. Mol. Biol. 15, 1235–1237 [DOI] [PubMed] [Google Scholar]
- 35.Maeder C., Kutach A. K., Guthrie C. (2008) ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C terminus of Prp8. Nat. Struct. Mol. Biol. 16, 42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brenner T. J., Guthrie C. (2005) Genetic analysis reveals a role for the C terminus of the S. cerevisiae GTPase Snu114 during spliceosome activation. Genetics 170, 1063–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Small E. C., Leggett S. R., Winans A. A., Staley J. P. (2006) The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol. Cell 23, 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Staley J. P., Guthrie C. (1998) Mechanical devices in the spliceosome: clocks, motors, springs and things. Cell 92, 315–326 [DOI] [PubMed] [Google Scholar]
- 39.Pleiss J. A., Whitworth G. B., Bergkessel M., Guthrie C. (2007) Rapid, transcript-specific changes in splicing in response to environmental stress. Mol. Cell 27, 928–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fitzgerald F. S. (1925) The Great Gatsby, Charles Scribner & Sons, New York [Google Scholar]
- 41.Daniell E. (2008) Every Other Thursday: Stories and Strategies from Successful Women Scientists, Yale University Press, New Haven, CT [Google Scholar]