

Abstract
The purpose of this work was to obtain information about conformational changes of the plasma membrane Ca2+-pump (PMCA) in the membrane region upon interaction with Ca2+, calmodulin (CaM) and acidic phospholipids. To this end, we have quantified labeling of PMCA with the photoactivatable phosphatidylcholine analog [125I]TID-PC/16, measuring the shift of conformation E2 to the auto-inhibited conformation E1I and to the activated E1A state, titrating the effect of Ca2+ under different conditions. Using a similar approach, we also determined the CaM-PMCA dissociation constant. The results indicate that the PMCA possesses a high affinity site for Ca2+ regardless of the presence or absence of activators. Modulation of pump activity is exerted through the C-terminal domain, which induces an apparent auto-inhibited conformation for Ca2+ transport but does not modify the affinity for Ca2+ at the transmembrane domain. The C-terminal domain is affected by CaM and CaM-like treatments driving the auto-inhibited conformation E1I to the activated E1A conformation and thus modulating the transport of Ca2+. This is reflected in the different apparent constants for Ca2+ in the absence of CaM (calculated by Ca2+-ATPase activity) that sharply contrast with the lack of variation of the affinity for the Ca2+ site at equilibrium. This is the first time that equilibrium constants for the dissociation of Ca2+ and CaM ligands from PMCA complexes are measured through the change of transmembrane conformations of the pump. The data further suggest that the transmembrane domain of the PMCA undergoes major rearrangements resulting in altered lipid accessibility upon Ca2+ binding and activation.
Keywords: Membrane Proteins, Plasma Membrane Calcium Pump, Equilibrium Constants, Photoactivatable Phospholipids, Conformational Changes
Introduction
Detailed structural information about the plasma membrane calcium pump (PMCA)2 is currently lacking. This pump is an integral part of the Ca2+ signaling mechanism (1) and is thus a crucial component of cell function. It is highly regulated by calmodulin (CaM), which activates this protein by binding to an auto-inhibitory region (2) and changes the conformation of the pump from an inhibited state to an activated one (2, 3).
Information about the structure and assembly of the transmembrane domain of an integral membrane protein can be obtained from an analysis of the lipid-protein interactions. In this work, we have used a hydrophobic photolabeling method to study the noncovalent interactions between the membrane domain of the PMCA and surrounding phospholipids under different experimental conditions that lead to known conformations. It has been previously demonstrated that both the conformation and the activity of the pump protein are preserved in either solubilized or reconstituted purified preparations compared with the native pump located in the erythrocyte (4).
In recent work, we assessed changes in the overall exposure of PMCA to surrounding lipids by quantifying the extent of protein labeling by the photoactivatable phosphatidylcholine analog [125I]TID-PC/16 under different conditions (5). This showed that labeling of PMCA incubated with Ca2+ and calmodulin decreases by 25% and incubation with Ca2+ alone increases labeling by more than 50% compared with control labeling of the PMCA in the absence of Ca2+ and CaM. These results suggest that the PMCA assumes two different E1 conformations: one that is auto-inhibited and in which the membrane domain is in contact with a higher amount of lipids (incubating with Ca2+ alone, E1I) and one in which the enzyme is fully active (incubating with Ca2+-calmodulin, E1A) and exhibits a more compact transmembrane arrangement with lesser exposure to lipids. These data provide the first evidence that there is an auto-inhibited conformation in P-type ATPases involving both the cytoplasmic regions and the transmembrane segments.
Activation of PMCA is caused by the binding of CaM to the C-terminal tail of the pump, leading to dissociation of the auto-inhibitory domain from its close proximity to the active site. This removes the self-inhibition of the enzyme and stimulates the PMCA-mediated Ca2+ transport severalfold (6). Previously, the binding of CaM to PMCA was measured indirectly by determining the PMCA activity (7). Recently, Liyanage et al. (8) reported a fluorescence polarization method to measure the binding of CaM modified with Oregon Green 488. This was an evident improvement because it evaluates CaM binding in equilibrium instead of at steady-state with PMCA activity. However, it does not evaluate the binding of CaM itself but of a modified probe. In this report, we introduce a method that allows calculation of the dissociation constant for binding of non-labeled CaM to PMCA.
We employed the photoactivatable phosphatidylcholine analog 1-palmitoyl-2-[9-[2′-[125I]iodo-4′-(trifluoromethyldiazirinyl)-benzyloxycarbonyl]-nonaoyl]-sn-glycero-3-phosphocholine [125I]TID-PC/16 that has been previously used to analyze lipid-protein interfaces (9–11). This reagent partitions in the phospholipidic milieu and upon photolysis reacts indiscriminately with its molecular environment. It is thus possible to directly analyze the interaction between the hydrophobic membrane-spanning domain of a membrane protein and lipids belonging to its immediate environment (12–14). Applying this technique on the PMCA, we were able to measure equilibrium constants for the dissociation of ligands from PMCA complexes and to draw structural conclusions about the regulation of the transport of Ca2+ in the presence of different modulators.
EXPERIMENTAL PROCEDURES
Reagents
All chemicals used in this work were of analytical grade. Calmodulin was obtained from Calbiochem. Recently drawn human blood for the isolation of PMCA was obtained from the Hematology Section of the Hospital de Clínicas General San Martín and from Fundación Fundosol (Argentina). Blood donation in Argentina is voluntary, and therefore the donor must provide informed consent for the donation of blood and the subsequent legitimate use of the blood by the transfusion service.
Purification of PMCA from Human Erythrocytes
PMCA was isolated from CaM-depleted erythrocyte membranes by the CaM-affinity chromatography procedure (4) and stored in 20% (w/v) glycerol, 0.005% C12E10, 120 mm KCl, 1 mm MgCl2, 10 mm MOPS-K, pH 7.4 at 4 °C, 2 mm EGTA, 2 mm dithiothreitol. Protein concentration after purification was about 10 μg/ml. No phospholipids were added at any step along the purification procedure. The PMCA preparation thus obtained is devoid of natural phospholipids, as assessed by the failure to detect inorganic phosphate by the method described below (see “Phospholipid Quantification”). Solubilization and purification of PMCA preserve transport activity and maintain the kinetic properties and regulatory characteristics of the enzyme in its native milieu (4). [Ca2+] was calculated by the program Max Chelator and controlled with an Orion electrode (OR-9720BNWP).
Preparation of [125I] TID-PC/16
TTD-PC/16 (tin precursor) was a kind gift from Dr. J. Brunner (ETH Zurich, Switzerland). [125I]TID-PC was prepared by radioiodination of its tin precursor according to Weber and Brunner (11). After the reaction was completed, the mixture was extracted with chloroform/methanol (2:1, v/v) and [125I]TID-PC/16 was purified by passage through a silica gel column (2.5 ml) using chloroform/methanol: water/acetic acid (65:25:4:1, v/v) as solvent. The elution was monitored by TLC/autoradiography, and the fractions containing the product were dried and stored at −20 °C.
Phospholipid Quantification
Phospholipid concentration was measured according to Chen et al. (15) with some modifications. Samples and standards containing 10–100 nmol of phosphorus were dried by heating at 100 °C. Mineralization was carried out by adding 0.1 ml of HNO3, 0.9 ml of HClO4, and incubating at 190 °C for 30 min. Inorganic phosphate was determined after Fiske and Subbarow (16).
Measurement of Ca2+-ATPase Activity
ATPase activity was measured at 37 °C by following the release of inorganic phosphate from ATP as described previously (4). The incubation medium was: 80 μm DMPC, 120 μm C12E10, 120 mm KCl, 30 mm MOPS-K (pH 7.4), 3.75 mm MgCl2, 1 mm EGTA, and enough CaCl2 to give the desired final free [Ca2+]. The reaction was started by the addition of ATP (final concentration 2 mm). Release of Pi was estimated according to the procedure of Fiske and Subbarow (16).
Labeling Procedure
A dried film of the photoactivatable reagent was suspended in DMPC/C12E10 mixed micelles (80 and 120 μm, respectively) containing 10 μg/ml of the membrane protein, 120 mm KCl, 30 mm MOPS-K (pH 7.4), 3.75 mm MgCl2, 1 mm EGTA, and enough CaCl2 to give the desired final free [Ca2+]. The samples were incubated for 20 min at 37 °C before being irradiated for 15 min with light from a filtered UV source (λ ≈360 nm).
Radioactivity and Protein Determination
Electrophoresis was performed according to the Tris-Tricine SDS-PAGE method (17). Polypeptides were stained with Coomassie BlueR, the isolated bands were excised from the gel, and the incorporation of radioactivity was directly measured on a γ-counter. The amount of protein was quantified by eluting each stained band as previously described (18), including bovine serum albumin in each gel as a standard for protein quantification. Specific incorporation was calculated as the ratio between measured radioactivity and amount of protein determined for each band.
Proteolysis of PMCA
Proteolysis of PMCA was performed for 5 min in the presence of 25 mm Tris-HCl, pH 7.4 at 37 °C, 2 mm EGTA, and 0.22 μg/ml of TLCK-treated chymotrypsin in water. The reaction was stopped by a 10-fold excess of ovomucoid trypsin inhibitor solution at 4 °C.
Data Analysis
All measurements were performed in triplicate to quintuplicate unless specified otherwise in the figures.
RESULTS
Incorporation of [125I]TID-PC/16 as a Conformation Marker
We first wished to determine the extent of [125I]TID-PC/16 labeling of the PMCA in its major known conformational states. The E2 state is attained by incubating the PMCA in the absence of Ca2+ (1 mm EGTA). The incorporation of [125I]TID-PC/16 in this condition was considered as the control and was set as 100% (Fig. 1). The other two conformers are E1I, which is obtained by incubating the enzyme in the presence of Ca2+ and binds the maximum concentration of [125I]TID-PC/16 (180% at optimal concentration of Ca2+, Fig. 1) and E1A, a conformation attainable in the presence of Ca2+ and CaM or with a CaM-like effector such as phosphatidic acid, oleic acid or after removal of the C terminus of PMCA. This conformation binds the least amount of [125I]TID-PC/16 (∼75% of the control for E1A obtained with Ca2+ and CaM) (Fig. 1). These data are as reported earlier (5) and illustrate the sensitivity of the incorporation of labeled [125I]TID-PC/16 on the different PMCA conformations.
FIGURE 1.
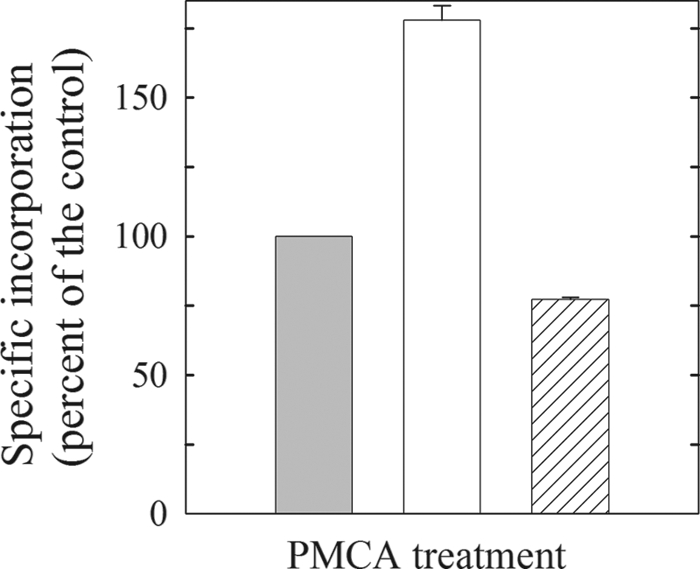
Relative specific incorporation of [125I]TID-PC/16 to PMCA under different conditions. Incorporation in the presence of 1 mm EGTA is taken as 100% control (E2, left bar). Middle, 15 μm Ca2+ (E1I). Right, 5 μm Ca2+ and 200 nm calmodulin (E1A). A similar result for E1A was obtained with phosphatidic acid, oleic acid, or removing the PMCA C-terminal domain with chymotrypsin. Data are the mean ± S.E. of 6–9 independent experiments.
Titration of the E2 to E1I Conformer Shift with Ca2+
To determine the [Ca2+] dependence of the conformational shift from the E2 to the E1I state, we performed a titration experiment (Fig. 2A) in which we measured the specific incorporation of [125I]TID-PC/16 to PMCA when the enzyme was incubated in the presence of increasing concentrations of Ca2+. In this experiment, we titrate the concentration of [125I]TID-PC/16 that binds to PMCA as it shifts from the E2 to the E1I conformer, i.e. as [125I]TID-PC/16 incorporation increases from 100% to near 180%. As illustrated in Fig. 2A, [125I]TID-PC/16 incorporation increases hyperbolically with low concentrations of Ca2+ (up to ∼40 μm) and then decreases to a level that cannot be easily evaluated with the available information. However, the data are well described by Equation 1.
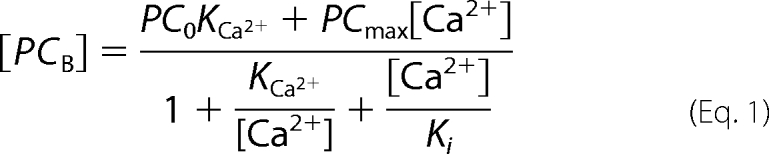 |
FIGURE 2.

[Ca2+] dependence of incorporation of [125I]TID-PC/16 to PMCA and of PMCA activity. A, purified PMCA devoid of CaM was incubated in the presence of different amounts of Ca2+, and after 3 min [125I]TID-PC/16 was added as described under “Experimental Procedures.” The inset shows the incorporation of TID-PC at low concentration of Ca2+. B, Ca2+-dependent ATPase activity as a function of [Ca2+]. The inset shows the data at low concentrations of Ca2+.
This empirically derived equation aims to characterize the effect of Ca2+ on the amount of [125I]TID-PC/16 bound to PMCA ([PCB]). PCmax is the maximal value of [PCB] attainable if [Ca2+] ≪ Ki (the constant describing the half-maximal concentration of Ca2+ for inhibition of [125I]TID-PC/16 binding to PMCA). The term [Ca2+]/Ki was included to account for the inhibitory effect by excess Ca2+ (see Fig. 2) and thus to allow a better estimation of KCa2+ (the concentration of Ca2+ for half-maximal binding of [125I]TID-PC/16 to PMCA) and of the maximal increase of [PCB], i.e. PCmax− PC0, (where PC0 is the amount of [125I]TID-PC/16 bound in the absence of Ca2+). Fitting the experimental data points to this equation yielded PC0 = 99.1 ± 4.8%, PCmax = 176.7 ± 4.3%, KCa2+ = 0.52 ± 0.14 μm, and Ki = 1212 ± 319 μm.
The inset in Fig. 2A shows the specific incorporation of [125I]TID-PC/16 to PMCA at very low concentrations of Ca2+ to illustrate the rapid increase in binding of [125I]TID-PC/16 at submicromolar [Ca2+]. It is also worth pointing out that after reaching a maximum near 40–50 μm Ca2+, [125I]TID-PC/16 binding decreases slowly with [Ca2+] in a similar way as is observed when measuring the Ca2+-ATPase activity (Fig. 2B). It was reported earlier that excess [Ca2+] in SERCA also induced a conformation different from that attained at lower concentrations of Ca2+ (19).
Fig. 2B shows the Ca2+-ATPase activity as a function of Ca2+. The data were quantitatively evaluated using Equation 2,
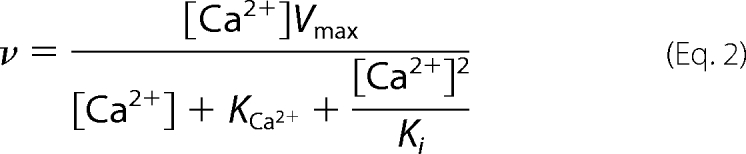 |
where Vmax is the maximum velocity, KCa2+ is the apparent dissociation constant for Ca2+ (Ca2+ concentration at half-maximal ATPase activity), and Ki is the concentration of Ca2+ for half-maximal inhibition of the enzyme at high [Ca2+]. Equation 2 is an empirical equation, which describes the effect of Ca2+ on Ca2+-ATPase activity and takes into account the inhibitory effect by excess Ca2+. It is analogous to Equation 1, which describes the effect of Ca2+ on the incorporation of [125I]TID-PC/16 into PMCA. It can be seen from Fig. 2B that the Ca2+-ATPase activity increases hyperbolically with low concentrations of Ca2+ (up to about 50 μm) and then decreases at higher [Ca2+] reflecting excess substrate inhibition. Applying Equation 2 to the data yielded Vmax = 13.4 ± 1.1 μmol Pi mg−1·min−1, KCa2+ = 11.0 ± 1.7 μm, and Ki = 248 ± 65 μm.
The most interesting finding resulting from a comparison of the Ca2+ dependence of [125I]TID-PC/16 incorporation (Fig. 2A) and ATPase activity (Fig. 2B) is that the K0.5 for the binding of [125I]TID-PC/16 is much lower (∼0.5 μm) than the KCa2+ for Ca2+ activation of the PMCA (11 μm). This strongly suggests that the intrinsic Ca2+ affinity of the PMCA in equilibrium is very different from its apparent Ca2+ affinity for activation by Ca2+.
Titration of the Ca2+ Dependence of the E2 to E1A Conformer Shift in the Presence of Calmodulin and Calmodulin-like Treatments
We next wished to compare the Ca2+ dependence of the E2 − E1I shift with that of the shift from E2 to the activated E1A conformation of the PMCA. To this end, we performed a series of titrations of [125I]TID-PC/16 incorporation in the presence of various activators of the pump. We chose the well-known “prototypical” PMCA activator CaM, the lipid activators phosphatidic acid (PA) and oleic acid (OA), as well as limited proteolytic treatment using TLCK-chymotrypsin. The latter treatment results in a C-terminally truncated and fully active pump lacking its auto-inhibitory tail (20). These activators thus reflect different mechanisms and pathways of PMCA activation.
Activation in the Presence of CaM
Fig. 3A shows the incorporation of [125I]TID-PC/16 as a function of concentration of Ca2+ in the presence of 1 μm CaM. Initially, incorporation of [125I]TID-PC/16 decreases rapidly with [Ca2+] and then slowly decreases to a constant value. The experimental data are well described by Equation 3,
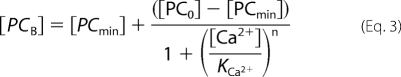 |
where [PCB] is the [125I]TID-PC/16 bound to PMCA at a given concentration of ionic calcium; [PCmin] is the final minimal concentration (%) of [125I]TID-PC/16 bound to PMCA (at non-limiting concentration of Ca2+), [PC0] is the initial concentration (%) of PC bound to PMCA (at zero [Ca2+]), “n” is the Hill coefficient, and KCa2+ is the concentration of Ca2+ for half-maximal binding to PMCA. This empirical equation describes the binding of [125I]TID-PC/16 to PMCA as a function of Ca2+. It can be seen from Fig. 3A that the amount of [125I]TID-PC/16 bound to PMCA decreases in a sigmoidal fashion with [Ca2+] from 100% (PC0, in the absence of Ca2+) to a constant minimal value (PCmin, at non-limiting concentration of Ca2+), which indicates that the PMCA conformation in this latter condition is more compact than that in the absence of Ca2+. Evaluating the data with Equation 3 yielded the following experimental parameters: [PCmin] = 76.7 ± 0.5%, [PC0] = 100.2 ± 0.5%, n = 1.7 ± 0.2, and KCa2+ = 0.46 ± 0.04 μm.
FIGURE 3.

[Ca2+] dependence of the incorporation of [125I]TID-PC/16 to PMCA in various activating conditions. A, purified PMCA was incubated in the presence of different amounts of Ca2+ and 1 μm CaM, and after 3 min, [125I]TID-PC/16 was added as described under “Experimental Procedures.” Inset, Ca2+-dependent ATPase activity as a function of Ca2+ in the presence of CaM. B, Ca2+ dependence of the incorporation of [125I]TID-PC/16 to PMCA in the presence of 58 μm phosphatidic acid. Inset, Ca2+-ATPase activity as a function of [Ca2+] in the presence of phosphatidic acid. C, [Ca2+] dependence of the incorporation of [125I]TID-PC/16 to PMCA in the presence of 10 μm oleic acid. Inset, Ca2+-ATPase activity as a function of [Ca2+] in the presence of oleic acid. D, [Ca2+] dependence of the incorporation of [125I]TID-PC/16 to PMCA submitted to proteolysis with chymotrypsin as described under “Experimental Procedures.” Inset, Ca2+-ATPase activity of chymotrypsin-truncated PMCA as a function of [Ca2+].
The inset in Fig. 3A shows the CaM-stimulated Ca2+-ATPase activity as a function of concentration of Ca2+. The experimental points were adjusted to a simple hyperbolic function with a Vmax of 12.8 ± 0.2 μmol Pi mg−1·min−1, and KCa2+ = 0.75 ± 0.07 μm.
In contrast to the large difference between the equilibrium Ca2+ affinity and the steady state (apparent) Ca2+ affinity for ATPase activation by Ca2+ alone (E2-E1I shift), the Ca2+ affinity for CaM stimulation of [125I]TID-PC/16 incorporation during the E2-E1A conformational shift is much closer to the true equilibrium Ca2+ affinity of the pump.
Activation in the Presence of Phosphatidic Acid
Fig. 3B shows the incorporation of [125I]TID-PC/16 as a function of concentration of Ca2+ in the presence of 58 μm PA. Incorporation of [125I]TID-PC/16 decreases with [Ca2+] in a sigmoidal manner and then reaches a constant minimal value. Applying Equation 3 yielded [PCmin] = 91.2 ± 0.4%, [PC0] = 99.8 ± 0.5, n = 3.7 ± 0.9, and KCa2+ = 0.67 ± 0.05 μm.
Whereas the values for most parameters are similar for the PA- and CaM-stimulated E2-E1A transition, the value of n describing the sigmoidicity of the Ca2+ dependence is much higher for PA than for any of the other treatments. Equation 3 is a Hill equation that shows in the denominator term the exponent n for the Ca2+ concentration, where n can be higher than one. In fact the value for n is near 2 for the effect of Ca2+ on [125I]TID-PC/16 binding in the presence of CaM (as well as of OA and for the enzyme that lacks the C-terminal region; see below), indicating that more than one molecule of Ca2+ is needed for the E2 to E1A shift. However, in the presence of PA, n is near 4 for the effect of Ca2+ on [125I]TID-PC/16 binding. This may be caused by a more complex behavior between PA and Ca2+: (i) PA could be at a non-optimal concentration and (ii) PA could associate with free Ca2+. However, when we measured the concentration of Ca2+ with a Ca2+-sensitive electrode in the absence and in the presence of 60 μm of PA, we found the value for free [Ca2] to be similar. We also determined the [125I]TID-PC/16 binding to PMCA as a function of PA, and the results indicate that the concentration used in the experiments of this work is sufficient to obtain the maximal effect, i.e. to reach PCmin (data not shown).
Measuring the PA-stimulated Ca2+-ATPase activity as a function of concentration of Ca2+ (inset in Fig. 3B) yielded a Vmax of 11.3 ± 0.2 μmol Pi mg−1·min−1, and KCa2+ = 0.83 ± 0.07 μm. These values are comparable to those determined for the activation of the PMCA by CaM and indicate that the apparent Ca2+ affinity for activation by either mechanism approaches the equilibrium Ca2+ affinity of the PMCA.
Activation in the Presence of Oleic Acid
The incorporation of [125I]TID-PC/16 as a function of concentration of Ca2+ in the presence of 10 μm OA is shown in Fig. 3C. As in the case of PA, incorporation of [125I]TID-PC/16 decreases with Ca2+ and then reaches a constant value. Using Equation 3 to evaluate the experimental points resulted in [PCmin] = 83.5 ± 3.6%, [PC0] = 100.3 ± 2.3%, n = 1.8 ± 1.1, and KCa2+ = 0.75 ± 0.28 μm.
The inset in Fig. 3C shows the Ca2+-ATPase activity in the presence of 10 μm OA as a function of Ca2+. The experimental points were adjusted to a simple hyperbolic function yielding a Vmax of 11.1 ± 0.3 μmol Pi mg−1·min−1, and KCa2+ = 0.96 ± 0.14 μm.
Removal of the PMCA C-terminal Domain by Proteolysis with TLCK-chymotrypsin
As mentioned above, proteolysis of PMCA by chymotrypsin yields a pump that is fully active, lacks the C-terminal region, and hence does not bind CaM. We used TLCK-chymotrypsin to avoid digestion of the N-terminal domain, which remains intact based on the reaction of the truncated PMCA with the specific antibody JA9 (5). Fig. 3D shows the incorporation of [125I]TID-PC/16 to chymotrypsin-treated PMCA as a function of [Ca2+]. Equation 3 was again fitted to the experimental points, yielding [PCmin] = 83.6 ± 0.6%, [PC0] = 100.2 ± 1.2%, n = 2.3 ± 0.5, and KCa2+ = 0.79 ± 0.10 μm.
The Ca2+-ATPase activity as a function of [Ca2+] of the C-terminally truncated pump is shown in the inset of Fig. 3D. Adjusting the experimental points to a simple hyperbolic function gave a Vmax of 13.9 ± 0.4 μmol Pi mg−1·min−1, and KCa2+ = 0.53 ± 0.07 μm.
Comparison of Apparent Ca2+ Affinities for PMCA Measured in Equilibrium and in the Steady State
Table 1 summarizes the results of the experiments described above and compares the Ca2+ affinities obtained through binding of [125I]TID-PC/16 and through the determination of Ca2+-ATPase activity in similar experimental conditions. From an inspection of this table we can draw the following conclusions: 1) Treatments that alter the equilibrium between E1 and E2 are recognized by a change in the incorporation of the probe. 2) Ca2+ affinities correlate well between the two methods employed when CaM or CaM-like treatments modified the PMCA. However, 3) The affinity for Ca2+ alone is similar to that in the presence of CaM or CaM-like treatments only when this parameter is evaluated according to [125I]TID-PC/16 incorporation on PMCA undergoing the shift between E2 to E1A or E2 to E1I. Taken together, these results indicate that the affinity for Ca2+ of the PMCA is not regulated at the Ca2+ site in the membrane but rather through the auto-inhibitory cytoplasmic domain.
TABLE 1.
Apparent Ca2+ affinities determined by enzyme activity and by specific [125I]TID-PC/16 incorporation under equilibrium conditions
Results represent the mean ± S.E. of 4–9 independent experiments by triplicate. E1A: an activated state of E1; E1I: an inhibited state of E1. PA: phosphatidic acid; OA: oleic acid; Chym: a 120-kDa PMCA devoid of the C-terminus obtained after chymotrypsin digestion.
| Condition | Conformation | Ca2+-ATPase activity |
Specific incorporation of [125I]TID-PC/16 |
||
|---|---|---|---|---|---|
| KCa2+ | Vmax | KCa2+ | [PC0]-[PCmax] or [PC0]-[PCmin]a | ||
| μm | μmol Pi mg−1.min−1 | μm | % | ||
| Ca2+ | E1I | 11.0 ± 1.7 | 13.4 ± 1.0 | 0.52 ± 0.14 | −77 ± 4b |
| Ca2++ CaM | E1A | 0.75 ± 0.07 | 12.8 ± 0.2 | 0.46 ± 0.04 | 24 ± 1 |
| Ca2+ + PA | E1A | 0.83 ± 0.07 | 11.3 ± 0.2 | 0.67 ± 0.05 | 9 ± 1 |
| Ca2+ + OA | E1A | 0.96 ± 0.14 | 11.1 ± 0.3 | 0.75 ± 0.28 | 17 ± 1 |
| Ca2+ + Chym | E1A | 0.53 ± 0.07 | 13.9 ± 0.4 | 0.79 ± 0.10 | 17 ± 1 |
a [125I]TID-PC/16 bound to PMCA increases or decreases with [Ca2+] from 100% (PC0, [125I]TID-PC/16 bound in the absence of Ca2+) to a new maximum or minimum value (PCmax or PCmin, [125I]TID-PC/16 bound at non-limiting concentration of Ca2+) as indicated by Eq. 1 and 3.
b The negative value reflects the fact that for Ca2+ alone, PCmax is higher than PC0.
Titration of the E1I to E1A Conformer Shift with Calmodulin
Activation of the PMCA is due to the binding of CaM, which dissociates the auto-inhibitory domain, removes the self-inhibition by the enzyme and thus stimulates PMCA transport severalfold (6). One way to measure the binding of CaM to the PMCA has been to follow pump activity as a function of the CaM concentration (7). We measured the binding of CaM to PMCA by quantifying the amount of bound [125I]TID-PC/16 to PMCA as a function of [CaM] in the presence of a saturating concentration of Ca2+. Fig. 4 shows the CaM-dependent incorporation of [125I]TID-PC/16 in the presence of 100 μm Ca2+. Equation 3 was fitted to the experimental points, but now the variable was the concentration of CaM instead of the concentration of Ca2+. Accordingly, KCa2+ of Equation 3 is replaced by KD(CaM) and [PC0] reflects the initial amount of [125I]TID-PC/16 bound in the absence of CaM rather than in the absence of Ca2+. This resulted in the following values: [PCmin] = 71.9 ± 1.8%, [PC0] = 154.8 ± 2.6%, n = 1.9 ± 0.2, and KD(CaM) = 9.6 ± 0.8 nm.
FIGURE 4.

Calmodulin dependence of the incorporation of [125I]TID-PC/16 to PMCA in the presence of saturating concentration of Ca2+. Incorporation of [125I]TID-PC/16 was determined in a medium containing 100 μm Ca2+ at 37 °C. Inset, Ca2+-ATPase activity as a function of the CaM concentration.
The inset shows the Ca2+-ATPase activity as a function of [CaM]. The experimental points were fitted to a hyperbolic function plus a term Vo that indicates the ATPase activity in the absence of CaM, yielding Vo = 4.9 ± 0.5 μmol Pi mg−1·min−1, Vmax = 12.5 ± 0.6 μmol Pi mg−1·min−1, and KD(CaM) = 7.2 ± 1.4 nm. These results show that the values for the dissociation constant of CaM calculated from the change in [125I]TID-PC/16 incorporation and from enzyme activation are very similar, validating the use of [125I]TID-PC/16 incorporation to follow the conformational shift from E1I to E1A.
DISCUSSION
[125I]TID-PC/16 has previously been used to identify and characterize regions within membrane proteins that interact with lipids (11, 14). Its physicochemical behavior in terms of mobility in thin layer chromatography is indistinguishable from PC, and its interaction with the transmembrane region of integral membrane proteins also appears to be identical to that of PC (12, 13). PC is generally chosen as the reference lipid against which relative lipid association constants of integral membrane proteins are compared, because these proteins show no selectivity for this phospholipid (21). This fact simplifies the interpretation of our results, as it allows a direct correlation between level of reagent incorporation and amount of protein surface exposed to surrounding lipids.
By using [125I]TID-PC/16 as a sensitive probe of the hydrophobic protein environment, we were able to study the transmembrane region of the PMCA in different conformations. Quantification of the amount of labeling by [125I]TID-PC/16 then allowed us to calculate the apparent affinity of the PMCA for Ca2+ and CaM as the pump shifts between different conformers in equilibrium with surrounding lipids.
Activation of the PMCA by CaM is commonly explained by the binding of CaM to the CaM binding domain in the C-terminal tail of the pump, followed by release of the inhibitory interactions from the cytosolic core. It should be noted that this hypothesis for the mechanism of auto-inhibition and activation does not predict changes in the transmembrane region. However, in a previous report, we demonstrated that the auto-inhibited conformation is distinct in its membrane domain, and that the conformational changes induced by auto-inhibition do expose additional hydrophobic surfaces of the protein to phospholipids. The auto-inhibited conformation was also obtained by adding the CaM-binding peptide C28 to an E1CaCaM state of PMCA (corresponding to the activated pump), showing the reversibility of this conformational transition (5). On the basis of these observations we postulated that the PMCA possesses two different E1-Ca2+ conformations: one that is auto-inhibited and is in contact with a higher amount of lipids (incubating with Ca2+ alone, E1I) and one in which the enzyme is fully active (incubating with Ca2+-calmodulin, E1A) and that exhibits a more compact transmembrane arrangement with a smaller surface area exposed to lipids.
According to kinetic enzyme activity measurements the activated conformer E1A can be obtained in the presence of CaM, acidic phospholipids, unsaturated fatty acids, protein oligomerization, or after removing the C-terminal tail by controlled proteolysis, e.g. with chymotrypsin (2, 4, 6, 22). In this report, we used several of these activating modes to compare the apparent affinity of the PMCA for Ca2+ as determined in two entirely different ways: (a) by evaluating the K0.5(Ca2+) for activation of the Ca2+-ATPase activity, i.e. in steady-state conditions of the enzyme, and (b) by measuring the equilibrium constants that drive the conformer E2 to E1A as reflected in a change in [125I]TID-PC/16 incorporation. The results clearly show that the PMCA possesses a high-affinity site for Ca2+ (KD ∼0.5 μm) regardless of the presence or absence of activators. Therefore, modulation of the pump activity is exerted through the C-terminal domain, which induces an auto-inhibited conformation but does not modify the affinity for Ca2+ at the site where it binds for transport into the transmembrane domain. This finding is analogous to an earlier study on the cardiac muscle SERCA (SERCA2a), which showed that the equilibrium Ca2+ binding affinity of the pump was unaffected by the inhibitory protein phospholamban (23). Similar to CaM activation of the PMCA, phospholamban phosphorylation stimulates SERCA activity by releasing the inhibitory interaction, which is reflected in an increase in the apparent Ca2+ affinity of the pump.
Ca2+-ATPase activation yields an “apparent” affinity for Ca2+, which is not the same as the actual binding affinity under equilibrium conditions. Our approach measures the real substrate affinity for Ca2+ in different conditions through the binding of [125I]TID-PC/16, which is directly proportional to the transmembrane surface of PMCA exposed to surrounding lipids. These two affinities should not be compared directly because they are determined in different states of the system. The experiments of this paper were designed to show that the true affinity of PMCA for Ca2+ is high, a fact that is masked by the steady-state condition, which shows a low affinity site for Ca2+.
Our findings raise interesting questions concerning the structural features of the transmembrane region of the PMCA in its auto-inhibited versus activated E1-Ca conformation. In the SERCA pump, for which atomic-resolution structural information is available for several conformers in both the E1 and E2 state (24–28), calculations of the membrane-embedded accessible surface area revealed only relatively modest differences between different Ca2+-bound E1-conformers (5). Major rearrangements involving the membrane-spanning region are, however, observed upon the E1 to E2 (or E2-E1) transition of the enzyme cycle. Because of the gross architectural similarity of the PMCA to the SERCA, it is assumed that comparable structural rearrangements accompany the reaction cycle in the PMCA, including large domain movements of the A (actuator) and N (nucleotide-binding) domains with respect to each other and the P (phosphorylation) cytosolic domain. Our data based on the changes in [125I]TID-PC/16 lipid binding to the PMCA clearly suggest that a major difference exists in the membrane domain of the pump in the auto-inhibited E1-Ca conformation (E1I) compared with the activated E1-Ca (E1A) conformation where the auto-inhibitory tail is dissociated from the core of the enzyme. The most salient difference between the SERCA and PMCA pumps is the presence of the extended C-terminal tail in the PMCA that encompasses the auto-inhibitory region. How could the presence of this tail, which is thought to be cytosolic, have such a dramatic effect on the membrane portion of the PMCA? Cross-linking studies have shown that in the auto-inhibited state, the tail is in close proximity to sequences in both the A- and N-domain (29, 30) and FRET studies indicate that the N and C termini of the pump are separated by less than 50 Å in the inhibited state (3). Thus, in the inhibited E1I conformation where the C-tail is “clamped-down” on the A- and N-domains, the transmembrane domain of the PMCA may be uniquely different from that in the E1-Ca state of SERCA and of the activated E1A state of the PMCA. Indeed, we found that any of the activating treatments (CaM, phosphatidic or oleic acid, and limited chymotrypsin proteolysis) resulted in a comparable (albeit not identical) change in [125I]TID-PC/16 incorporation to PMCA, suggesting that the common denominator is the release of the auto-inhibitory tail to allow a shift to the E1A conformer. Although a better understanding of the differences in the membrane domain of the E1I and E1A conformers of the PMCA will require higher resolution structural information, the data strongly suggest that substantial differences exist in the arrangement of the transmembrane domain in the PMCA in comparison with the SERCA pump, at least in the auto-inhibited E1 state of the PMCA. We cannot exclude the possibility that CaM, PA, OA, and partial proteolysis affect the membrane domain of the PMCA in a different manner and to a different degree. In fact, incorporation of [125I]TID-PC/16 to PMCA is different in the presence of CaM or PA. Experiments involving [125I]TID-PC/16 labeling and proteolysis with V8 protease show that labeling of PMCA with CaM affects transmembrane segments 3 and 4 and the bundle of TM 5 to 10, whereas in the presence of PA transmembrane segments 1–2 and 3–4 are mostly responsible for the incorporation of [125I]TID-PC/16.3 Therefore, the definition of the characteristics of the conformer E1A is purely operational and involves a series of structurally different conformers with a similar apparent high affinity for Ca2+.
Using a similar approach as for the determination of Ca2+ affinities we followed the transition from E1I to E1A by titrating this conformational shift with CaM. This allowed us to calculate the dissociation constant for binding of native non-labeled CaM to PMCA in equilibrium. The resulting value of 9.6 ± 0.8 nm is similar to the KD for CaM obtained in steady state conditions by evaluating the half-maximal activation of Ca2+-ATPase (7.2 ± 1.4 nm measured in this study (Fig. 4), 11.6 ± 2.4 nm, reported by Penheiter et al. (31)). This indicates that the apparent affinity for CaM is indistinguishable in steady state and in equilibrium experiments. Recently, Liyanage et al. (8), using a fluorescence polarization assay to evaluate the binding of a modified CaM to the PMCA, determined a KD of 5.8 ± 0.5 nm, a value comparable to the one obtained in this work. However, it is possible that the small difference in these KD values is attributable to different sources of CaM or to the Oregon Green 488 modified CaM used in their method. Regardless, our approach allows an independent calculation of the dissociation constant of non-labeled CaM from the PMCA.
The data presented here represent the first example where equilibrium constants for the dissociation of ligands from PMCA complexes are measured through the change of transmembrane conformations of the pump. The method is sensitive, precise, and requires a very low amount of protein. This should enable its application to other membrane proteins that cycle through different conformations, such as all members of the P-type ATPase family. Experiments are currently underway using this methodology to evaluate the affinity constants of PMCA for ATP under different experimental conditions.
Acknowledgments
We thank Dr. J. Brunner, Dept. of Biochemistry, Swiss Federal Institute of Technology Zurich (ETHZ) for the kind gift of TTD-PC/16 (tin precursor) and Dr. Rolando Rossi for helpful comments.
This work was supported, in whole or in part, by the National Institutes of Health, Fogarty International Center Grant R03TW006837 and by Agencia Nacional de Promoción Científica y Tecnológica, CONICET, and Universidad de Buenos Aires Ciencia y Técnica from Argentina.
I. Mangialavori, A. Villamil-Giraldo, M. Ferreira-Gomes, A. Caride, and J. P. Rossi, unpublished observation.
- PMCA
- plasma membrane calcium pump
- [125I]TID-PC/16
- 1-O-hexadecanoyl-2-O-[9-[[[2-[125I]iodo-4-(trifluoromethyl-3H-diazirin-3-yl)benzyl]oxy]carbonyl] nonanoyl]-sn-glycero-3-phosphocholine
- MOPS
- 3-(N-morpholino)propanesulfonic acid
- DMPC
- 1,2-dimyristoyl-sn-glycero-3-phosphocholine
- CaM
- calmodulin
- C12E10
- polyoxyethylene (10) dodecyl ether/3,6,9,12,15,18,24,27,30-decaoxadotetracontan-1-ol
- OA
- oleic acid
- PA
- phosphatidic acid
- TM
- transmembrane
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1.Strehler E. E., Caride A. J., Filoteo A. G., Xiong Y., Penniston J. T., Enyedi A. (2007) Ann. N. Y. Acad. Sci. 1099, 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarkadi B., Enyedi A, Földes-Papp Z., Gárdos G. (1986) J. Biol. Chem. 261, 9552–9557 [PubMed] [Google Scholar]
- 3.Corradi G. R., Adamo H. P. (2007) J. Biol. Chem. 282, 35440–35448 [DOI] [PubMed] [Google Scholar]
- 4.Filomatori C. V., Rega A. F. (2003) J. Biol. Chem. 278, 22265–22271 [DOI] [PubMed] [Google Scholar]
- 5.Mangialavori I., Giraldo A. M., Buslje C. M., Gomes M. F., Caride A. J., Rossi J. P. (2009) J. Biol. Chem. 284, 4823–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niggli V., Adunyah E., Carafoli E. (1981) J. Biol. Chem. 256, 8588–8592 [PubMed] [Google Scholar]
- 7.Enyedi A., Vorherr T., James P., McCormick D. J., Filoteo A. G., Carafoli E., Penniston J. T. (1989) J. Biol. Chem. 264, 12313–12321 [PubMed] [Google Scholar]
- 8.Liyanage M. R., Zaidi A., Johnson C. K. (2009) Anal. Biochem. 385, 1–6 [DOI] [PubMed] [Google Scholar]
- 9.Brunner J., Semenza G. (1981) Biochemistry 20, 7174–7182 [DOI] [PubMed] [Google Scholar]
- 10.Brunner J. (1993) Annu. Rev. Biochem. 62, 483–514 [DOI] [PubMed] [Google Scholar]
- 11.Weber T., Brunner J. (1995) J. Am. Chem. Soc. 117, 3084–3095 [Google Scholar]
- 12.Villamil Giraldo A. M., Castello P. R., González Flecha F. L., Moeller J. V., Delfino J. M., Rossi J. P. F. C. (2006) FEBS Lett. 580, 607–612 [DOI] [PubMed] [Google Scholar]
- 13.Villamil Giraldo A. M., Castello P. R., González Flecha F. L., Delfino J. M., Rossi J. P. F. C. (2006) Cell Biochem. Biophys. 44, 431–437 [DOI] [PubMed] [Google Scholar]
- 14.Durrer P., Galli C., Hoenke S., Corti C., Glück R., Vorherr T., Brunner J. (1996) J. Biol. Chem. 271, 13417–13421 [DOI] [PubMed] [Google Scholar]
- 15.Chen P. S., Toribara T. Y., Warner H. (1956) Anal. Chem. 28, 1756–1758 [Google Scholar]
- 16.Fiske C. H., Subbarow Y. (1925) J. Biol. Chem. 66, 375–400 [Google Scholar]
- 17.Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 18.Ball E. H. (1986) Anal. Biochem. 155, 23–27 [DOI] [PubMed] [Google Scholar]
- 19.Picard M., Toyoshima C., Champeil P. (2005) J. Biol. Chem. 280, 18745–18754 [DOI] [PubMed] [Google Scholar]
- 20.Penniston J. T., Enyedi A. (1998) Adv. Mol. Cell. Biol. 23, B249–274 [Google Scholar]
- 21.Marsh D., Horváth L. I. (1998) Biochim. Biophys. Acta 1376, 267–296 [DOI] [PubMed] [Google Scholar]
- 22.Kosk-Kosicka D., Bzdega T., Wawrzynow A. (1989) J. Biol. Chem. 264, 19495–19499 [PubMed] [Google Scholar]
- 23.Cantilina T., Sagara Y., Inesi G., Jones L. R. (1993) J. Biol. Chem. 268, 17018–17025 [PubMed] [Google Scholar]
- 24.Takahashi M., Kondou Y., Toyoshima C. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5800–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toyoshima C., Nakasako M., Nomura H., Ogawa H. (2000) Nature 405, 647–655 [DOI] [PubMed] [Google Scholar]
- 26.Toyoshima C., Nomura H. (2002) Nature 418, 605–611 [DOI] [PubMed] [Google Scholar]
- 27.Toyoshima C., Mizutani T. (2004) Nature 430, 529–535 [DOI] [PubMed] [Google Scholar]
- 28.Olesen C., Picard M., Winther A. M., Gyrup C., Morth J. P., Oxvig C., Møller J. V., Nissen P. (2007) Nature 450, 1036–1042 [DOI] [PubMed] [Google Scholar]
- 29.Falchetto R., Vorherr T., Brunner J., Carafoli E. (1991) J. Biol. Chem. 266, 2930–2936 [PubMed] [Google Scholar]
- 30.Falchetto R., Vorherr T., Carafoli E. (1992) Protein Sci. 1, 1613–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Penheiter A. R., Bajzer Z., Filoteo A. G., Thorogate R., Török K., Caride A. J. (2003) Biochemistry 42, 12115–12124 [DOI] [PubMed] [Google Scholar]