

Abstract
Poly(A)-specific ribonuclease (PARN) is a mammalian 3′-exoribonuclease that degrades poly(A) with high specificity. To reveal mechanisms by which poly(A) is recognized by the active site of PARN, we have performed a kinetic analysis using a large repertoire of trinucleotide substrates. Our analysis demonstrated that PARN harbors specificity for adenosine recognition in its active site and that the nucleotides surrounding the scissile bond are critical for adenosine recognition. We propose that two binding pockets, which interact with the nucleotides surrounding the scissile bond, play a pivotal role in providing specificity for the recognition of adenosine residues by the active site of PARN. In addition, we show that PARN, besides poly(A), also quite efficiently degrades poly(U), ∼10-fold less efficiently than poly(A). The poly(U)-degrading property of PARN could be of biological significance as oligo(U) tails recently have been proposed to play a role in RNA stabilization and destabilization.
Keywords: RNA/Messenger RNA/mRNA, RNA/Metabolism, RNA/Processing, RNA/Turnover, Deadenylation, mRNA Degradation, mRNA Poly(A) Tail
Introduction
Removal of the poly(A) tail at the 3′ end of the eukaryotic mRNA is a critical step in mRNA degradation (1, 2). Poly(A)-specific ribonuclease (PARN)5 (3–6) is one of the few known mammalian nucleases that degrade poly(A) with high specificity (7). Among the ribonucleases, PARN has the unique property of interacting with the 5′ end-located cap-structure in addition to the poly(A) tail (8–11). The interaction with the cap-structure enhances the rate of degradation (8–10) and amplifies the processivity of PARN action (12). PARN is a divalent metal ion-dependent enzyme and belongs to the DEDD family of nucleases (13, 14). Structural studies (15, 16) have revealed that PARN is composed of at least three structural domains. The active site is located within the nuclease domain, where four acidic amino acids Asp-28, Glu-30, Asp-292, and Asp-382 are believed to coordinate the catalytically important divalent metal ions (17, 18). In addition to the nuclease domain, two RNA binding domains, referred to as the RNA-recognition motif (RRM) and the R3H domain, have been identified. The RRM (19, 20) consists of four anti-parallel β-strands and two α-helixes located behind. Interestingly, amino acids important for PARN cap binding are located within the RRM (11, 16, 21, 22), and the RRM by itself shows similar RNA-binding properties as full-length PARN (11).
A crystal structure of PARN in complex with m7GpppG cap analogue has recently been solved, and it revealed that the cap binding and active sites overlap both structurally and functionally (16). Based on this structure and two previously determined structures (15), a homodimeric model for PARN has been reconstructed (16). In this model, the position of the first transcribed guanosine residue of the cap structure overlaps with the position of the penultimate adenosine residue of the poly(A) substrate, suggesting that the guanosine residue of the cap structure and the penultimate nucleotide of the poly(A) tail to some extent are recognized by the same set of amino acid residues.
Here, we have investigated the mechanisms behind the recognition of adenosine residues by PARN. Most importantly, we demonstrate that PARN has specificity for recognition of adenosine residues in its active site and that the nucleotides surrounding the scissile bond are critical for adenosine recognition.
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant Polypeptides
Human PARN or mutants thereof (16) were expressed and purified from the Escherichia coli strain BL21 (DE3) as described previously (23). The protein concentrations of purified PARN or mutants thereof were measured using Bio-Rad protein assay kit, and the purity was analyzed by gel electrophoresis (SDS-PAGE) followed by Coomassie staining. Purified polypeptides were either snap-frozen directly in TALON elution buffer (20 mm HEPES, pH 7.9, 500 mm KCl, 10% glycerol, and 150 mm imidazole) or dialyzed overnight against Buffer A (32 mm KH2PO4/K2HPO4, pH 7.0, 0.2 mm dithiothreitol, 100 mm KCl, 0.2 mm EDTA) or Buffer B (125 mm HEPES, pH 7.0, 500 mm NaCl) and subsequently aliquoted and snap-frozen. Aliquots were stored at −70 °C until used.
Preparation of RNA Substrates
The RNA substrates A5 to A20, G20, C20, U20, AAA, GGG, CCC, UUU, AXX, XXA, XAX, AXA, AAX, and XAA (where X denotes G, C, or U) were purchased from Dharmacon Research, Inc. Before use, the RNA oligonucleotides were deprotected according to the instructions from the manufacturer. 10 pmol of RNA substrate was 5′-labeled with 20 pmol of [γ-32P]ATP (3000 Ci/mmol, GE Healthcare or PerkinElmer Life Sciences) by 10–15 units of T4 polynucleotide kinase (United States Biochemical Corp. or Fermentas) in 30-μl reactions at 37 °C for 45 min. Reaction conditions were as follows: 50 mm Tris-HCl, pH 7.6, 10 mm MgCl2, and 10 mm β-mercaptoethanol (when using T4 polynucleotide kinase purchased from United States Biochemical Corp.) or 50 mm Tris-HCl, pH 7.6, 10 mm MgCl2, 5 mm dithiothreitol, 0.1 mm spermidine, and 1 mm EDTA (when using T4 polynucleotide kinase purchased from Fermentas). The labeled nucleotides were then fractionated by electrophoresis in 25% polyacrylamide gels (19:1 acrylamide/bisacrylamide), and bands were cut out and eluted in water. Poly(A), poly(U), poly(G), and poly(C) were purchased from Sigma. The 44-nucleotide heteropolymeric RNA (5′-CCA UCU CAU CCC UGC GUG UCC CAU CUG UUC CCU CCC UGU CUC AG-3′) was purchased from Metabion.
Electrophoretic Mobility Shift Assay
10-μl reactions were performed in Buffer A using 10 nm U20 RNA-oligo and 2.5–40 μm PARN, respectively. The reactions were incubated for 15 min at room temperature. 5 μl of loading dye (8% glycerol, 0.15% bromphenol blue/xylene cyanol) was added to the reaction prior to loading the samples to nondenaturing gels (0.5× TBE, 6% 19:1 acrylamide/bisacrylamide, v/v) pre-run at 200 V, 10 watts for 30 min at 4 °C. The gels were run for 1 h, 30 min at 10 watts at 4 °C and dried in a Bio-Rad gel dryer for 1 h and finally exposed and scanned by a 400S PhosphorImager (GE Healthcare).
Filter Binding Assay
Reactions using 32P-labeled oligo(A), varying in length between A5 and A20 or U20 RNA were performed as described under “Electrophoretic Mobility Shift Assay.” The RNA was titrated between 1 and 30 nm when U20 was used and between 2.5 and 1000 nm when oligo(A) substrates of different lengths were used. The concentration of PARN (between 1 and 7 nm) was experimentally determined for each complex and was kept fixed during each titration series. After 15 min of incubation, the reaction was applied to a Protran BA 85 cellulose nitrate membrane (catalogue no. 10 401191, Schleicher & Schuell) preincubated in Buffer A and mounted on a filter device with no vacuum applied. The membrane was washed with 0.75 ml of Buffer A with vacuum applied and dried, and finally the amount of bound protein-RNA complex was quantified in a Beckman Coulter LC6500 scintillation counter. The dissociation constant, KD, was obtained by plotting experimental data and fitting curves with nonlinear regression (Origin 7 software, OriginLab Corp.) using binding Equation 1
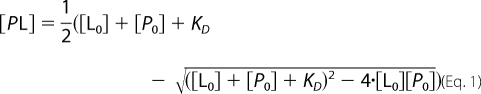 |
when oligo(A) substrates of different lengths were used or the binding Equation 2
 |
when U20 was used. [P0] is the active concentration of PARN polypeptides.
PARN Activity Assay
Conditions for the activity assays were as follows: 25 mm HEPES, pH 7.0, 100 mm NaCl, 0.1 μg/μl bovine serum albumin, 2 mm MgCl2, 2 nm to 20 μm RNA substrate (A20, U20, G20, C20, A3, U3, C3, G3, AXA, XAX, XXA, AXX, AAX, or XAA (where X denotes G, C, or U)) as indicated and 0.88–256 nm PARN as indicated. When 20-nucleotide-long substrates were used, the reaction volume was 10 μl, and incubations were performed at 30 °C for 10 min. Reactions were stopped by adding 10 μl of stop solution (83% formamide, 10 mm Tris-HCl, pH 7.9, 80 mm EDTA, 0.05% xylene cyanol, 0.05% bromphenol blue). For time course assays, reaction volume was 120 μl, and incubations were performed at 30 °C for the indicated times. For each time point, 10 μl of the reaction volume was removed and mixed with 10 μl of stop solution. The resulting products were analyzed by electrophoresis in 25% polyacrylamide gels (19:1 acrylamide/bisacrylamide). The gels were run at 7.5 watts for 2 h, 15 min at room temperature and visualized using a 400S PhosphorImager (GE Healthcare). When trinucleotides were used as substrates, the same reaction conditions, as described above, were used. The reaction was stopped by adding 2 μl of 0.5 m EDTA to the reaction. Subsequently, 1 μl of the reaction was applied to a 20 × 20-cm TLC plate using 0.5 m LiCl as solvent. The plate was dried before being exposed overnight to a PhosphorImager screen (GE Healthcare). The screen was scanned, and the result was analyzed using ImageQuant software (GE Healthcare). Data were plotted using the Origin 7 software (OriginLab Corp.).
For time course assays, the reaction volume was 40–90 μl, and incubations were performed at 30 °C for the indicated time. For each time point, 5–10 μl of the reaction volume was removed and stopped by adding 1–2 μl of 0.5 m EDTA. The reaction products were then separated by TLC (20 × 30 cm for ACC and AAC, 20 × 20 cm for the rest of the substrates) and quantified by PhosphorImager analysis as described above.
Kinetic Analysis of PARN Activity
Reaction conditions were as described above. The concentrations of the substrate and PARN or mutants thereof were adjusted so multiple turnover conditions were valid. AAA, UUU, CCC, GGG, ACA, CAC, CCA, ACC, AAC, and CAA were titrated between 0.5 and 400, 20 and 1000, 20 and 1000, 25 and 1000, 20 and 400, 20 and 400, 20 and 400, 3 and 300, 10 and 400, and 2–40 μm, respectively. The concentrations of PARN, PARN(L291A), or PARN(M425A) were between 4 and 209 nm depending on the substrate. The reaction volume was 10 μl, and incubations were performed at 30 °C for 1–30 min depending on the substrate, ensuring that the quantification was done in the linear range of the reaction. For AAA, UUU, GGG, ACA, CCA, CAA, and CCC, the reaction was stopped by adding 2 μl of 0.5 m EDTA to the reaction mixture before applying 1 μl of the solution onto a 20 × 20-cm plate for TLC and proceeded as described above. For CAC, ACC, and AAC, the reaction was stopped by the addition of 10 μl of stop solution (83% formamide, 10 mm Tris-HCl, pH 7.9, 80 mm EDTA, 0.05% xylene cyanol, 0.05% bromphenol blue). The resulting products were analyzed by electrophoresis in 25% polyacrylamide gels (19:1 acrylamide/bisacrylamide). The gels were run at 10 watts for 1 h, 30 min at room temperature and visualized using a 400S PhosphorImager (GE Healthcare). The kinetic constants were calculated by curve fitting to the Michaelis-Menten Equation 3,
 |
using the Origin 7.0 software (OriginLab Corp.).
RESULTS
Degradation of Homopolymeric RNA Substrates
To investigate molecular mechanisms behind the high specificity for PARN in degrading poly(A), we compared the hydrolytic activity when using 20-nucleotide-long homopolymeric RNA substrates, i.e. A20, U20, C20, or G20 in the presence of increasing amounts of PARN (Fig. 1). In keeping with earlier studies (3–5, 10), A20 was the preferred substrate over U20, whereas C20 and in particular G20 were poor substrates for PARN. At higher concentrations of PARN and when using A20 as the substrate, we detected reaction products corresponding to A4 and A2 in addition to the final mononucleotide product A1 (Fig. 1A). Short oligonucleotide products were also observed when we used the polypyrimidine substrates U20 and C20 (Fig. 1, B and C). Interestingly, the length of the short oligopyrimidine reaction products differed compared with the oligonucleotide reaction products that were generated when we used A20 as the substrate. In the case of U20, we could observe three oligonucleotide products being five, four, and two nucleotides in lengths (Fig. 1B), although only the C5 and C4 products were detected when the C20 substrate was used at the two highest tested concentrations of PARN (Fig. 1C). The reason for not detecting the C2 product was probably due to the poorer performance of this substrate relative to the A20 and U20 substrates (see also Table 1). To investigate if the detected oligonucleotide reaction products behaved as true reaction intermediates, we followed the hydrolysis of A20 and U20 during time course experiments. Fig. 1 shows that the oligonucleotide reaction products for both substrates appeared and turned over during the duration of the reaction (Fig. 1, E and F), just as predicted for bona fide reaction intermediates.
FIGURE 1.

Degradation of homopolymeric substrates. 10 nm A20 (A), U20 (B), C20 (C), or G20 (D) was incubated with 1 nm (lane 3), 2 nm (lane 4), 4 nm (lane 5), 8 nm (lane 6), 16 nm (lane 7), 32 nm (lane 8), 64 nm (lane 9), 132 nm (lane 10), and 256 nm (lane 11) PARN, respectively, for 10 min at 30 °C. 10 nm A20 (E) or U20 (F) was incubated with 10 nm PARN for the indicated times at 30 °C. The reacted 5′ end-labeled RNA was fractionated by electrophoresis in 25% polyacrylamide gels (polyacrylamide/bisacrylamide 19:1). Lane 1 in each panel shows the migration of indicated 5′ end-labeled marker oligonucleotides. In lane 2 in each panel, the indicated RNA substrate was incubated in the absence of PARN for 10 min at 30 °C before fractionation by electrophoresis. The positions of the reaction intermediates and products are indicated at the right-hand side of each panel.
TABLE 1.
Summary of kinetic parameters
The kinetic parameters were determined as described under “Experimental Procedures” using the indicated trinucleotide substrate and monitoring the accumulation of dinucleotide product. The given values are average ± experimental error of at least three independent titration series.
| PARN polypeptidea | Substrate | Km | Vmaxb | Vmax/Km | Relative Vmax/Km | Ratioc AAA/ACA | Ratiod AAA/AAC |
|---|---|---|---|---|---|---|---|
| mm | μmol min−1mg−1 | ||||||
| Wild type | AAA | 0.010 ± 0.004 | 0.55 ± 0.23 | 54 ± 31 | 1.0 | 5.4 | 7.8 |
| CCC | 0.16 ± 0.08 | 0.31 ± 0.12 | 1.9 ± 1.2 | 0.035 | |||
| GGG | 1.7 ± 0.1 | 7.2 ± 1.7 | 4.2 ± 1.0 | 0.078 | |||
| UUU | 0.093 ± 0.037 | 0.66 ± 0.31 | 7.1 ± 4.3 | 0.13 | |||
| AAC | 0.098 ± 0.018 | 0.67 ± 0.03 | 6.9 ± 1.3 | 0.13 | |||
| ACA | 0.094 ± 0.027 | 0.96 ± 0.41 | 10 ± 5.3 | 0.19 | |||
| ACC | 0.12 ± 0.01 | 0.24 ± 0.06 | 2.0 ± 0.6 | 0.037 | |||
| CAA | 0.016 ± 0.004 | 0.72 ± 0.20 | 46 ± 17 | 0.85 | |||
| CAC | 0.11 ± 0.01 | 1.1 ± 0.4 | 10 ± 4.1 | 0.19 | |||
| CCA | 0.14 ± 0.07 | 1.7 ± 0.7 | 12 ± 7.4 | 0.22 | |||
| L291A | AAA | 0.041 ± 0.011 | 0.40 ± 0.12 | 9.6 ± 3.8 | 0.18 | 2.2 | 11 |
| AAC | 0.33 ± 0.08 | 0.28 ± 0.05 | 0.85 ± 0.27 | 0.016 | |||
| ACA | 0.12 ± 0.04 | 0.52 ± 0.18 | 4.3 ± 2.1 | 0.080 | |||
| M425A | AAA | 0.017 ± 0.005 | 0.11 ± 0.05 | 6.6 ± 3.7 | 0.12 | 1.8 | 6.6 |
| AAC | 0.12 ± 0.02 | 0.12 ± 0.02 | 1.0 ± 0.3 | 0.019 | |||
| ACA | 0.081 ± 0.028 | 0.29 ± 0.07 | 3.6 ± 1.5 | 0.067 | |||
a PARN mutants were generated by site-directed mutagenesis.
b The given Vmax values are normalized to the used amount of PARN polypeptide or mutant thereof and represent the measured maximum reaction rate for the conversion of the trinucleotide substrate into a dinucleotide product.
c The values represent the calculated ratios between the Vmax/Km values for the AAA and ACA substrates for each of the investigated PARN polypeptides.
d The values represent the calculated ratios between the Vmax/Km values for the AAA and AAC substrates for each of the investigated PARN polypeptides.
The hydrolysis of A20 and U20 implies that PARN can interact with and form complexes with poly(A) and poly(U). It has previously been demonstrated that PARN interacts with poly(A) (11). To investigate if PARN also could interact with poly(U), we performed electrophoretic mobility shift assays using U20 as the probe. Fig. 2 (lanes 1–6) shows that PARN forms a stable complex with U20. The addition of increasing amounts of unlabeled poly(U), poly(G), poly(C), and heteropolymeric single-stranded RNA showed that the complex was competed efficiently by the addition of poly(U) (Fig. 2, lanes 7–11) or heteropolymeric RNA (lanes 22–26), to some extent by poly(G) (lanes 12–16), and very poorly by poly(C) (lanes 17–21). Subsequently, we determined, using a filter binding assay (see under “Experimental Procedures”), the equilibrium dissociation constant (KD) for the PARN-U20 complex and found it to be 4.3 ± 0.8 nm, in the same range as the KD value for the PARN-A20 complex (11).
FIGURE 2.

Identification of a PARN/U20 complex. In lanes 1–6, 10 nm U20 was incubated with 0, 2.5, 5, 10, 20, and 40 μm of PARN, respectively. In lanes 7–26, 10 nm U20 was incubated with 10 μm PARN. In lanes 7–11, 0.0001, 0.001, 0.01, 0.1, and 1 g/liter, respectively, of unlabeled poly(U) was included in the reactions. In lanes 12–16, 0.0001, 0.001, 0.01, 0.1, and 1 g/liter, respectively, of unlabeled poly(G) was included in the reactions. In lanes 17–21, 0.0001, 0.001, 0.01, 0.1, and 1 g/liter, respectively, of unlabeled poly(C) was included in the reactions. In lanes 22–26, 0.0001, 0.001, 0.01, 0.1, and 1 g/liter, respectively, of an unlabeled 44-nucleotide-long heteropolymeric RNA was included in the reactions. Buffer conditions were as described under “Experimental Procedures.” Formed complexes were analyzed by electrophoretic mobility shift assays. O, C, and S denote the locations of origin of electrophoresis, RNA-protein complex, and free RNA, respectively.
Taken together, these sets of analyses confirmed earlier studies (for example Ref. 10) that poly(A) is the preferred substrate for PARN and that poly(U) is a reasonable good substrate. The analyses also revealed that PARN degraded long and short substrates with different efficiencies, as reaction intermediates were detected during the course of the hydrolytic reaction. In the case of A20, we could detect two size classes of oligonucleotide intermediates, and we could detect three size classes of intermediates when we used U20 as the substrate.
One obvious explanation for the appearance of short reaction intermediates could be that short substrates bind less efficiently to PARN than longer substrates. To investigate this possibility, we investigated if the KD value for the PARN-oligo(A) complex was dependent on the length of oligo(A). The results are summarized in Fig. 3 and show that the KD values increased sharply when the substrates were six residues or shorter. We were not able to establish KD values for oligonucleotides being four residues or shorter. Thus, the appearance of the reaction intermediates is in keeping with the proposal that the short intermediates accumulated, at least to some extent, because of their poor interaction with PARN.
FIGURE 3.

RNA length requirement for binding oligo(A) to PARN. RNA-binding properties of PARN were investigated using filter binding assay. KD values for the complexes between PARN and oligo(A) RNA of different lengths (as indicated) were determined as detailed under “Experimental Procedures.”
Active Site of PARN Harbors Specificity for Recognition of Adenosine
The appearance of the short reaction intermediates suggests that A20 and U20 are degraded through at least three and four kinetically distinct reaction phases, respectively. The presence of kinetically distinct reaction phases that depend on the length of the substrate will undoubtedly affect a kinetic analysis with the aim to understand the molecular mechanisms behind the interaction and the specificity for recognition of adenosine residues in the active site of PARN. It is, for example, very likely that substrate-binding properties outside the active site will affect the overall catalytic performance of PARN when long substrates are used relative shorter substrates, as a long substrate besides interacting with the active site also very likely will interact with either or both of the RNA binding regions, i.e. the RRM or the R3H, that are present in PARN. To avoid this particular problem, we therefore used trinucleotides as the substrates in our kinetic analysis of PARN specificity.
Typical results from PARN titration assays, where homotrinucleotides A3, G3, U3, and C3 were used as the substrates, are shown in Fig. 4 and indicate that A3 is the preferred substrate relative to the other three homotrinucleotide substrates. To quantify this observation, we determined the Michaelis-Menten parameters for the four homotrinucleotides. The results are summarized in Table 1 and show that the adenosine-containing trinucleotide substrate was the preferred substrate for PARN. Furthermore, and in accordance with the hydrolytic performance when using 20-nucleotide-long homopolymeric substrates (Fig. 1), U3 was hydrolyzed quite efficiently, whereas the C3 trinucleotide was a poor substrate for PARN. However, and in contrast to the homopolymeric G20 substrate, which was left almost untouched by PARN (Fig. 1D), the G3 substrate was almost as efficiently hydrolyzed by PARN as the U3 substrate. This shows that the active site of PARN has the capacity to accept and hydrolyze guanosine-containing substrates and suggests that the inability of PARN to digest G20 was not caused by properties within the active site. We have not investigated any further the reasons behind the inability to degrade G20, but it seems likely that the formation of G-quartets, which are quadruplex structures formed by guanosine-containing oligonucleotides (reviewed in Ref. 24), could be the main cause for the inability of PARN to degrade the G20 homopolymer. Taken together, the kinetic analysis of the homotrinucleotide substrates revealed that the adenosine trinucleotide substrate was ∼10-fold more efficiently hydrolyzed than the guanosine or uridine trinucleotide substrates and ∼30-fold more efficiently used than the C3 substrates. Thus, we conclude that the active site of PARN per se harbors specificity for recognition of adenosine.
FIGURE 4.

Degradation of homopolymeric trinucleotide substrates. 2 nm of indicated 5′ end-labeled AAA, GGG, UUU, or CCC trinucleotide substrate was incubated with the indicated amount of PARN for 10 min at 30 °C. An aliquot of the reacted RNA was analyzed by one-dimensional TLC, as described under “Experimental Procedures.” The percent of reacted trinucleotide substrate was calculated and plotted against the concentration of PARN.
Importance of the Penultimate and 3′ End-located Nucleotides
To further characterize the mechanisms behind adenosine recognition in the active site of PARN, we compared the catalytic performance of PARN when using trinucleotide substrates containing either one or two adenosine residues, i.e. the trinucleotides AXX, XAX, XXA, AAX, AXA, and XAA, where X denotes G, U, or C. Results from such analyses are shown in supplemental Fig. S1 and indicate the following. (i) The two nucleotides surrounding the scissile bond are primary determinants for adenosine specificity in the active site of PARN. (ii) Substrates containing at least one cytosine residue either at the penultimate or 3′-terminal positions in general are poorer substrates than the corresponding substrates containing at least one guanosine or uridine residue at these positions. (iii) The 5′ end-located nucleotide of the trinucleotide substrate does not appear to play any critical role for adenosine recognition by the active site of PARN.
To investigate in greater detail if these proposals are correct, we focused our studies on the eight adenosine/cytosine-containing trinucleotides, because our initial screening analysis presented in supplemental Fig. S1 revealed larger differences in catalytic performance among such substrates than among the corresponding adenine/guanosine or adenine/uridine containing trinucleotide substrates. First, we investigated the degradation of the eight substrates during a time course experiment (Fig. 5), and subsequently we determined the Michaelis-Menten parameters for the eight selected substrates (Table 1). Based on these two analyses, we could identify three categories of substrates as follows: an efficient group consisting of the CAA and AAA trinucleotides; an intermediate group wherein all the members contained one adenosine residue either at the penultimate or at the terminal 3′ end position; and finally a very inefficient group of substrates wherein neither of the two trinucleotides, i.e. ACC and CCC, contained an adenosine residue at the penultimate or the terminal 3′ end position. An analysis of the kinetic parameters in Table 1 revealed that the poorer performance of the intermediate group relative to AAA and CAA was primarily due to an increased Km value and that the poorer performance of the inefficient group, i.e. ACC and CCC, relative to the intermediate group was caused by a drop in the Vmax value. Taken together, the kinetic analysis confirmed that the two nucleotides located at the penultimate and at the terminal 3′ end positions, i.e. the two nucleotides surrounding the scissile bond, play an important role as key residues required for the high specificity of PARN in degrading adenosine-containing substrates.
FIGURE 5.

Nucleotides surrounding the scissile bond are primary determinants for adenosine specificity. 20 μm of indicated 5′ end-labeled trinucleotide substrate was incubated with 20 nm PARN at 30 °C, as detailed under “Experimental Procedures.” At each time point, a sample of the reaction was taken out, stopped by the addition of EDTA and fractionated by one-dimensional TLC, as detailed under “Experimental Procedures.” The amount of mononucleotides released was calculated and plotted against time.
Identification of Amino Acids in the Active Site That Interact with Adenosine Residues
Two amino acid residues, i.e. Phe-115 and Ile-34, located in the active site of PARN have recently been shown to participate both structurally and functionally in the recognition of adenosine residues (15). The residue Phe-115 stacks against the 3′ end-located adenosine base, whereas the residue Ile-34 stacks against the base of the penultimate nucleotide. Furthermore, it was observed in the structure of PARN in complex with the cap analogue m7GpppG (16) that the first transcribed guanosine nucleotide of the cap structure was located in a hydrophobic pocket comprising amino acid residues Ile-34, Leu-57, Leu-290, Leu-291, and Met-425 and that the guanosine base in the closed conformation of the active site co-localized with the adenosine base of the penultimate nucleotide when poly(A) is bound to the active site. Of these residues, Leu-291 and Met-425 clamp the base of the first transcribed guanosine residue and thus must contribute significantly to the interaction between PARN and the guanosine base. Because of this, we decided to investigate if any of these two amino acid residues, in addition to the previously identified Ile-34 residue, could play a role in the recognition of the penultimate adenosine base. For this purpose, we investigated the catalytic performance of the two mutant polypeptides, PARN(L291A) and PARN(M425A), using three different substrates, i.e. AAA, ACA, and AAC. The reason for using these three substrates was that we would expect a smaller relative difference in catalytic performance between the two substrates AAA and ACA if any of the two residues Leu-291 and Met-425 played a role in the recognition of the adenosine base at the penultimate position, whereas a correspondingly smaller relative difference would not be observed between the two substrates AAA and AAC unless any of the two amino acid residues Leu-291 or Met-425 played a role in the recognition of the 3′ end-located adenosine base. The results from these comparisons are shown in Table 1, and it can be concluded that both mutant polypeptides were defective in their catalytic performance relative to the wild type PARN polypeptide. Interestingly, both mutant polypeptides showed a defect in their specificity toward the ACA substrate as they could not select as well as the wild type PARN polypeptide could against the presence of a cytosine residue located at the penultimate position of the substrate, i.e. wild type PARN showed a 5-fold preference for the AAA substrate versus ACA, whereas both mutants showed a 2-fold preference for AAA versus ACA. In contrast, both mutant PARN polypeptides could still select against a cytosine residue at the terminal 3′ end position. Taken together, we conclude that both amino acid residues Leu-291 and Met-425 most likely participate in providing specificity toward the recognition of an adenosine residue at the penultimate position.
DISCUSSION
In this study, we have investigated molecular mechanisms behind the high specificity by PARN to degrade poly(A). Most importantly, our study revealed that the active site of PARN per se harbors specificity for the recognition of adenosine residues (Fig. 4, Fig. 5, and Table 1). We also found that the two nucleotides surrounding the scissile bond were critical residues behind the high specificity (Table 1, Fig. 5, and supplemental Fig. S1), suggesting that both these two residues are specifically recognized by amino acid residues in the active site of PARN.
Two amino acid residues of PARN, i.e. Phe-115 and Ile-34, have previously been identified as critical residues required for efficient hydrolysis of poly(A) and recognition of the 3′ end-located adenosine residue (15). In addition to these two amino acid residues, we identified two residues, i.e. Leu-291 and Met-425 (Table 1), that were required for efficient hydrolysis and very likely also participated in the recognition of the penultimate adenosine residue of the substrate. Thus, four amino acid residues have so far experimentally been identified as candidate residues that participate in the recognition of adenosine residues in the active site of PARN. At present, it is not obvious how these four residues would discriminate an adenosine base from the other nucleotide bases because none of them appears to form base-specific hydrogen bonds with the adenosine base. This implies that other amino acid residues beside the four that have been identified so far must play a role in the recognition of adenosine residues by the active site of PARN. Despite this uncertainty, this study has nevertheless revealed the presence of two nucleotide binding pockets in the active site of PARN that interact and recognize the bases of the two nucleotides surrounding the scissile phosphodiester bond that PARN hydrolyzes. It is obvious that further studies, and in particular a crystal structure of the binary PARN-poly(A) complex wherein amino acid residues that interact with the adenosine bases in the active site are visualized, are required to fully understand how the active site of PARN specifically recognizes adenosine residues.
We have recently reported that the position of the penultimate base of the poly(A) substrate partially overlaps with the binding site of the first transcribed nucleotide of the cap structure (16). This suggests and is in agreement with our current study that this binding pocket can accommodate other bases than adenosine and yet provide enough specificity to preferentially hydrolyze adenosine-containing substrates. In light of this, it is interesting to note that a recent structural study of the related Schizosaccharomyces pombe DEDDh deadenylase Pop2p (25, 26) has revealed the presence of a highly dynamic active site that participates in providing both specificity for substrate recognition and movements required for catalytic action. The dynamic nature of the Pop2p active site could very well be a property that is shared by the active site of PARN and thereby provides a possibility for the PARN active site on one subunit to accommodate nucleotides belonging to one end of the mRNA, i.e. either the cap structure or the poly(A) tail, whereas the other subunit at the same time could accommodate the other end of the mRNA substrate.
One interesting observation in this study was the identification of the three kinetically distinct reaction phases by which PARN degraded an oligonucleotide consisting of 20 adenosine residues (Fig. 1). The fast phase resulted in the accumulation of an intermediate product being 4 nucleotides long. The accumulation of such an intermediate could be due to the presence of an RNA-binding domain located 5 nucleotides away from the active site. In the most recently determined structure of PARN (16), the RRM was located at approximately a distance from the active site of PARN that corresponds to the length of a 5-nucleotide-long oligonucleotide. It is therefore tempting to speculate that the RRM of PARN fulfills the role as an RNA-binding site that docks and even participates in pushing the poly(A) substrate into the active site of PARN. It is worthwhile to mention that a similar scenario has been proposed and supported by structural evidence for RNase II (27, 28). The accumulation of the intermediate being two adenosine residues in length is most likely due to differences in substrate binding properties within the active site per se.
Recently, several poly(U) polymerases that add short oligo(U) tails to the 3′ end of RNA have been discovered in eukaryotic cells (29). Direct evidence for the presence of oligo(U) tails at the 3′ ends of histone mRNA (30) and to polyadenylated actin mRNA (31) has been obtained. The functional significance of the addition of oligo(U) tails is not yet fully understood, although it appears likely that the oligo(U) tails could play a role in both RNA stabilization and destabilization (reviewed in Refs. 32, 33). Most recently, it has been shown that the 3′-terminal oligo(U) tract mediates stimulation of decapping (34). In a novel pathway of bulk mRNA turnover in S. pombe, adding uridines to the poly(A) tail of mRNA (3′-uridylation) has been shown to initiate the degradation pathway by stimulating decapping (35). These results indicated that the oligo(U) tails at the 3′ end of mRNA play an important role in mRNA decay. Exonucleolytic degradation of poly(U) suggests that poly(U)-degrading enzymes are present in eukaryotic cells. In view of this, it will be interesting in the future to investigate if the poly(U) degrading activity of PARN could play a role in mRNA decay involving oligo(U) tails.
Supplementary Material
Acknowledgments
We thank Måns Ehrenberg, Pontus Larsson, Lei Liu Conze, Jens Schuster, Susanne Stier, Assad Alhaboub, and Kyriakos Kokkoris for valuable suggestions throughout the completion of this work.
This work was supported in part by the Swedish Research Council, the Linneus Support from the Swedish Research Council to the Uppsala RNA Research Centre, and the Lennanders Foundation at Uppsala University.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- PARN
- poly(A)-specific ribonuclease
- RRM
- RNA recognition motif.
REFERENCES
- 1.Garneau N. L., Wilusz J., Wilusz C. J. (2007) Nat. Rev. Mol. Cell Biol. 8, 113–126 [DOI] [PubMed] [Google Scholar]
- 2.Parker R., Song H. (2004) Nat. Struct. Mol. Biol. 11, 121–127 [DOI] [PubMed] [Google Scholar]
- 3.Aström J., Aström A., Virtanen A. (1991) EMBO J. 10, 3067–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aström J., Aström A., Virtanen A. (1992) J. Biol. Chem. 267, 18154–18159 [PubMed] [Google Scholar]
- 5.Körner C. G., Wahle E. (1997) J. Biol. Chem. 272, 10448–10456 [DOI] [PubMed] [Google Scholar]
- 6.Körner C. G., Wormington M., Muckenthaler M., Schneider S., Dehlin E., Wahle E. (1998) EMBO J. 17, 5427–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstrohm A. C., Wickens M. (2008) Nat. Rev. Mol. Cell Biol. 9, 337–344 [DOI] [PubMed] [Google Scholar]
- 8.Dehlin E., Wormington M., Körner C. G., Wahle E. (2000) EMBO J. 19, 1079–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao M., Fritz D. T., Ford L. P., Wilusz J. (2000) Mol. Cell 5, 479–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez J., Ren Y. G., Thuresson A. C., Hellman U., Astrom J., Virtanen A. (2000) J. Biol. Chem. 275, 24222–24230 [DOI] [PubMed] [Google Scholar]
- 11.Nilsson P., Henriksson N., Niedzwiecka A., Balatsos N. A., Kokkoris K., Eriksson J., Virtanen A. (2007) J. Biol. Chem. 282, 32902–32911 [DOI] [PubMed] [Google Scholar]
- 12.Martînez J., Ren Y. G., Nilsson P., Ehrenberg M., Virtanen A. (2001) J. Biol. Chem. 276, 27923–27929 [DOI] [PubMed] [Google Scholar]
- 13.Moser M. J., Holley W. R., Chatterjee A., Mian I. S. (1997) Nucleic Acids Res. 25, 5110–5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuo Y., Deutscher M. P. (2001) Nucleic Acids Res. 29, 1017–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu M., Reuter M., Lilie H., Liu Y., Wahle E., Song H. (2005) EMBO J. 24, 4082–4093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu M., Nilsson P., Henriksson N., Niedzwiecka A., Lim M. K., Cheng Z., Kokkoris K., Virtanen A., Song H. (2009) Structure 17, 276–286 [DOI] [PubMed] [Google Scholar]
- 17.Ren Y. G., Kirsebom L. A., Virtanen A. (2004) J. Biol. Chem. 279, 48702–48706 [DOI] [PubMed] [Google Scholar]
- 18.Ren Y. G., Martínez J., Virtanen A. (2002) J. Biol. Chem. 277, 5982–5987 [DOI] [PubMed] [Google Scholar]
- 19.Cléry A., Blatter M., Allain F. H. (2008) Curr. Opin. Struct. Biol. 18, 290–298 [DOI] [PubMed] [Google Scholar]
- 20.Maris C., Dominguez C., Allain F. H. (2005) FEBS J. 272, 2118–2131 [DOI] [PubMed] [Google Scholar]
- 21.Monecke T., Schell S., Dickmanns A., Ficner R. (2008) J. Mol. Biol. 382, 827–834 [DOI] [PubMed] [Google Scholar]
- 22.Nagata T., Suzuki S., Endo R., Shirouzu M., Terada T., Inoue M., Kigawa T., Kobayashi N., Güntert P., Tanaka A., Hayashizaki Y., Muto Y., Yokoyama S. (2008) Nucleic Acids Res. 36, 4754–4767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nilsson P., Virtanen A. (2006) Int. J. Biol. Macromol. 39, 95–99 [DOI] [PubMed] [Google Scholar]
- 24.Davis J. T. (2004) Angew. Chem. Int. Ed. Engl. 43, 668–698 [DOI] [PubMed] [Google Scholar]
- 25.Andersen K. R., Jonstrup A. T., Van L. B., Brodersen D. E. (2009) RNA 15, 850–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jonstrup A. T., Andersen K. R., Van L. B., Brodersen D. E. (2007) Nucleic Acids Res. 35, 3153–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amblar M., Barbas A., Fialho A. M., Arraiano C. M. (2006) J. Mol. Biol. 360, 921–933 [DOI] [PubMed] [Google Scholar]
- 28.Frazão C., McVey C. E., Amblar M., Barbas A., Vonrhein C., Arraiano C. M., Carrondo M. A. (2006) Nature 443, 110–114 [DOI] [PubMed] [Google Scholar]
- 29.Kwak J. E., Wickens M. (2007) RNA 13, 860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullen T. E., Marzluff W. F. (2008) Genes Dev. 22, 50–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rissland O. S., Mikulasova A., Norbury C. J. (2007) Mol. Cell. Biol. 27, 3612–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rissland O. S., Norbury C. J. (2008) Biochim. Biophys. Acta 1779, 286–294 [DOI] [PubMed] [Google Scholar]
- 33.Wilusz C. J., Wilusz J. (2008) Genes Dev. 22, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song M. G., Kiledjian M. (2007) RNA 13, 2356–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rissland O. S., Norbury C. J. (2009) Nat. Struct. Mol. Biol. 16, 616–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.