

Abstract
The differentiation of monocytes into macrophages and dendritic cells is accompanied by induction of cell-surface neuraminidase 1 (Neu1) and cathepsin A (CathA), the latter forming a complex with and activating Neu1. To clarify the biological importance of this phenomenon we have developed the gene-targeted mouse models of a CathA deficiency (CathAS190A) and a double CathA/Neu1 deficiency (CathAS190A-Neo). Macrophages of CathAS190A-Neo mice and their immature dendritic cells showed a significantly reduced capacity to engulf Gram-positive and Gram-negative bacteria and positively and negatively charged polymer beads as well as IgG-opsonized beads and erythrocytes. Properties of the cells derived from CathAS190A mice were indistinguishable from those of wild-type controls, suggesting that the absence of Neu1, which results in the increased sialylation of the cell surface proteins, probably affects multiple receptors for phagocytosis. Indeed, treatment of the cells with purified mouse Neu1 reduced surface sialylation and restored phagocytosis. Because Neu1-deficient cells showed reduced internalization of IgG-opsonized sheep erythrocytes whereas binding of the erythrocytes to the cells at 4 °C persisted, we speculate that the absence of Neu1 in particular affected transduction of signals from the Fc receptors for immunoglobulin G (FcγR). Indeed the macrophages from the Neu1-deficient mice showed increased sialylation and impaired phosphorylation of FcγR as well as markedly reduced phosphorylation of Syk kinase in response to treatment with IgG-opsonized beads. Altogether our data suggest that the cell surface Neu1 activates the phagocytosis in macrophages and dendritic cells through desialylation of surface receptors, thus, contributing to their functional integrity.
Keywords: Cell/Phagocytosis, Glycoproteins/Lysosomal, Immunology/Cellular response, Subcellular Organelles/Lysosomes, macrophage, sialidase/neuraminidase
Introduction
Sialic acids are abundantly expressed on the surface of immune cells and implicated in mediating recognition between the cells and between the cells and extracellular matrix as well as between the cells and a range of pathogenic viruses, bacteria, and protozoa during the inflammatory and immune reactions (for review, see Ref. 1). For example, members of the Siglecs (sialic acid binding immunoglobulin-like lectins) superfamily are known to mediate many of these interactions contributing, in particular, to scavenging function of macrophages, pathogen uptake, and antigen presentation (for review, see Ref. 2). Similarly, it was established that desialylation of the cell surface with sialidases (neuraminidases) substantially enhances the capacity of resting B cells to stimulate the proliferation of allogeneic-and antigen-specific syngeneic T cells (3–7), suggesting the important role of mammalian sialidases and sialotransferases in the immune function.
Previous data showed that mammalian neuraminidase 1 (Neu1),5 in addition to its role in the intralysosomal catabolism, may be also involved in cellular signaling during the immune response. In particular, during the activation of mouse T cells, Neu1 is expressed on the plasma membrane and is required for the early production of interleukin-4 and for the interaction of T cells with the antigen-presenting cells (8–12). In addition, Neu1 of T cells converts the group-specific component (Gc protein) into a factor necessary for the inflammation-primed activation of macrophages (13, 14). T cells derived from SM/J or B10.SM strains of mice with the reduced Neu1 activity, due to a missense mutation in the Neu1 gene (15), fail to convert Gc and synthesize interleukin-4, whereas B cells of these mice cannot produce IgG1 and IgE after immunization with pertussis toxin (8, 14, 16). Strikingly, surface desialylation of macrophages by viral sialidase from Arthrobacter ureafaciens significantly increases their capacity for phagocytosis of influenza virus-infected HeLa cells (17), providing the direct link between the surface sialylation of antigen-presenting cells and their biological activity.
Previously we showed that Neu1 increased 14-fold during the differentiation of human monocytes into macrophages (18). We further showed that a majority of Neu1 in macrophages is targeted to the cell surface (19). Activation and stabilization of Neu1 in the lysosome requires its association with the lysosomal multienzyme complex, also containing the lysosomal carboxypeptidase A (cathepsin A/protective protein, CathA), β-galactosidase, and N-acetylgalactosamine-6-sulfate sulfatase (for review, see Ref. 20). During the differentiation of macrophages, part of CathA is sorted to the plasma membrane similarly to Neu1. Both proteins are first targeted to the lysosome and then to LAMP-2-negative, major histocompatibility complex class II-positive vesicles later merged with the plasma membrane (19).
To clarify the physiological role of Neu1 and CathA in the antigen-presenting immune cells, we have compared essential functional properties of macrophages (MØ) and immature dendritic cells (DC) from the genetically targeted mice having either a single CathA or a double CathA/Neu1 deficiency (21). The obtained data suggest that the cell surface Neu1 activates the phagocytosis in MØ and DC presumably through desialylation of multiple receptors, including Fc receptors for immunoglobulin G (FcγR).
EXPERIMENTAL PROCEDURES
Animals
CathAS190A mice carrying the point c.571AGC>GCA (S190A) mutation in the CathA gene and CathAS190A-Neo mice carrying, in addition, a PGK-Neo cassette in intron 7 were generated through the targeted disruption of the CathA gene as described (21). The CathAS190A-Neo and CathAS190A mice ranging from 4 to 12 weeks of age were compared with the appropriate wild-type (WT) littermate controls. All mice were bred and maintained in the Canadian Council on Animal Care (CCAC)-accredited animal facilities of the Ste. Justine Hospital Research Center according to the CCAC guidelines. Approval for the experiments was granted by the Animal Care and Use Committee of the Ste. Justine Hospital Research Center.
Isolation and Culturing of Murine Peritoneal MØ
To obtain inflammatory MØ, 1.0 ml of 3% Brewer thioglycollate medium (Sigma) was injected into peritoneum of mice. Peritoneal macrophages were harvested 5–7 days later by washing the peritoneal cavity of mice with 10 ml of sterile PBS. The harvested cells were cultured at a density of 2 × 106 cells/ml in RPMI medium 1640 (Invitrogen). After 24 h non-adherent cells were removed by two washes with PBS, and the adherent MØ were used for different purposes as described.
Isolation and Culturing of Murine Splenocytes, Splenocyte-derived MØ, and Splenocyte-derived DC
Fresh splenocytes were obtained by teasing the mouse spleens under aseptic conditions. Erythrocytes were removed by centrifugation over Ficoll-Paque gradient, and the remaining unfractionated nucleated spleen cells were washed twice with PBS and adjusted to a density of 7.5 × 105 of cells/ml of RPMI 1640 medium containing 10% of normal mouse serum (EMD Chemicals).
To obtain splenocytes-derived MØ, 4 × 106 splenocytes/well in 6-well tissue culture plates (Costar) were suspended in RPMI 1640 medium and incubated for 2 h at 37 °C in a humidified atmosphere containing 5% CO2. After 2 h non-adherent cells were removed by 2 washes with PBS, and the adherent monocytes were maintained for 7 days to differentiate into MØ (larger and more granular than monocytes as seen by light microscopy) in RPMI 1640 medium containing 10% fetal calf serum and 5% normal mouse serum. The cells were confirmed to have characteristic macrophage cell surface phenotypic markers (CD14+, CD206+) by flow cytometry.
Immature splenocyte-derived DC were obtained as described (22). Purified mouse splenocytes were suspended at a density of 7.5 × 105 of cells/ml in Iscove's modified Dulbecco's medium (Invitrogen) supplemented with 12.5% heat-inactivated mouse serum, nonessential amino acids (0.1 mm/ml), sodium pyruvate (1 mm/ml), 2-mercaptoethanol (50 μm), penicillin (5000 units/ml), streptomycin (50 mg/ml), and Fungizone (0.5%). To generate immature DC, 2.0 ml of this suspension was plated per 35-mm tissue culture dish (Nunclon) and cultured for the first 6 days in the presence of interleukin-6 (25 ng/ml), flt-3L (25 ng/ml), and granulocyte-macrophage colony-stimulating factor (1000 units/ml) (R&D Systems). On day 7 the cells were washed, counted, and diluted to a concentration of 1.5 × 105 of cells per ml in complete medium and cultured in the presence of flt-3L (25 ng/ml) and granulocyte-macrophage colony-stimulating factor (1000 units/ml) during the next 4 weeks. The cells were split again on days 10, 14, 20, and 26 by replating at 4–6 × 105 cells/ml. To obtain mature splenocyte-derived DC, the cells were cultured for an additional 48 h in the presence of 10 μg/ml lipopolysaccharides (Escherichia coli 026:B6, Sigma).
Isolation and Culturing of Murine Monocyte-derived MØ
Murine blood was drawn by cardiac puncture and peripheral blood mononuclear cells were isolated by subsequent centrifugations over Ficoll-Paque (Amersham Biosciences) gradients as described (19). To isolate monocytes, the peripheral blood mononuclear cells were suspended in RPMI 1640 medium and incubated for 2 h at 37 °C in a humidified atmosphere containing 5% CO2. After 2 h non-adherent cells were removed by 2 washes with PBS. To obtain monocyte-derived MØ, the adherent murine monocytes were maintained for 5–7 days in RPMI 1640 medium containing 10% fetal calf serum and 5% heat-inactivated mouse serum. The harvested cells were confirmed to have characteristic macrophage cell surface phenotypic markers (CD14+, CD206+) by flow cytometry as described below.
Immunofluorescent Labeling and Flow Cytometry
Monoclonal antibodies conjugated with fluorescein isothiocyanate (FITC) or phycoerythrin (PE) against mouse major histocompatibility complex class II (anti-I-A), HL3-PE (anti-CD11c), RMMP-1 (anti-CD86-PE, B7–2), CD45, and CD14 were purchased from eBioscience. Cell surface phenotype was studied by flow cytometry. Adherent cells were detached by pipetting after 3–5 min of incubation at 37 °C in PBS containing 3 mm EDTA. Aliquots of the cells (1–2 × 106) were washed with cold PBS and incubated on ice for 30 min with the FITC or phycoerythrin-conjugated antibodies in the dilutions recommended by the manufacturer. The cells were then washed 3 times with PBS and fixed with 4% paraformaldehyde for 10 min. Flow cytometry was performed on a FACSCalibur instrument (BD Biosciences). At least 1 × 105 cells were acquired in each run, and the results were analyzed using CellQuest Pro Software.
Enzyme Assays
Sialidase activity in cellular lysates was assayed using the synthetic fluorogenic substrate 2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid (4MU-NeuAc; Sigma) as described (23) with the following modifications. Freshly isolated cells (2–5 × 106) were homogenized by sonication in 0.20 ml of a solution containing 0.5% (v/v) Triton X-100, 0.05 m sodium acetate, pH 4.4, and incubated with 0.125 mm 4MU-NeuAc at 37 °C for 1 h. The reaction was terminated by the addition of 1.9 ml of 0.4 m glycine buffer, pH 10.5. Liberated 4-methylumbelliferone was measured using a Shimadzu RF-5301 spectrofluorometer with excitation at 355 nm and emission at 460 nm. Protein concentration was measured using a protein assay kit (Bio-Rad).
β-Hexosaminidase activity in cellular homogenates was assayed with 1.25 mm 4-methylumbelliferyl-2-acetamido-2-deoxy-β-d-glucopyranoside as previously described (24). After the incubation at 37 °C for 15 min, the reaction was terminated with 1.9 ml of 0.4 m glycine buffer, pH 10.4, and the fluorescence of the liberated 4-methylumbelliferone was measured as described above. The carboxypeptidase activity of CathA was measured as follows. Freshly isolated cells (2–5 × 106) were homogenized in 0.20 ml of water containing 0.5% (v/v) Triton X-100 and incubated with 100 μl of 1.5 mm benzyloxycarbonyl-Phe-Leu (Sigma) in 100 mm sodium acetate buffer, pH 5.2, at 37 °C for 30 min. The reaction was terminated by the addition of 20 μl of trichloroacetic acid. Precipitate was removed by 10 min of centrifugation at 12,000 × g, and 100 μl of the supernatant was mixed with 1.9 ml of the 0.05 m sodium borate buffer, pH 9.5, containing phthaldialdehyde (0.15 mg/ml) and β-mercaptoethanol (0.075 mg/ml). After 5 min of incubation, the concentration of leucine was measured using a Shimadzu RF-5301 spectrofluorometer with excitation at 340 nm and emission at 455 nm (25).
Phagocytosis of Bacteria, Polymer Beads, and IgG-opsonized Polymer Beads
MØ or immature DC seeded on coverslips were incubated for 3 h with fluorescein-labeled E. coli (K-12 strain) of Staphylococcus aureus (Wood strain) bioparticles (both from Molecular Probes) in a ratio of 50 particles/cell at 37 or 4 °C, washed 3 times with cold PBS, fixed with 4% paraformaldehyde for 1 h, permeabilized with 0.5% Triton X-100, and washed twice with 0.05% Tween 20 in PBS. Cells were counterstained with 3 μm DAPI and washed again with 0.05% Tween 20 in PBS. Slides were examined using the Nikon Eclipse E6000 epifluorescence microscope. The ingestion index was calculated as a percentage of cells with engulfed particles multiplied by the average number of particles engulfed per cell. The background level measured at 4 °C was subtracted from all values. For quantitative analysis, after the incubation with bacteria, cells were washed with ice-cold PBS, fixed in 4% paraformaldehyde, and analyzed by flow cytometry as described above.
To study phagocytosis of polymer beads, peritoneal macrophages were incubated at 37 °C or at 4 °C with fluorescent polystyrene latex beads (∼2-μm diameter) modified with either positively charged amino groups (L0905, Sigma) or negatively charged carboxylate groups (LB30, Sigma) at a ratio of 50 beads/cell. After the 3-h incubation with beads, cells were washed with ice-cold PBS, fixed in 4% paraformaldehyde, and analyzed by flow cytometry.
To study the FcγR-mediated phagocytosis, 1.5 × 106 carboxylate-modified beads were covalently opsonized with 100 μg of mouse IgG1 (Sigma) by a carbodiimide coupling reaction for 1 h at room temperature in 50 μl of 50 mm MES buffer, pH 6.1. Beads were washed 3 times and resuspended in PBS. MØ (106/well, plated in 6-well pates) were exposed to IgG-opsonized beads at a ratio of 10 beads/cell at 37 °C. After the 3-h incubation with the beads, cells were washed with ice-cold PBS, fixed in 4% paraformaldehyde, and analyzed by flow cytometry.
To study the effect of exogenous mouse Neu1 on phagocytosis of IgG-opsonized beads, the cells were pretreated overnight by the medium supplemented with Neu1 (1 milliunits/ml of medium) isolated from mouse kidney tissue by the affinity chromatography on concanavalin A and p-aminophenyl-β-d-thiogalactopyranoside-agarose (Sigma) columns as previously described (26). The cells were washed twice with culture medium, and the assay proceeded as described above.
FcγR-mediated Signaling
After 20 min of incubation at 37 °C with IgG-opsonized beads at a ratio of 10 beads/cell, mouse-cultured macrophages were washed with ice-cold PBS and harvested by gentle scraping with a polyethylene cell scraper and centrifugation at 800 × g for 10 min. Cells were lysed by sonication in ice-cold RIPA buffer containing complete cocktails of protease inhibitors (Sigma) and phosphatase inhibitors (PhosSTOP, Roche Diagnostics). After the removal of cell debris by 5 min of centrifugation at 13,000 × g, proteins from cell lysates were resolved by SDS-polyacrylamide gel electrophoresis using NuPAGE 4–12% Bis-Tris gels (Invitrogen) and electrotransferred to nitrocellulose membranes. Syk and phospho-Tyr346 Syk detection was performed with rabbit polyclonal antibodies (Cell Signaling, dilution 1:1000). Detection was performed using Lumi-Light Western blotting Substrate (Roche Diagnostics) in accordance with the manufacturer's protocol.
For detection of phosphorylated FcγR, macrophage lysates were processed to immunoprecipitate the receptor using polyclonal anti-mouse FcγRI/CD64 antibodies (RD Systems) specific against both α and γ chains of the receptor at a final concentration of 10 μg/ml lysate essentially as described (12). Immunoprecipitated receptors were resolved by SDS-polyacrylamide gel electrophoresis using NuPAGE 4–12% gels and electrotransferred to nitrocellulose membrane. Detection was performed with either anti-phosphotyrosine mouse monoclonal antibodies (Tyr(P)-100 from Cell Signaling; dilution 1:2000) or anti-mouse FcγRI/CD64 antibodies (dilution 1:10,000) as described above.
Binding and Phagocytosis of Antibody-coated Sheep Erythrocytes
Sheep erythrocytes (SE, Innovative Research) were coated with anti-sheep blood cells antibodies (Anti-Sheep Red Blood Cell Stroma antibody, Sigma S8014) by incubating them with antibodies in sub-agglutinating concentrations in magnesium-free PBS. Mouse peritoneal macrophages grown on glass coverslips were incubated with SE for 30 min either at 37 °C or on ice. Unbound SE were washed away, and coverslips were treated with 2% glutaraldehyde in PBS and mounted to observe the binding of SE to macrophages. To observe internalization, some coverslips were incubated in 1.4% NH4Cl at room temperature for 5 min to lyse uninternalized SE before fixation. Then the cells were fixed as above, mounted, and studied by phase-contrast microscopy on a Nikon Eclipse E6000 microscope.
Lectin Fluorescence Microscopy and Lectin Blot of FcγR
The splenocyte-derived murine MØ or peritoneal MØ seeded on coverslips were incubated for 15 min at 37 °C with 100 μg/ml fluorescein-labeled Maackia amurensis lectin (EY Laboratories, Inc.). Then the cells were washed 3 times with cold PBS and fixed with 4% paraformaldehyde for 1 h. Cells were counterstained with 3 μm DAPI and washed again with 0.05% Tween 20 in PBS. Slides were examined using the Nikon Eclipse E6000 epifluorescence microscope.
To perform a lectin blot of FcγR, immunoprecipitated receptors were resolved by SDS-PAGE and transferred to a polyvinylidene fluoride membrane as described above. The blots were probed with biotinylated M. amurensis lectin II (MAL-2; Vector Laboratories Inc., Burlington, Ontario, Canada, dilution 1:2000 in 5% bovine serum albumin in 0.1% Tris-buffered saline, Tween) for 3 h at room temperature followed by horseradish peroxidase-conjugated streptavidin and Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences). To determine protein loading, the blot was stained with Ponceau S (Sigma).
RESULTS
Macrophages and DC Derived from CathAS190A-Neo but Not from CathAS190A Mice Show the Reduced Induction of Sialidase Activity during the Differentiation
We have previously generated animal models of single CathA deficiency and double CathA/Neu1 deficiency by gene targeting in mice (21). In the first strain (CathAS190A) we replaced the nucleophil of the CathA active site, Ser190, with Ala, which abolished the enzymatic activity, whereas the mutant protein retained its ability to activate Neu1. The second strain, CathAS190A-Neo, with a PGK-Neo cassette inserted in the noncoding region of CathA gene, had significantly reduced (∼10% of normal) Neu1 tissue activity due to dramatically decreased CathA mRNA levels, consistent with previously reported hypomorphic effects of the Neo gene (27–29). Both CathAS190A-Neo and CathAS190A mice were vital and fertile and had a normal growth progression and a normal lifespan.
To find whether the immune cells of CathAS190A-Neo and CathAS190A mice showed a decrease in the CathA and sialidase activities similar to that observed in non-lymphatic tissues, we have assayed both activities in the blood monocytes and in the monocyte-derived MØ. We have also assayed CathA and sialidase activities in the monocytes purified from the spleen tissues as well as in splenocyte-derived MØ, immature, and mature DC. Only background levels (10–15%) of the carboxypeptidase activity measured with the specific CathA substrate benzyloxycarbonyl-Phe-Leu could be detected in blood monocytes and MØ, both from CathAS190A-Neo and CathAS190A mice (Fig. 1A). This activity does not change upon the differentiation and reflects the background level of benzyloxycarbonyl-Phe-Leu conversion by other cellular carboxypeptidases. In the cells from WT mice, CathA activity increased but not significantly (Fig. 1A). Similar results were obtained also for the spleen-derived monocytes, MØ, and DC (Fig. 1A). In contrast, a drastic increase of the sialidase activity was observed during differentiation of the monocytes derived from blood or spleen of WT and CathAS190A mice (Fig. 1B). As compared with both spleen and blood-derived monocytes, the sialidase activity was increased at least 2-fold in MØ, at least 4-fold in immature DC, and at least 5-fold in the mature DC. MØ from CathAS190A-Neo mice (both blood- and spleen-derived) showed sialidase activity almost similar to that in the corresponding non-differentiated monocytes, confirming our previous report that the increased sialidase activity at the cell surface of MØ results from the induced expression of the Neu1 gene (18, 19). Although the sialidase activity in immature and mature DC from CathAS190A-Neo mice increased as compared with monocytes, it still remained much lower than in DC from WT or CathAS190A mice. The control lysosomal enzyme, β-hexosaminidase, showed the same activity in the cells from all mice (not shown).
FIGURE 1.

Enzyme activity in the mouse immune cells. Bars show specific CathA activity against benzyloxycarbonyl-Phe-Leu (A) and specific sialidase activity against the synthetic substrate, 4MU-NeuAc (B) in total lysates of blood-derived and spleen-derived monocytes (MC) and macrophages (MØ) as well as of the splenocyte-derived immature (IDC) and mature (MDC) dendritic cells. Values represent the means ± S. D. of three independent experiments. PBMC, peripheral blood mononuclear cells.
Macrophages and Immature DC from CathAS190A-Neo Mice Show Impaired Phagocytosis
The immunophenotypic profiling of total splenocytes obtained from WT, CathAS190A, and CathAS190A-Neo mice and double-stained with anti- major histocompatibility complex class II and anti-CD14 monoclonal antibodies did not reveal any difference in the splenic population of monocytes (data not shown). At the same time, ex vivo studies revealed drastic difference in the phagocytic activity of MØ. Peritoneal as well as splenocyte-derived MØ from WT, CathAS190A-Neo, and CathAS190A mice were incubated for 3 h at 37 or 4 °C with the fluorescein-labeled E. coli and S. aureus, washed, fixed, and analyzed by flow cytometry. After incubation with fluorescein-labeled bacteria at 37 °C, 80–90% of peritoneal MØ from WT or CathAS190A mice were positive for fluorescein compared with only 30–40% of the cells from CathAS190A-Neo mice (Fig. 2A). Similarly, the number of splenocyte-derived MØ-positive for E. coli was reduced from 45 to 48% for the cells from WT or CathAS190A mice to 16% for the cells from CathAS190A-Neo mice. The number or S. aureus-positive cells was, respectively, reduced from 85–95 to 45% (Fig. 2B). At the same time the number of bacteria-positive MØ measured at 4 °C, when the phagocytosis is blocked, was similar for all mice, indicating that the changes observed at 37 °C are related to partially impaired internalization of bacteria in the Neu1-deficient cells.
FIGURE 2.

Reduced pathogen association with MØ and immature DC from CathAS190A-Neo mice. MØ obtained from peritoneal cavity (A) and spleens (B) of WT, CathAS190A, or CathAS190A-Neo mice were incubated for 3 h with FITC-conjugated E. coli and S. aureus at 37 or 4 °C at a 1:50 ratio, washed with ice-cold PBS, fixed in 4% paraformaldehyde, and analyzed by flow cytometry. The panels show overlap of the histograms of the cells incubated with (white) and without (black) FITC-conjugated bacteria. The numbers on the panels show the percent of cells that are positive for FITC-conjugated bacteria. C, splenocyte-derived macrophages from WT, CathAS190A-Neo, and CathAS190A mice were incubated for 3 h at 4 or 37 °C in the presence of FITC-labeled E. coli at a 50:1 ratio, washed, fixed, and stained with DAPI. Slides were examined on the Nikon Eclipse E6000 direct epifluorescence microscope. Magnification, 200×. Panels represent typical images obtained in triplicate experiments; from 70 to 150 cells (10 panels) were studied for each condition in each experiment. The ingestion index was calculated for each type of cells as the percentage of cells with engulfed particles multiplied by the average number of particles engulfed per cell. The control level measured at 4 °C was subtracted from all values. The results show an average ± S.D. of three independent experiments. *, statistically different (p < 0.01) from the cells of WT and CathAS190A mice. D, splenocyte-derived immature DC from WT, CathAS190A, and CathAS190A-Neo mice were incubated for 3 h at 4 or 37 °C in the presence of FITC-labeled E. coli at a 50:1 ratio, fixed, and stained with DAPI. Slides were prepared and examined as described above. Panels represent typical images obtained in triplicate experiments; from 70 to 150 cells (10 panels) were studied for each condition in each experiment. Ingestion index was calculated as described for macrophages. The results show an average ± S.D. of three independent experiments. *, statistically different (p < 0.01) from the cells of WT and CathAS190A mice.
In a separate experiment the cells were stained with DAPI and studied using an epifluorescence microscope. No statistical difference was observed between the ingestion index of MØ (a percentage of cells with engulfed particles multiplied by the average number of particles per cell) derived from CathAS190A (21.5 ± 4.9) and WT (19.3 ± 5.3) mice. In contrast, the ingestion index for the MØ derived from CathAS190A-Neo mice (9.2 ± 2.0) was statistically reduced as compared with the cells from both WT and CathAS190A (Fig. 2C).
As compared with the MØ, the splenocyte-derived immature DC showed higher phagocytic activity. When immature DC derived from the splenocytes of WT or CathAS190A mice were incubated for 3 h with the fluorescein-labeled E. coli bioparticles, their ingestion index ranged between 44 and 48, whereas the ingestion index of the immature DC from CathAS190A-Neo mice was reduced to 26 ± 5 (Fig. 2D).
Similarly to the partial impairment of phagocytosis of Gram-positive and Gram-negative bacteria, macrophages from Neu1-deficient mice showed significant inhibition of FcγR-mediated phagocytosis specific for internalization of apoptotic cells. When peritoneal (Fig. 3) or splenocyte-derived (supplemental Fig. 1) MØ were incubated for 3 h with fluorescent latex beads covalently opsonized with murine IgG either at 4 or at 37 °C, washed, fixed, and analyzed by flow cytometry, the number of cells positive for the beads was reduced from 75–95% for WT and CathA-deficient mice to 35–40% for Neu1-deficient mice (Fig. 3). The phagocytosis of non-opsonized latex beads was also reduced, but the degree of inhibition was less then that of IgG-coated beads (Fig. 3).
FIGURE 3.

Reduced phagocytosis of IgG-opsonized latex beads by MØ from CathAS190A-Neo mice. MØ obtained from the peritoneal cavity of WT, CathAS190A, or CathAS190A-Neo mice were incubated for 3 h with fluorescent polystyrene latex beads opsonized or not with mouse IgG1 at 37 or 4 °C at a 1:50 ratio, washed with ice-cold PBS, fixed in 4% paraformaldehyde, and analyzed by flow cytometry. The panels show overlap of the histograms of the cells incubated with opsonized beads (solid line), non-opsonized beads (dashed line), and without the beads (gray histogram). The numbers on the panels show the average percent of beads-positive cells.
Impairment of Phagocytosis in Macrophages Is Associated with the Neu1 Deficiency and Sialylation but Not with the Charge of the Cell Surface
To verify whether impairment of phagocytosis in macrophages from CathAS190A-Neo mice is associated with the deficiency of Neu1 potentially involved in processing the sialylated macromolecules present on the cell surface, we have stained MØ derived from CathAS190A-Neo and CathAS190A mice or their WT siblings with the fluorescein-labeled lectin from M. amurensis (MAL-2), specific against 2,3-linked sialic acid residues. The cells were incubated with the lectin, washed, fixed with paraformaldehyde, stained with DAPI, and studied using an epifluorescent microscope. We found that the MØ from CathAS190A-Neo mice showed intensive peripheral staining with the lectin (Fig. 4A), consistent with the high sialylation of their surface. The cells from the CathA-deficient CathAS190A mice showed weak staining similar to that of the cells from WT mice. Increased sialylation of multiple cellular proteins in the MØ from CathAS190A-Neo mice was also observed when the cellular homogenates were resolved by SDS-PAGE, blotted to polyvinylidene fluoride membranes, and hybridized with biotinylated MAL-2 (supplemental Fig. 2).
FIGURE 4.

Impaired phagocytosis in MØ from CathAS190A-Neo mice is associated with Neu1 deficiency and hypersialylation of cell proteins. A, induced cell surface staining of MØ from CathAS190A-Neo mice by the lectin from M. amurensis. MØ from WT, CathAS190A-Neo, and CathAS190A mice and those from CathAS190A-Neo mice treated overnight with exogenous Neu1 were incubated for 15 min at 37 °C in the presence of fluorescein-labeled M. amurensis lectin II, fixed, and stained with DAPI. Slides were examined on the Nikon Eclipse E6000 epifluorescence microscope equipped with AxioCam camera (Zeiss). The graph shows mean intensity of fluorescence ± S.D. in the green region (500 nm < λem < 570 nm) measured for 15 randomly chosen microscope fields using the AxioVision 3.0 software. Control shows fluorescence of unstained cells. Magnification 100×. ***, statistically different (p < 0.001) from the cells of both WT and CathAS190A mice. Panels represent typical images obtained in triplicate experiments; at least 30 cells were studied for each condition in each experiment. Magnification, 1000×. B, exogenous mouse Neu1 restores phagocytosis of IgG-opsonized latex beads by MØ from CathAS190A-Neo mice. MØ from CathAS190A-Neo mice treated or not with exogenous Neu1 as indicated under “Experimental Procedures” were incubated for 3 h at 37 °C with fluorescent polystyrene latex beads opsonized with mouse IgG at 1:50 ratio, washed with ice-cold PBS, fixed in 4% paraformaldehyde, and analyzed by flow cytometry. The panels show overlap of the histograms of the cells incubated with opsonized beads and without the beads (black histograms). The figures on the panels show the average percent of beads-positive cells. C, phagocytosis of negatively and positively charged latex bead beads by MØ from WT, CathAS190A, and CathAS190A-Neo mice. MØ obtained from the peritoneal cavity of WT, CathAS190A, or CathAS190A-Neo mice were incubated for 3 h with fluorescent polystyrene latex beads modified with either positively charged amino groups or negatively charged carboxylate groups at a ratio of 50 beads/cell at 37 or 4 °C, washed with ice-cold PBS, fixed in 4% paraformaldehyde, and analyzed by flow cytometry. Black histograms represent cells incubated without the beads.
To understand if the proper sialylation of the cell surface and possibly the impaired phagocytosis in the Neu1-deficient cells can be restored by the treatment of cells with the exogenous enzyme, we have treated the peritoneal MØ from CathAS190A-Neo mice with Neu1 affinity-purified from mouse kidney. The cells were treated overnight by the medium supplemented with Neu1 (1 milliunits/ml of medium), which in a separate experiment was shown to reduce the overall sialylation of the cell surface to the level of the wild-type cell (Fig. 4A). Treated and non-treated cells were rinsed with culture medium and exposed for 3 h to fluorescent latex beads covalently opsonized with murine IgG, washed, fixed, and quantified by fluorescence-activated cell sorter as above. Remarkably, treatment with exogenous Neu1 restored the phagocytosis in Neu1-deficient cells from CathAS190A-Neo to the level of WT cells (Fig. 4B).
Because the hypersialylation of the cell surface should significantly increase its bulk negative charge, one could hypothesize that the inhibition of phagocytosis could happen due to the simple charge repulsion of the negatively charged beads or bacteria. To verify this hypothesis, we have compared the phagocytosis of negatively charged (carboxylate-modified) beads with that of positively charged (modified with amino groups) beads. The binding of the positively charged beads with the cells should be increased by sialylation had this hypothesis been correct. Our results (Fig. 4C) show that the engulfment of both positively and negatively charged beads was reduced in Neu1-deficient mice to the same extent, which rules out the electrostatic effect but points to the existence of the specific molecular mechanism potentially relevant to the sialylation of surface proteins involved in the phagocytosis.
Macrophages Derived from CathAS190A-Neo Mice Show Impaired FcγR-mediated Signaling
To distinguish between the inhibition of the adhesion and internalization of FcγR-mediated phagocytosis in the Neu1-deficient cells, we have loaded mouse macrophages with SE coated with mouse anti-sheep erythrocyte antibodies. After 30 min of incubation either at 4 or at 37 °C, the cells were washed to remove unbound SE. The cells incubated at 4 °C were fixed and studied by phase contrast microscopy for counting the bound SE. The cells incubated at 37 °C were treated with hypotonic buffer (1.4% NH4Cl) to lyse uninternalized SE. Then the cells were fixed and studied to count the internalized SE. We found that the amount of IgG-coated SE bound to macrophages at 4 °C was approximately similar for all mouse genotypes, but the amount of internalized SE was significantly reduced in the macrophages from CathAS190A-Neo mice, suggesting that the Neu1 deficiency affected transduction of signals from the Fc receptors (Table 1). Ligation of FcγR by IgG in MØ and DC is known to elicit activation and phosphorylation of the ITAM domains on the γ-subunit of the receptor followed by recruitment and phosphorylation of spleen tyrosine kinase (Syk) (30, 31). This activates signaling pathways leading to cytoskeletal changes and the phagocytosis of IgG-coated particles as well as to granule secretion (for review, see Ref. 32). Both events were studied in cultured splenocytes-derived and peritoneal MØ from WT, CathAS190A-Neo, and CathAS190A mice. Cells were treated for 20 min with murine IgG-opsonized latex beads, lysed, and studied by Western blot with antibodies against total Syk or Syk phosphorylated at Tyr346 (Tyr352 in human Syk) residue. Alternatively, cell lysates were processed to immunoprecipitate FcγR receptor, in which phosphorylation was further studied by Western blot using monoclonal antibodies against phospho-Tyr. In the MØ obtained from WT or CathA-deficient CathAS190A mice, 20-min treatments with IgG-coated particles resulted in selective increase of phosphorylation of both Syk and the γ-chain of FcγR receptor (Fig. 5). Similar results were obtained for peritoneal and splenocyte-derived MØ. In contrast, in the cells of Neu1-deficient CathAS190A-Neo mice both Syk and γ-chain of FcγR receptor showed a significantly reduced reactivity toward phosphospecific antibodies, consistent with down-modulated phosphorylation of tyrosine-based activation motifs on the receptor and Syk recruitment (Fig. 5). Because both events are triggered by the binding of the ligands to the receptors and cross-linking of the receptors (30, 31), the reduced FcγR signaling cannot be explained by the different internalization rate in wild-type and Neu1-deficient cells.
TABLE 1.
Ingestion and binding of IgG-coated sheep erythrocytes to peritoneal MØ from WT, CathAS190A, or CathAS190A-Neo mice
MØ obtained from the peritoneal cavity of WT, CathAS190A, or CathAS190A-Neo mice were incubated for 30 min with IgG-coated sheep erythrocytes at 37 or 4 °C as described under “Experimental Procedures,” rinsed with ice-cold PBS, washed or not with 1.4% NH4Cl for 5 min, fixed, and analyzed by phase contrast microscopy. The results show an average number of erythrocytes ± S.D. ingested or bound to the cells from 50–75 cells (5 panels) were studied for each condition in 2 independent experiments.
| Genotype | WT | CathAS190A | CathAS190A-Neo |
|---|---|---|---|
| Internalization at 37 °C | 8.05 ± 3.9 | 9.8 ± 3.0 | 3.78 ± 2.0a |
| Binding at 4 °C | 19.8 ± 4.1 | 20.2 ± 3.1 | 18.2 ± 2.8 |
a Statistically different (p < 0.001) from the cells of WT and CathAS190A mice.
FIGURE 5.

Impaired FcγR signaling in MØ from CathAS190A-Neo mice. A, reduced Tyr phosphorylation (pTyr) of the γ-subunit of FcγR is shown. MØ from WT, CathAS190A-Neo, and CathAS190A mice were incubated for 20 min at 37 °C in the presence of latex beads covalently opsonized with mouse IgG1. FcγR immunopurified from cell lysates were resolved by SDS-PAGE, and phosphorylation of its γ-subunits was analyzed by Western blotting using monoclonal antibodies against phospho-Tyr. B, reduced Tyr phosphorylation of Syk. Total lysates of MØ treated or not with mouse IgG-opsonized latex beads were analyzed by Western blot using antibodies against total Syk or Syk phosphorylated at the Tyr246 residue. Relative intensity of phosphorylation (bar graph) was measured as the ratio of the intensities of the signals obtained with phospho-specific antibodies and antibodies against the total protein. Values represent the means ± S.D. of triplicate measurements. *, statistically different (p < 0.01) from the cells of WT and CathAS190A mice.
FcγR in MØ from CathAS190A-Neo Mice Show Higher Staining with the Sialic Acid Binding Lectin as Compared with the Cells from WT or CathAS190A Mice
If FcγR is one of the molecular targets of Neu1, then we should observe its increased sialylation in the Neu1-deficient cells. To test this, we have resolved immunoprecipitated FcγR from MØ of all three mouse strains by SDS-PAGE, transferred it to polyvinylidene fluoride membranes, and probed the blot with biotinylated MAL-2. We observed that MAL-2 showed substantially higher reactivity with FcγR immunoprecipitated from the cells from CathAS190A-Neo mice when compared with the control or CathAS190A mice (Fig. 6). These results suggest that FcγR had retained 2,3-linked sialyl residues in the Neu1-deficient cells, consistent with it being the molecular target of Neu1 sialidase.
FIGURE 6.
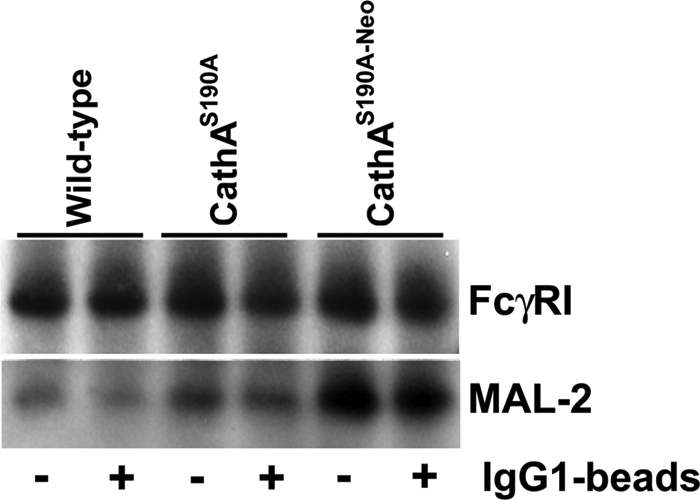
Hypersialylation of FcγR in macrophages from CathAS190A-Neo mice. FcγR immunopurified from the lysates of splenocyte-derived macrophages from WT, CathAS190A-Neo, and CathAS190A mice were resolved by SDS-PAGE and analyzed by lectin blot using biotinylated M. amurensis lectin II.
DISCUSSION
A growing body of evidence suggests that in addition to their role in catabolism of macromolecules, lysosomal enzymes have important physiological functions outside the lysosome, especially on the cell surface or outside the cell. In particular, the components of the multienzyme complex, Neu1, CathA, and β-galactosidase (or its alternatively spliced elastin-binding form), participate in processing of endothelin-1 (21, 33), assembly of the elastic fibers (21, 34, 35), pro-inflammatory response in macrophages (36), migration, invasion, and adhesion of cancer cells (37), proliferation of aortic smooth muscle cells (38), and exocytosis (39). In humans, genetic defects in CathA cause disruption of the complex and trigger galactosialidosis (MIM 256540), a severe multisystemic disease characterized by combined deficiency of Neu1, β-galactosidase, and CathA (for review, see Ref. 40). A clinically similar disease, sialidosis (MIM 256550), is caused by mutations directly affecting the Neu1 gene (for reviews, see Refs. 20 and 41). Interestingly, both sialidosis and galactosialidosis patients are predisposed to repeated pulmonary infections with bacterial and viral pathogens.
Our previous data showed that THP-1-derived MØ treated with sialidase inhibitors, Neu1 siRNA, or neutralizing antibodies against Neu1 have compromised ability to engulf bacteria or to produce cytokines interleukin-1b and -6 and tumor necrosis factor-α in response to ionomycin (19). Now we provide the first ex vivo evidence that the Neu1-deficient immune cells have a significantly reduced functional capacity. We found that the peritoneal-, splenocyte-, and peripheral blood mononuclear cell-derived MØ from Neu1-deficient CathAS190A-Neo mice are compromised in their ability to engulf Gram-positive and Gram-negative bacteria as well as IgG-opsonized and non-opsonized particles and IgG-coated red blood cells, suggesting that all types of phagocytosis are affected.
The observed effect is relevant to the deficiency of Neu1 activity as the treatment of the cells from CathAS190A-Neo mice with the exogenous mouse Neu1 sialidase, which reduced the sialylation of the cell surface to the normal levels, completely restored the phagocytosis. At the same time, because Neu1 deficiency affected phagocytosis of both negatively and positively charged polymer beads, the effect most likely is not relevant to simple repulsion of the cells and beads from the negatively charged hypersialylated surface of macrophages.
Because no single receptor is shared between the pathways for phagocytosis of Gram-negative bacteria, Gram-positive bacteria, and apoptotic cells, we speculate that Neu1 deficiency changes sialylation of multiple phagocytic receptors, affecting their activity. This conclusion is indirectly supported by our observation of the increased cross-reactivity of MAL-2 lectin specific for sialic acid residues with multiple proteins in the macrophages from Neu1-deficient mice, but further experiments are needed to identify the affected proteins. Also, we cannot rule out the possibility that Neu1 deficiency and/or over-sialylation of the cell surface proteins affects the internalization of phagosomes, considering that Neu1 deficiency has been shown to promote excessive lysosomal exocytosis by increasing the sialylation and decreasing the turnover rate of exocytic receptor LAMP-1 (39).
This study illustrates the proposed mechanism for one of the phagocytic pathways using FcγR receptors as an example. First, FcγR from Neu1-deficient mice but not from WT control or CathA-deficient mice had increased sialylation, as shown by staining with the MAL-2 lectin. Second, Neu1-deficient cells showed reduced internalization of IgG-opsonized sheep erythrocytes, whereas binding of the erythrocytes to the cells at 4 °C persisted, suggesting partial inhibition of FcγR-mediated signaling. Indeed both FcγR and the key signaling kinase Syk from the Neu1-deficient cells also showed impaired phosphorylation in response to latex beads opsonized with murine IgG, indicating that the hypersialylation of the receptor attenuates signaling pathways for the regulation of phagocytosis. The understanding of the exact molecular mechanisms by which FcγR becomes inhibited by oversialylation or is activated by Neu1 requires additional studies, but one can speculate that sialic acid residues may affect the conformation and membrane topology of the receptors as well as their ability to interact with other proteins. Together with recently published data showing the involvement of Neu1 in the functionally important desialylation of other receptors including integrin β-4 (37), TLR-2, -3, and -4 (36), insulin-like growth factor-1 receptor (38), and LAMP-1, (39) our data suggest that this enzyme plays an important role in maintaining activity of membrane receptors and identifies sialylation as a new important parameter controlling interactions between receptors, their ligands, and signaling proteins.
Supplementary Material
Acknowledgments
We thank Eva Lacroix for help in the preparation of the manuscript, Drs. Sergio Grinstein and Mila Ashmarina for helpful advice, and Audrey Pincemin for help in studying phagocytosis of sheep erythrocytes.
This work was supported in part by Canadian Institutes of Health Research Operating Grants MOP 15079 and GOP 38107 and by an equipment grant from Canadian Foundation for Innovation (to A. V. P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
- Neu1
- neuraminidase 1
- CathA
- cathepsin A
- MØ
- macrophage(s)
- WT
- wild type
- FITC
- fluorescein isothiocyanate
- 4MU-NeuAc
- 2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid
- SE
- sheep erythrocytes
- MAL-2
- M. amurensis lectin II
- DC
- dendritic cells
- PBS
- phosphate-buffered saline
- DAPI
- 4′,6-diamidino-2-phenylindole
- MES
- 4-morpholineethanesulfonic acid
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2- (hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1.Kelm S., Schauer R. (1997) Int. Rev. Cytol. 175, 137–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munday J., Floyd H., Crocker P. R. (1999) J. Leukocyte Biol. 66, 705–711 [DOI] [PubMed] [Google Scholar]
- 3.Frohman M., Cowing C. (1985) J. Immunol. 134, 2269–2275 [PubMed] [Google Scholar]
- 4.Kearse K. P., Cassatt D. R., Kaplan A. M., Cohen D. A. (1988) J. Immunol. 140, 1770–1778 [PubMed] [Google Scholar]
- 5.Krieger J., Jenis D. M., Chesnut R. W., Grey H. M. (1988) J. Immunol. 140, 388–394 [PubMed] [Google Scholar]
- 6.Baum L. G., Derbin K., Perillo N. L., Wu T., Pang M., Uittenbogaart C. (1996) J. Biol. Chem. 271, 10793–10799 [DOI] [PubMed] [Google Scholar]
- 7.Bagriaçik E. U., Miller K. S. (1999) Glycobiology 9, 267–275 [DOI] [PubMed] [Google Scholar]
- 8.Landolfi N. F., Cook R. G. (1986) Mol. Immunol. 23, 297–309 [DOI] [PubMed] [Google Scholar]
- 9.Chen X. P., Enioutina E. Y., Daynes R. A. (1997) J. Immunol. 158, 3070–3080 [PubMed] [Google Scholar]
- 10.Chen X. P., Ding X., Daynes R. A. (2000) Cytokine 12, 972–985 [DOI] [PubMed] [Google Scholar]
- 11.Nan X., Carubelli I., Stamatos N. M. (2007) J. Leukocyte Biol. 81, 284–296 [DOI] [PubMed] [Google Scholar]
- 12.Lukong K. E., Seyrantepe V., Landry K., Trudel S., Ahmad A., Gahl W. A., Lefrancois S., Morales C. R., Pshezhetsky A. V. (2001) J. Biol. Chem. 276, 46172–46181 [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto N., Kumashiro R. (1993) J. Immunol. 151, 2794–2802 [PubMed] [Google Scholar]
- 14.Naraparaju V. R., Yamamoto N. (1994) Immunol. Lett. 43, 143–148 [DOI] [PubMed] [Google Scholar]
- 15.Carrillo M. B., Milner C. M., Ball S. T., Snoek M., Campbell R. D. (1997) Glycobiology 7, 975–986 [DOI] [PubMed] [Google Scholar]
- 16.Landolfi N. F., Leone J., Womack J. E., Cook R. G. (1985) Immunogenetics 22, 159–167 [DOI] [PubMed] [Google Scholar]
- 17.Watanabe Y., Shiratsuchi A., Shimizu K., Takizawa T., Nakanishi Y. (2004) Microbiol. Immunol. 48, 875–881 [DOI] [PubMed] [Google Scholar]
- 18.Stamatos N. M., Liang F., Nan X., Landry K., Cross A. S., Wang L. X., Pshezhetsky A. V. (2005) FEBS J. 272, 2545–2556 [DOI] [PubMed] [Google Scholar]
- 19.Liang F., Seyrantepe V., Landry K., Ahmad R., Ahmad A., Stamatos N. M., Pshezhetsky A. V. (2006) J. Biol. Chem. 281, 27526–27538 [DOI] [PubMed] [Google Scholar]
- 20.Pshezhetsky A. V., Ashmarina M. (2001) Prog. Nucleic Acid Res. Mol. Biol. 69, 81–114 [DOI] [PubMed] [Google Scholar]
- 21.Seyrantepe V., Hinek A., Peng J., Fedjaev M., Ernest S., Kadota Y., Canuel M., Itoh K., Morales C. R., Lavoie J., Tremblay J., Pshezhetsky A. V. (2008) Circulation 117, 1973–1981 [DOI] [PubMed] [Google Scholar]
- 22.Berthier R., Martinon-Ego C., Laharie A. M., Marche P. N. (2000) J. Immunol. Methods 239, 95–107 [DOI] [PubMed] [Google Scholar]
- 23.Potier M., Mameli L., Bélisle M., Dallaire L., Melançon S. B. (1979) Anal. Biochem. 94, 287–296 [DOI] [PubMed] [Google Scholar]
- 24.Rome L. H., Garvin A. J., Allietta M. M., Neufeld E. F. (1979) Cell 17, 143–153 [DOI] [PubMed] [Google Scholar]
- 25.Roth M. (1971) Anal. Chem. 43, 880–882 [DOI] [PubMed] [Google Scholar]
- 26.Pshezhetsky A. V., Potier M. (1996) J. Biol. Chem. 271, 28359–28365 [DOI] [PubMed] [Google Scholar]
- 27.Meyers E. N., Lewandoski M., Martin G. R. (1998) Nat. Genet. 18, 136–141 [DOI] [PubMed] [Google Scholar]
- 28.Carmeliet P., Ferreira V., Breier G., Pollefeyt S., Kieckens L., Gertsenstein M., Fahrig M., Vandenhoeck A., Harpal K., Eberhardt C., Declercq C., Pawling J., Moons L., Collen D., Risau W., Nagy A. (1996) Nature 380, 435–439 [DOI] [PubMed] [Google Scholar]
- 29.Moran J. L., Levorse J. M., Vogt T. F. (1999) Nature 399, 742–743 [DOI] [PubMed] [Google Scholar]
- 30.Crowley M. T., Costello P. S., Fitzer-Attas C. J., Turner M., Meng F., Lowell C., Tybulewicz V. L., DeFranco A. L. (1997) J. Exp. Med. 186, 1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sedlik C., Orbach D., Veron P., Schweighoffer E., Colucci F., Gamberale R., Ioan-Facsinay A., Verbeek S., Ricciardi-Castagnoli P., Bonnerot C., Tybulewicz V. L., Di Santo J., Amigorena S. (2003) J. Immunol. 170, 846–852 [DOI] [PubMed] [Google Scholar]
- 32.Swanson J. A., Hoppe A. D. (2004) J. Leukocyte Biol. 76, 1093–1103 [DOI] [PubMed] [Google Scholar]
- 33.Itoh K., Kase R., Shimmoto M., Satake A., Sakuraba H., Suzuki Y. (1995) J. Biol. Chem. 270, 515–518 [DOI] [PubMed] [Google Scholar]
- 34.Hinek A., Rabinovitch M., Keeley F., Okamura-Oho Y., Callahan J. (1993) J. Clin. Invest. 91, 1198–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinek A., Pshezhetsky A. V., von Itzstein M., Starcher B. (2006) J. Biol. Chem. 281, 3698–3710 [DOI] [PubMed] [Google Scholar]
- 36.Amith S. R., Jayanth P., Franchuk S., Siddiqui S., Seyrantepe V., Gee K., Basta S., Beyaert R., Pshezhetsky A. V., Szewczuk M. R. (2009) Glycoconj. J., in press [DOI] [PubMed] [Google Scholar]
- 37.Uemura T., Shiozaki K., Yamaguchi K., Miyazaki S., Satomi S., Kato K., Sakuraba H., Miyagi T. (2009) Oncogene 28, 1218–1229 [DOI] [PubMed] [Google Scholar]
- 38.Hinek A., Bodnaruk T. D., Bunda S., Wang Y., Liu K. (2008) Am. J. Pathol. 173, 1042–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yogalingam G., Bonten E. J., van de Vlekkert D., Hu H., Moshiach S., Connell S. A., d'Azzo A. (2008) Dev. Cell 15, 74–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas G. H. (2001) in Metabolic and Molecular Bases of Inherited Disease (Scriver C. R., Beaudet A. L., Sly W. S., Valle D. eds) pp. 3507–3534, McGraw-Hill Inc., New York [Google Scholar]
- 41.d'Azzo A., Andria G., Strisciuglio G., Galjaard H. (2001) in Metabolic and Molecular Bases of Inherited Disease (Scriver C. R., Beaudet A. L., Sly W. S., Valle D. eds) pp. 3811–3826, McGraw-Hill Inc., New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.