

Abstract
Retroviral nucleocapsid (NC) proteins are molecular chaperones that facilitate nucleic acid (NA) remodeling events critical in viral replication processes such as reverse transcription. Surprisingly, the NC protein from human T-cell leukemia virus type 1 (HTLV-1) is an extremely poor NA chaperone. Using bulk and single molecule methods, we find that removal of the anionic C-terminal domain (CTD) of HTLV-1 NC results in a protein with chaperone properties comparable with that of other retroviral NCs. Increasing the ionic strength of the solution also improves the chaperone activity of full-length HTLV-1 NC. To determine how the CTD negatively modulates the chaperone activity of HTLV-1 NC, we quantified the thermodynamics and kinetics of wild-type and mutant HTLV-1 NC/NA interactions. The wild-type protein exhibits very slow dissociation kinetics, and removal of the CTD or mutations that eliminate acidic residues dramatically increase the protein/DNA interaction kinetics. Taken together, these results suggest that the anionic CTD interacts with the cationic N-terminal domain intramolecularly when HTLV-1 NC is not bound to nucleic acids, and similar interactions occur between neighboring molecules when NC is NA-bound. The intramolecular N-terminal domain-CTD attraction slows down the association of the HTLV-1 NC with NA, whereas the intermolecular interaction leads to multimerization of HTLV-1 NC on the NA. The latter inhibits both NA/NC aggregation and rapid protein dissociation from single-stranded DNA. These features make HTLV-1 NC a poor NA chaperone, despite its robust duplex destabilizing capability.
Introduction
Nucleic acid (NA)5 chaperones are proteins that facilitate NA remodeling and annealing (1). Retroviral nucleocapsid proteins (NC) are essential NA chaperones (2–4) that facilitate many steps in the retroviral life cycle, including dimerization of the RNA genome (5–10), reverse transcription (11–14), annealing of the tRNA primer to the primer-binding site (7, 15–22), and integration of viral DNA into the host genome (23–27). Previous work has shown that NCs from different retroviruses display a wide range of NA chaperone activities (28). A model-annealing reaction involving complementary trans-activation response element (TAR) RNA and DNA hairpins derived from the R region of the HIV-1 genome was used to characterize NCs from human immunodeficiency virus, type 1 (HIV-1), Rous sarcoma virus, murine leukemia virus, and human T-cell leukemia virus, type 1 (HTLV-1) (28). Surprisingly, the annealing activity of these NCs varies by 5 orders of magnitude; HIV-1 NC was the most efficient chaperone and HTLV-1 NC was the least efficient. A single molecule (SM) Förster resonance energy transfer (FRET) study confirmed the observation that HTLV-1 NC is a very poor NA chaperone (29).
To determine the physical origin of these differences in retroviral NC chaperone activity, we measured the NA aggregation and duplex destabilization activity for each protein (28). Both of these capabilities, together with rapid protein/NA interaction kinetics, are known to be major factors contributing to chaperone activity (2, 4, 30, 31). Surprisingly, the duplex destabilizing ability of all four NC proteins studied was similar (28). The ability of HTLV-1 NC to effectively destabilize DNA duplexes was also observed in the SM-FRET study (29). Rous sarcoma virus and murine leukemia virus NC were able to effectively aggregate NA at concentrations comparable with that of HIV-1 NC, whereas HTLV-1 NC was unable to do so, even at high concentrations. Although this result could explain the poor chaperone activity of the HTLV-1 NC, the aggregation data were unable to explain the relatively poor annealing activity of murine leukemia virus NC compared with HIV or Rous sarcoma virus NC. SM λ-DNA stretching experiments revealed an excellent correlation between the overall chaperone function of each NC and its NA dissociation kinetics (28, 30, 32). The slow dissociation kinetics of HTLV-1 NC relative to that of other retroviral NCs was particularly striking, and the DNA stretching behavior resembled that of a single-stranded (ss) DNA-binding protein.
In this study, we set out to understand the mechanistic basis for the slow NA dissociation kinetics and weak NA aggregation activity of HTLV-1 NC, which are the main factors contributing to its overall poor chaperone function. In addition to measuring TAR RNA/DNA annealing kinetics in the presence of WT and mutant HTLV-1 NC under varying conditions, we also used a number of other biophysical approaches to characterize the interaction of these proteins with NA. Specifically, we investigated the NA aggregating ability of the proteins, as well as their ability to destabilize NA duplexes. SM DNA stretching was used to characterize their NA interaction kinetics (30, 32). Taken together, the results of this investigation lead to a proposed model in which electrostatic interaction between the anionic C-terminal domain (CTD) and cationic N-terminal domain (NTD) within a single HTLV-1 NC protein in solution, or between DNA-bound neighbors, results in slow NA association and dissociation, respectively, thereby negatively regulating the chaperone activity of this protein.
EXPERIMENTAL PROCEDURES
Protein and NA Preparation
The genes encoding HTLV-1 (33, 34) and -2 (GenBankTM accession number NC_001488) NC were PCR-amplified from full-length HTLV-1 (pCS-HTLV-1) and -2 proviral plasmids (generous gifts from David Derse, NCI-Frederick, National Institutes of Health) and cloned into pET32a, generating plasmids pDR2559 (for HTLV-1 NC) and pDR2560 (for HTLV-2 NC). These plasmids allow expression of NC as a thioredoxin fusion with a TEV protease cleavage site (ENLYFQ) to liberate authentic HTLV-1 or -2 NC (28, 35, 36). Mutant NC sequences were constructed using standard mutagenesis/molecular biology techniques and cloned into pET32a as for the WT HTLV-1 NC. WT and mutant HTLV-1 and -2 NC proteins were expressed, isolated, and purified essentially as described previously (24, 28, 37). A plasmid encoding a chimeric NC was constructed that contained amino acids 1–56 of HIV-1 NC (from pNL4-3, GenBankTM accession number AF324493), and residues 57–85 from the CTD of HTLV-1 NC (33, 34). This construct, also a gift from David Derse (NCI-Frederick, National Institutes of Health), was PCR- amplified and cloned into pGEX5X-1 (GE Healthcare), along with the sequence encoding the TEV protease cleavage site followed by a methionine (ENLYFQM) at the N terminus of the chimeric NC (the Met residue was included to facilitate more extensive cleavage of the TEV protease site (28, 35, 36)). The resulting expression plasmid, pDB2703, was used for glutathione S-transferase-NC fusion protein expression, which was produced and affinity-purified according to the manufacturer's instructions. TEV cleavage of the purified fusion protein and final purification by HPLC were performed as described above for the HTLV-1 recombinant NC proteins. NCs were stored in lyophilized form with 1 eq of Zn2+ per finger at −80 °C. Prior to use, the proteins were resuspended in diethyl pyrocarbonate-treated water or NC storage buffer (20 mm HEPES, 5 mm β-mercaptoethanol, 0.1 mm tris(2-carboxyethyl) phosphine hydrochloride (TCEP-HCl), pH 7.5). NC concentrations were determined by measuring the absorbance at 280 nm (A280) and using an extinction coefficient of 11,740 m−1 cm−1 for WT and mutant HTLV-1 NC and 5,690 m−1 cm−1 for the chimeric NC.
The mini-TAR RNA and DNA constructs were obtained from Dharmacon (Lafayette, CO) and Integrated DNA Technologies (Coralville, IA), respectively. The oligonucleotides were purified on 16% denaturing polyacrylamide gels, dissolved in diethyl pyrocarbonate-treated water, and stored at −20 °C. Oligonucleotide concentrations were determined at A260 using the following extinction coefficients: mini-TAR RNA, 2.82 × 105 m−1 cm−1; mini-TAR DNA, 3.06 × 105 m−1 cm−1.
Mini-TAR RNA was radiolabeled with [γ-32P]ATP (PerkinElmer Life Sciences) and T4 polynucleotide kinase (New England Biolabs, Ipswich, MA) using standard protocols. The radiolabeled TAR constructs were purified on 16% denaturing polyacrylamide gels, dissolved in diethyl pyrocarbonate-treated water, and stored at −20 °C.
Prior to use, mini-TAR oligonucleotides were refolded in 25 mm HEPES, pH 7.5, and 100 mm NaCl at a concentration that was 100-fold greater than the final assay concentration. The oligonucleotides were incubated at 80 °C for 2 min, cooled to 60 °C for 2 min, followed by addition of MgCl2 to a final concentration of 10 mm and placement on ice.
Annealing Assays
Single time point annealing assays were performed to determine the amount of NC required to attain saturating annealing levels. Solutions containing 15 nm refolded 32P-labeled mini-TAR RNA and 90 nm refolded mini-TAR DNA in reaction buffer (20 mm HEPES, pH 7.5, 20 mm NaCl, 5 mm dithiothreitol, and 0.2 mm MgCl2) were incubated for 5 min at 37 °C. NC was then added to final concentrations varying from 0.25 to 6 μm, and reactions were incubated for 30 min at 37 °C. For assays testing the effect of the HTLV-1 NC CTD in trans, a peptide corresponding to residues 57–85 of HTLV-1 NC and labeled with fluorescein isothiocyanate at its N terminus (CTD-FL) was used. The peptide was purchased from GenScript (Piscataway, NJ). CTD-FL (0–15 μm) was added to 1 μm (3.3 nt:NC) HTLV-1 ΔC29 NC prior to addition of NA, and reactions were incubated at 37 °C in reaction buffer containing either 10 or 100 mm NaCl. Reactions were quenched and analyzed as described previously (38).
Annealing assays to determine the kinetics of mini-TAR RNA and DNA annealing were performed in the presence of 5.0 μm (0.66 nt:NC) WT or mutant HTLV-1 NC. Annealing reactions for salt dependence experiments were carried out using 3 μm (1.1 nt:NC) NC in low salt (20 mm NaCl) and 6 μm (0.55 nt:NC) NC in high salt (100 mm NaCl) to account for poorer binding. To determine the effect of zinc binding, annealing was performed in the presence of 3 μm apo-WT HTLV-1 NC prepared by pretreating with 1 mm EDTA for 30 min (30, 39, 40). Experiments to compare annealing of HIV-1 NC, HTLV-1 NC, and the HIV-1/HTLV-1 chimera were performed using 1.5 μm (2.2 nt:NC) NC in low salt (20 mm NaCl). Separate mixtures containing mini-TAR RNA (15 nm) and mini-TAR DNA (90 nm) in reaction buffer were prepared and preincubated at 37 °C. DNA and RNA were mixed immediately prior to addition of NC. Reactions were quenched at the specified time points and analyzed as for the single time point assays.
Sedimentation/Aggregation Assays
Solutions containing refolded 32P-labeled mini-TAR RNA (15 nm) and mini-TAR DNA (45 nm) were incubated with varying concentrations of WT and mutant HTLV-1 NC for 30 min at room temperature in reaction buffer (20 mm HEPES, pH 7.5, 20 mm NaCl, 5 mm dithiothreitol, and 0.2 mm MgCl2). The reactions were analyzed as described previously (38).
FA Assays
Equilibrium binding of WT and mutant HTLV-1 NC to a 6-carboxyfluorescein (FAM)-labeled 20-nt ssDNA oligonucleotide (5′-FAM-CTTCTTTGGGAGTGAATTAG-3′) was examined using an HPLC-purified DNA oligomer (5′-FAM DNA20) from TriLink Biotechnologies (San Diego). Binding to the corresponding 20-nt DNA/RNA hybrid duplex was also measured. The HPLC-purified RNA oligomer (5′-CUAAUUCACUCCCAAAGAAG-3′) was from Dharmacon. FA measurements were performed on an Analyst AD or a Spectramax M5 plate reader system (Molecular Devices, Sunnyvale, CA) using Corning 3676 low volume 384-well black nonbinding surface polystyrene plates. Reaction mixtures contained 20 nm 5′-FAM DNA20 or the DNA/RNA duplex, varying concentrations of NC, and a buffer consisting of 20 mm HEPES, pH 7.5, 50 mm NaCl, 5 mm β-mercaptoethanol, 1 μm ZnCl2, and 100 μm TCEP (final concentrations). Reactions were incubated for 30 min at room temperature in the dark to allow samples to reach equilibrium. Samples were excited at 485 nm, and the emission intensities at 530 nm from the parallel and perpendicular planes were measured. Equilibrium dissociation constants (Kd) were obtained by fitting the binding curves as described previously (28). To determine the free energy per base pair (41), Equation 1 was used,
where R is the molar gas constant; n is the binding site size in bp; Kdss and Kdds are the protein dissociation constants from ssDNA and dsDNA, respectively, and T is temperature in Kelvin. At room temperature (23 °C), RT = 0.59 kcal/mol.
For FA measurements performed to characterize HTLV-1 NC ΔC29/CTD binding, the CTD-FL peptide described above was used. The HTLV-1 NC ΔC29 variant was titrated into a solution of CTD-FL and incubated for 30 min. The titrations were performed in buffer (20 mm HEPES, pH 7.5, 5 mm β-mercaptoethanol, 1 μm ZnCl2, and 100 μm TCEP) with variable [NaCl] (1–100 mm). The binding curves were fit to determine the dissociation constant for ΔC29/CTD-FL interaction as described previously (28). The measured salt dependence of the HTLV-1 NC NTD/CTD dissociation constant, Kd, NTD/CTD, was used to estimate the salt-dependent free energy of this interaction, ΔGNTD,CTD, using Equation 2,
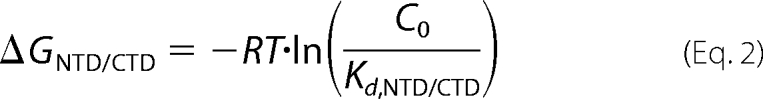 |
where C0 is the effective concentration of NTD and CTD domains in this intramolecular binding reaction. C0 is the reciprocal volume of the HTLV-1 NC molecule, 400 m. This value was estimated using the volume of a sphere of 2 nm in diameter to approximate the average size of HTLV-1 NC, which was assumed to be an 85-residue flexible polypeptide chain.
Time-resolved FRET Assays
TAR DNA (64 nt) labeled with AlexaFluor-488 at the 5′-end and 4-(4′-dimethylaminophenylazo)benzoic acid at the 3′-end was obtained HPLC-purified from TriLink Biotechnologies (San Diego) and refolded as described (38). Folded TAR DNA at a final concentration of 100 nm was incubated with and without NC in a buffer containing 20 mm HEPES, pH 7.5, 50 mm NaCl, 10 μm TCEP, 5 mm β-mercaptoethanol, and 1 μm zinc acetate for 30 min. Fluorescence measurements were performed using the time-correlated single photon counting technique (42) at room temperature on a LifeSpec Red time-resolved spectrometer (Edinburgh Instruments, Livingston UK) equipped with an EPL-475 picosecond diode laser (wavelength = 475 nm, repetition rate of 200 ns). Samples were excited using 475 nm vertically polarized pulses from the laser, and emission at 520 nm was detected at 90° with respect to the excitation axis with a miniature-PMT detector (Hamamatsu H7422) and a Glan Thompson polarizer set at the magic angle. The emission decay was fit using a noniterative nonlinear least squares fitting procedure by the reconvolution fit analysis module of the T900 software (Edinburgh Instruments), in which decay data were fit to a sum of exponential decays convoluted with the instrumental response function. Relative free energy ΔGT1 for the T1 state for each NC concentration was determined using Equation 3,
SM DNA Stretching
Dual beam optical tweezers were used to stretch bacteriophage λ DNA (Roche Applied Science), which was labeled with biotin on both 5′-ends. A single DNA molecule was caught between two streptavidin-coated polystyrene beads (Bangs Labs) with optical tweezers. All stretching experiments were performed at a pulling rate of ∼100 nm/s in a buffer consisting of 10 mm HEPES, pH 7.5, 50 mm Na+. Solutions of saturating NC concentrations were exchanged with buffer surrounding a single DNA molecule to investigate protein effects on DNA stretching curves. Experiments were performed in the presence of 700 and 200 nm WT and ΔC29 HTLV-1 NC, respectively. To measure DNA reannealing rate, we stretched λ-DNA through the DNA force-induced melting plateau and then relaxed to the mid-point of the transition. At a fixed DNA extension, the stretching force was recorded as function of time until equilibrium was achieved.
RESULTS
Fig. 1 compares the amino acid sequence of HTLV-1 NC (Fig. 1A) with that of HIV-1 NC (Fig. 1C, top). Both proteins possess two zinc binding domains, which contain many basic residues and two or three aromatic residues. A distinguishing feature is the highly anionic CTD of HTLV-1 NC, which is absent in mature HIV-1 NC. Whereas most retroviral NCs have an overall basic character (pI = 9–10), the overall pI of HTLV-1 NC is about 7. Here, we investigate the hypothesis that this unusual feature leads to poor chaperone function by comparing the chaperone properties of the WT protein with that of a truncated variant lacking 29 amino acids from its CTD (ΔC29, see Fig. 1A). We also prepared three additional HTLV-1 NC mutants that lack (either through substitution with Ala or deletion) one or more C-terminal acidic residues. The negative charge of the anionic CTD of these mutants is diminished to varying extents, and they are designated EED, in which three acidic residues are neutralized by changing them to Ala, LLD, which lacks residues 65–70 including one acidic residue, and point mutant D69A. These HTLV-1 NC mutants are similar to the ones shown in a recent study to have a significant effect on apolipoprotein B editing catalytic polypeptide 3G (APOBEC3G) packaging into HTLV-1 virions (34).
FIGURE 1.

Sequence and zinc finger structures of the WT and mutant NCs studied in this work, including HTLV-1 (A), HTLV-2 (B), HIV-1 (C), and HIV-1/HTLV-1 chimeric NC (D). The arrows mark the C-terminal deletion (ΔC29) or mutations or deletions of individual residues in the CTD. The corresponding names of all mutants are indicated in parentheses. C also shows the p1 and p6 domains of the HIV-1 precursor protein NCp15.
Nucleic Acid Chaperone Activity of HTLV-1 NC Is Negatively Modulated by Its Anionic C-terminal Domain
In this study, we used a mini-TAR DNA/RNA model system to characterize the overall chaperone activity of WT and mutant HTLV-1 NC (Fig. 2). The mini-TAR RNA sequence is derived from the TAR RNA hairpin present near the 5′-end of the 96-nt R region of the HIV-1 genome. The HTLV-1 R region contains 228 nt and folds into a complex secondary structure (43–45), whose 5′ region is predicted to contain several hairpins (e.g. nts 27–46 and 139–163) with similar thermodynamic stability as the mini-TAR RNA substrate used here (ΔG = −9.70 kcal/mol) (46). We compared the chaperone activities of these proteins by measuring the annealing time course in the presence of saturating amounts of NC. As shown previously, in the absence of NC this reaction is extremely slow and is characterized by a bimolecular rate constant of ∼100 m−1·s−1 in low salt (20 mm NaCl) (38). Addition of HIV-1 NC increases this rate by ∼103-fold. Presented in Fig. 3 is the mini-TAR annealing time course in 20 mm NaCl in the presence of saturating amounts of WT HTLV-1 NC (5 μm; 0.66 nt:NC ratio) and the following four mutants indicated in Fig. 1A: ΔC29, EED, D69A, and LLD. The ability of these proteins to facilitate the annealing reaction varies greatly. WT HTLV-1 NC results in only a 2-fold increase above the background rate of annealing, even at a concentration of 5 μm. The ΔC29 variant, which lacks 8 out of a total of 11 negatively charged residues, facilitates annealing ∼100-fold over the background rate but is still not quite as effective as HIV-1 NC (∼4-fold lower) (38). The next best chaperone is the EED variant, whose chaperone activity is only ∼3-fold reduced relative to ΔC29 NC. These data suggest that the three neighboring acidic residues that were neutralized in this mutant provide a major contribution to the inhibition of chaperone activity for HTLV-1 NC. The chaperone activities of D69A and LLD are ∼5-fold lower than the EED variant and indistinguishable from one another within our experimental error. This result is consistent with the fact that relative to WT HTLV-1 NC, both of these variants lack just a single negatively charged amino acid at position 69 (see Fig. 1A).
FIGURE 2.

A, schematic illustration of the annealing reaction between the TAR RNA and DNA hairpins or mini-TAR RNA and DNA hairpins. B, typical native polyacrylamide gel quantifying the fraction of bound (B) and free (F) RNA as a function of time indicated by each lane in minutes, in the presence of saturating HIV-1 NC.
FIGURE 3.

Annealing time courses for mini-TAR RNA/DNA hairpins in the absence of NC or in the presence of WT or mutant HTLV-1 NC as indicated by each curve. The lines are two-exponential fits performed as described previously (28).
Altering the Solution Ionic Strength Supports an Electrostatic Mechanism of CTD Inhibition of HTLV-1 NC Chaperone Function
Based on the results presented in Fig. 3, the anionic residues of the CTD of HTLV-1 NC are responsible for the poor chaperone function of the WT protein. We hypothesize that an electrostatic interaction between anionic CTD and cationic NTD of HTLV NC (see Fig. 1A) may be responsible for this effect. Because increasing salt is expected to weaken this electrostatic interaction, the difference between WT and ΔC29 HTLV-1 NC annealing activity is also expected to be reduced as a function of increasing salt concentration. To test this prediction, the mini-TAR RNA/DNA annealing activity was compared using saturating amounts of WT and ΔC29 HTLV-1 NC in low (20 mm NaCl) and high (100 mm NaCl) salt buffer.
To determine saturating conditions for each protein at the different solution ionic strengths tested, we first performed steady-state titration experiments, where the percent of mini-TAR RNA annealed to mini-TAR DNA was monitored at 30 min after mixing as a function of protein concentration. The annealing in the presence of ΔC29 NC is always higher than with the WT, but, as expected, these two proteins behave much more similarly at higher salt concentrations (Fig. 4A). In 100 mm NaCl, the effect of either protein on annealing saturates at ∼5 μm NC. Interestingly, for both proteins in 20 mm NaCl, the annealing first increases with [NC] up to ∼1–2 μm but declines upon increasing the protein concentration further (Fig. 4A). This phenomenon is likely due to significant NA duplex destabilizing activity in the presence of saturating amounts of each protein and the reversible nature of the annealing reaction. In other words, the mini-TAR RNA/DNA annealing equilibrium shifts toward reactants, thereby leading to less product formation at equilibrium, as observed previously for other retroviral NC proteins (28). This effect is only apparent in lower salt conditions under which the annealed duplex is much less stable than in higher salt. Based on these fixed time point results, we performed mini-TAR RNA/DNA annealing kinetic studies in the presence of 3 μm (1.1 nt:NC, low salt reactions) and 6 μm (0.55 nt:NC, high salt reactions) protein. Both proteins are near saturation at these concentrations, and the duplex destabilization observed in low salt is minimal (see Fig. 4A).
FIGURE 4.

A, percent of mini-TAR RNA (15 nm) annealed to mini-TAR DNA (90 nm) 30 min after mixing at 37 °C as a function of NC concentration. The filled symbols correspond to WT HTLV-1 NC and open symbols to ΔC29 in low salt (LS, 20 mm NaCl) or high salt (HS, 100 mm NaCl). B, time course of mini-TAR RNA annealing to mini-TAR DNA in the absence of NC (−NC) or in the presence of WT or mutant HTLV-1 NC. NC concentrations were 3 μm for low salt and 6 μm for high salt conditions. The experiment with WT HTLV-1 NC treated with EDTA as described in the text (WT + EDTA) was conducted under the low salt conditions. For all curves, the lines are not fits but connect the data points.
Kinetics of Mini-TAR RNA/DNA Annealing in the Presence of Saturating WT and ΔC29 HTLV-1 NC in Low and High Salt
Presented in Fig. 4B are the mini-TAR annealing time courses at low and high salt in the presence of either WT or ΔC29 HTLV-1 NC. The annealing time courses in 20 and 100 mm NaCl in the absence of protein are also shown. In the latter case, the extent of annealing after 30 min is slightly higher at 100 mm compared with 20 mm NaCl (see Fig. 4B). This result is in agreement with the facilitating effect of mono- and divalent cations on annealing (47). Interestingly, the annealing in the presence of ΔC29 HTLV-1 NC is equally efficient either at low or high salt. In contrast, annealing in the presence of WT HTLV-1 NC is very poor in low salt but is facilitated 2–3-fold in 100 mm NaCl. In low salt, the annealing times with WT and ΔC29 are ∼10-fold different, whereas in high salt they differ by less than 2-fold. This result supports our hypothesis that the electrostatic attraction between the anionic CTD and cationic NTD of HTLV-1 NC contributes to its poor chaperone function.
Assays presented in Fig. 4 were also performed with the closely related HTLV-2 NC protein and a CTD truncation mutant (Fig. 1B). The results for HTLV-1 and -2 are indistinguishable within experimental error (data not shown). This result supports our hypothesis that it is the overall cationic/anionic character of the proteins, rather than their exact amino acid sequence (compare Fig. 1, A and B), that determines their chaperone activity.
Elimination of Zinc Finger Structures Slightly Improves Chaperone Function of HTLV-1 NC
The zinc fingers of HIV-1 NC are known to be responsible for the ability of this protein to destabilize NA secondary structures (18, 19, 31, 37, 48–54). To test the potential role of the zinc finger structures in the chaperone function of HTLV-1 NC, the mini-TAR RNA/DNA annealing reaction was performed in the presence of saturating amounts of WT HTLV-1 NC (3 μm; 1.1 nt:NC), which had been preincubated for 30 min with 1 mm EDTA. Under these conditions, the EDTA is expected to fully chelate the zinc (30, 39, 40), thereby eliminating the zinc finger structures (55). The annealing rate was slightly increased (∼2.2-fold) by EDTA treatment (Fig. 4B). These results suggest that the zinc finger structures of HTLV-1 NC contribute only slightly to the poor chaperone function of this protein.
HTLV-1 NC CTD and NTD Domains Bind Each Other via Direct Electrostatic Interaction
To establish whether a direct electrostatic interaction occurs between the acidic CTD and basic NTD of HTLV-1 NC, the binding between the two separate domains was monitored by FA. The ΔC29 mutant was titrated into CTD-FL. At low ionic strength (1 mm NaCl), a binding interaction is clearly observed with an apparent Kd, NTD/CTD of 206 ± 40 nm (Fig. 5A). The Kd, NTD/CTD value increases with increasing [NaCl], and at 100 mm NaCl, binding can no longer be detected within the range of ΔC29 concentration studied (≤3 μm) (Fig. 5A). A log-log plot of Kd, NTD/CTD versus [NaCl] is shown in Fig. 5B. A linear fit of the data yields a slope S = 1 ± 0.3, suggesting that ΔC29/CTD-FL binding is associated with a net release of ∼1 Na+. This result implies that an effective negative charge of ∼1 exists on the CTD site that binds NTD (32), likely localized to a region created by several closely spaced anionic residues, such as the EED (see Fig. 1A). Although the binding data shown in Fig. 5 were obtained with separate NTD and CTD constructs in trans (i.e. an intermolecular interaction), these domains are present in cis in the full-length HTLV-1 NC. The effective concentration (C0) of NTD and CTD domains within a single molecule can be estimated as the reciprocal volume of a single HTLV-1 NC molecule as described under “Experimental Procedures” (C0 ≈ 400 m). The free energy of the ΔC29/CTD-FL interaction at a given salt concentration was then estimated using Equation 2. ΔGNTD/CTD changes with increasing salt concentration, from −12.6 kcal/mol at 1 mm NaCl to −8.5 kcal/mol at 100 mm NaCl, i.e. the CTD/NTD interaction weakens significantly. Although the absolute value of the actual NTD/CTD intramolecular interaction is most likely smaller due to steric constraints, the salt dependence of this interaction is expected to be close to these calculated trends.
FIGURE 5.

A, FA of labeled CTD-FL (19 nm) monitored as a function of ΔC29 concentration. [NaCl] was varied as indicated on each curve. The lines are fits of the data to a 1:1 binding isotherm. B, plot of the dissociation constant for the NTD/CTD interaction versus [NaCl]. The Kd,NTD/CTD was obtained by fitting the data in A as described under “Experimental Procedures.” The line is the power law fit of Kd,NTD/CTD versus [NaCl] with a log-log slope S = 1 ± 0.3.
Anionic CTD Domain Inhibits Chaperone Activity of HTLV-1 NTD in Trans and HIV-1 NC in Cis
Single time point gel-shift assays were used to characterize mini-TAR RNA/DNA annealing by ΔC29 HTLV-1 NC in the presence of increasing concentrations of CTD-FL. As expected, the presence of CTD-FL had no effect on ΔC29 HTLV-1 NC-facilitated annealing under high salt (100 mm) conditions (Fig. 6A), where binding between NTD and CTD domains was not detected by FA (see Fig. 5A). However, in low salt (10 mm), titration with the CTD-FL reduced annealing by ΔC29 HTLV-1 NC to 85% of the level achieved in the absence of CTD.
FIGURE 6.

A, relative percent mini-TAR RNA/DNA annealed at 30 min using 1 μm ΔC29 HTLV-1 NC in the presence of increasing amounts of CTD-FL in 10 or 100 mm NaCl. B, time course annealing of mini-TAR RNA/DNA in the presence of 1.5 μm HIV-1, HTLV-1, or HIV-1/HTLV-1 chimeric NC.
To further examine the inhibitory capability of the HTLV-1 NC CTD, a chimeric NC was constructed wherein the CTD of HTLV-1 NC was appended to the C terminus of WT HIV-1 NC (Fig. 1D). The annealing activity of the chimera was compared with that of WT HIV-1 and WT HTLV-1 NC. The chimeric NC showed chaperone activity that was greater than that of HTLV-1 NC but was impaired relative to HIV-1 NC (Fig. 6B). Curve fitting to a single exponential function yielded annealing rate constants of 1.74 and 0.20 min−1 for WT HIV-1 NC and chimeric HIV-1/HTLV-1 NC, respectively. Thus, addition of the acidic CTD of HTLV-1 NC to HIV-1 NC in cis resulted in an ∼9-fold decrease in chaperone activity.
NA Aggregating Ability of WT and Mutant HTLV-1 NC Correlates with the Charge of the CTD
A sedimentation assay was used to evaluate the NA aggregating ability of WT and mutant HTLV-1 NC constructs. A plot of the percent mini-TAR RNA aggregated under conditions similar to those used in annealing experiments is shown in Fig. 7 as a function of protein concentration. The NA aggregating ability of these proteins follows a similar trend as their NA chaperone activity (compare Figs. 3 and 7) and is correlated with the anionic character of their CTDs. Indeed, whereas the ΔC29 variant is a very effective aggregating agent, WT, LLD, and D69A HTLV-1 NC fail to aggregate RNA even at saturating concentrations of 5 μm. The EED mutant, with three negative charges removed, displays an intermediate level of aggregating ability.
FIGURE 7.

Percent of mini-TAR RNA aggregated from a solution of 15 nm32P-labeled mini-TAR RNA and 45 nm mini-TAR DNA at 37 °C as a function of WT and mutant HTLV-1 NC concentration.
Nucleic Acid Binding and Duplex Destabilizing Activities of HTLV-1 NC Are Only Weakly Affected by CTD Mutations
FA was used to determine the apparent dissociation constants (Kd) for WT and mutant HTLV-1 NC binding to a 20-nt ssDNA and a 20-bp DNA/RNA duplex. These data are summarized in Table 1, along with previous data obtained for other retroviral NCs (28). Notably, HTLV-1 NC binds ssDNA 3-fold more weakly than HIV-1 NC, and the affinity for the DNA/RNA duplex is ∼8-fold lower. The measured Kd values are in good agreement with the results of a recent fluorescence quenching binding study, which reported the following values for WT HTLV-1 NC: Kdss = 500 ± 300 nm (binding to poly(dT)) and Kdds = 3000 ± 2000 nm (binding to ds calf thymus DNA) (56). The latter studies were conducted in much lower salt (1 mm sodium phosphate buffer instead of 50 mm NaCl used here), and the agreement in Kd values is consistent with the weak salt dependence of HTLV-1 NC binding to NA (56).
TABLE 1.
Duplex destabilization free energies and apparent equilibrium dissociation constants for binding of WT and mutant HTLV-1 NC and other retroviral NCs to ss and ds NA
Kdss and Kdds values are dissociation constants measured using FA for binding to an ssDNA 20-mer and a 20-bp RNA/DNA hybrid duplex, respectively. The free energy of protein-induced destabilization per base pair (ΔG) was estimated from measured Kdss and Kdds values using Equation 1 and assuming that each protein binds to 4 bp (n = 4). RSV is Rous sarcoma virus, and MLV is murine leukemia virus.
| NC | Kdss | Kdds | ΔG |
|---|---|---|---|
| nm | nm | kcal/mol/bp | |
| HTLV-1 WT | 431 ± 87 | 1840 ± 400 | −(0.2 ± 0.05) |
| HTLV-1 ΔC29 | 276 ± 16 | 4366 ± 777 | −(0.4 ± 0.1) |
| HTLV-1 EED | 380 ± 15 | 4213 ± 1353 | −(0.3 ± 0.1) |
| HTLV-1 LLD | 252 ± 19 | 6059 ± 1453 | −(0.5 ± 0.05) |
| HTLV-1 D69A | 291 ± 30 | 4596 ± 1863 | −(0.4 ± 0.05) |
| HIV-1a | 123 ± 9 | 233 ± 11 | −(0.1 ± 0.02) |
| RSVa | 200 ± 49 | 660 ± 47 | −(0.2 ± 0.05) |
| MLVa | 334 ± 34 | 1092 ± 28 | −(0.2 ± 0.05) |
a Values were taken from Ref. 28.
Removal of the CTD or mutation of the acidic residues has only a very minor effect on ssDNA binding, whereas binding to a DNA/RNA hybrid duplex is reduced ∼2–3-fold (Table 1). Thus, the preference for ss over ds NA binding is increased upon CTD deletion or mutation.
WT and Mutant HTLV-1 NC Are Good NA Duplex Destabilizers
The relative ss and ds binding affinities reported in Table 1 suggest that the ΔC29 variant should display even better NA destabilization activity than WT HTLV-1 NC. The values of Kdss and Kdss together with the binding site size of a single NC protein can be used to estimate the protein-induced free energy of destabilization per bp, ΔG, using Equation 1 (see “Experimental Procedures”). Values for ΔG are summarized in Table 1 and suggest that HTLV-1 NCs are equally effective or better duplex destabilizers than the other retroviral NC proteins tested.
To confirm the predictions based on the DNA binding studies, we directly tested the duplex destabilizing ability of WT and mutant HTLV-1 NC using a time-resolved FRET-based approach (42). TAR DNA hairpins were labeled with a fluorophore and quencher pair (Fig. 8A), and fluorescence lifetimes were measured in the absence of NC and as a function of NC protein concentration. Under all conditions, the fluorescence decay is best described by a triple exponential, indicating at least three conformational states designated as T1 (open), T2 (semi-open), and T3 (closed hairpin) (Fig. 8B). A plot of the relative free energy of the open (T1) state as a function of NC protein concentration is shown in Fig. 8C. Increasing NC concentration leads to lower (i.e. more favorable) free energy, reflecting the duplex destabilizing ability of these proteins. Interestingly, the robust destabilization of TAR DNA observed upon HIV-1 NC titration saturates at ∼1 μm NC. In contrast, titration with WT HTLV-1 NC does not saturate even at the highest protein concentration tested (3.2 μm), consistent with the lower NA binding affinity of this protein (see Table 1). However, at high HTLV-1 NC concentrations (3.2 μm), more effective opening of the TAR DNA hairpin is achieved than in the presence of saturating amounts of HIV-1 NC, in agreement with the conclusion that HTLV-1 NC possesses stronger duplex destabilizing activity (see Table 1) (29). Interestingly, the HTLV-1 NC CTD mutants seem to destabilize TAR DNA even more effectively than the WT protein. This conclusion is consistent with the data in Table 1 and appears to be due to their greater preference for ssDNA over dsDNA.
FIGURE 8.

A, sequence and secondary structure of the fluorescently labeled TAR DNA hairpin. B, schematic illustration of the three conformational states of the TAR DNA hairpin monitored by time-resolved FRET. C, relative free energy of the T1 state of the TAR DNA hairpin calculated according to Equation 3, as a function of HIV-1 NC (thick line) and WT or mutant HTLV-1 NC concentration.
SM DNA Stretching Studies Reveal Slow Kinetics of WT HTLV-1 NC/ssDNA Dissociation
The differences in nucleic acid binding properties and destabilization activity of WT and mutant HTLV-1 NC are consistent with the improved chaperone function observed for the mutant proteins. However, rapid nucleic acid binding kinetics is another important component of chaperone function that has previously been used to explain differences in overall chaperone function of retroviral NCs (28). Protein-nucleic acid binding kinetics can be measured using SM DNA stretching by optical tweezers (30, 57–60). Here, this technique was used to probe the NA interaction kinetics of WT and mutant HTLV-1 NCs.
Fig. 9A shows a stretching and relaxation cycle for bacteriophage λ-DNA in the absence of protein (black) and in the presence of a saturating concentration of WT HTLV-1 NC (red). In the absence of protein, at extensions much less than the B-form contour length of dsDNA (0.34 nm/bp), very little force is required to stretch the DNA. As the contour length is approached, the force increases dramatically, reflecting the elasticity of the double helix. The plateau observed as DNA is stretched from 0.34 to 0.6 nm/bp at a constant force of about 60 piconewtons represents a change from dsDNA to ssDNA or a force-induced melting transition (49, 61–63). In the absence of protein, the transition is highly cooperative, and as the DNA is relaxed back to lower extensions, the force-extension curve is almost completely reversible, showing little hysteresis. In contrast, in the presence of WT HTLV-1 NC (Fig. 9A), the DNA relaxation force is significantly lower than forces observed during extension, resulting in large hysteresis. In other words, upon relaxation, the two strands do not re-anneal but rather relax as ssDNA bound by HTLV-1 NC, indicating that the protein does not dissociate from ssDNA on the time scale of the relaxation cycle, which is a few minutes. The stretching part of the DNA cycle is nonequilibrium and therefore depends on the rate of DNA pulling, with slower rates corresponding to lower forces, as was observed previously for the T4 and T7 single-stranded DNA-binding proteins (59, 64–66). The relaxation curves obtained with WT HTLV-1 NC are in sharp contrast to those obtained with HIV-1 NC, which display pulling rate independence and very little hysteresis due to rapid protein/NA dissociation and association (28, 49, 51).
FIGURE 9.

A, DNA stretching (solid line) and relaxation (dotted line) in the absence (black) and presence (red) of WT HTLV-1 NC (700 nm). B, DNA stretching (solid line) and relaxation (dotted line) in the absence (black) and presence (red) of ΔC29 HTLV-1 NC (200 nm).
To test the hypothesis that the large hysteresis (because of slow HTLV-1 NC dissociation from ssDNA) was caused by the anionic CTD, we also performed DNA stretching in the presence of the ΔC29 variant. Fig. 9B shows a representative DNA stretching and relaxation cycle in the presence of saturating ΔC29. At low force, jumps were observed on the stretching curve, during which the force increases at extensions below the B-form contour length of 0.34 nm but then drops suddenly. These jumps represent self-aggregation of the single DNA molecule induced by the protein. This result is consistent with the observation that in contrast to WT HTLV-1 NC, the CTD deletion mutant does aggregate NA (see Fig. 7). This type of aggregation has been observed in the presence of several NA chaperone proteins, including HIV-1 NC (30, 38, 47, 67, 68), the retrotransposon chaperone LINE-1 ORF1p (69, 70), and other retroviral NCs (28). In the presence of ΔC29, the ssDNA stretching curve above the strand separation transition (i.e. above 60 piconewtons) shifted to shorter contour lengths, which indicates that protein-bound ssDNA is shorter than ssDNA in the absence of protein. The hysteresis observed upon relaxation in the presence of the ΔC29 variant is much smaller than that observed in the presence of WT HTLV-1 NC. This indicates that the protein was able to dissociate from the ssDNA, and the DNA was able to almost completely re-anneal on the time scale of the relaxation cycle.
As discussed above, the different levels of hysteresis observed upon DNA relaxation after force-induced melting reflect differences in the DNA re-annealing time. This difference in DNA re-annealing time, in turn, reflects the difference in the protein dissociation time from ssDNA. To more directly monitor the DNA re-annealing time in the presence of WT and ΔC29 HTLV-1 NC, the DNA was stretched in the presence of each protein completely through the melting plateau until reaching the ssDNA stretching region, located at about 0.6 nm/bp. During the process of stretching, DNA base pairs are opened, and ssDNA is exposed for protein binding (71). Next, the DNA was relaxed to a position halfway through the transition at an extension of 0.46 nm/bp. At this point, the DNA extension was held constant, and the force was measured as a function of time. As shown in Fig. 10, the force increased exponentially with time until saturation, reflecting the dissociation of the protein from ssDNA and the subsequent re-annealing of the two protein-free complementary DNA strands. A single exponential fit of the force relaxation curves yields the DNA re-annealing time constant, T↑, which differs by a factor of ∼10 between the two proteins (44.8 ± 6.6 s for WT and 5.3 ± 2 s for ΔC29). As will be discussed in detail elsewhere, the relaxation time, T↑, depends on the stretched DNA length and is related to the off time of the protein, τoff (the inverse of the off rate), as follows: T↑ ∼ τoffC/Kd,6 where τoff is the protein dissociation time constant at concentration C. The equilibrium dissociation constants for these two proteins differed by about a factor of 2 (see Table 1), and we used about three times more WT protein. Therefore, ΔC29 HTLV-1 NC dissociates about 10 times faster than WT HTLV-1 NC. These results are consistent with the hypothesis that the electrostatic interaction of the anionic CTD of HTLV-1 NC with the cationic NTD of a neighboring protein bound to ssDNA is responsible for the slow dissociation of WT HTLV-1 NC.
FIGURE 10.

Time-dependent relaxation data showing protein dissociation and subsequent DNA re-annealing for WT (700 nm, blue line) and ΔC29 HTLV-1 NC (200 nm, red line). The black solid and dotted lines show single exponential fits of the ΔC29 and WT data, respectively. The inset shows the data points and fit obtained for ΔC29 on a shorter time scale.
DISCUSSION
HTLV-1 NC displays extremely poor overall chaperone activity in vitro, despite the fact that it is an effective duplex destabilizer (see also Table 1 and Fig. 8) (28, 29). The highly basic nature of most retroviral NCs is a major factor that contributes to their high NA binding affinity. By contrast, the overall pI of HTLV-1 NC is ∼7 due to a negatively charged CTD extension. In accord with its less basic character, the NA binding affinity of HTLV-1 NC is lower than that of other retroviral NC proteins studied to date (Table 1). Interestingly, although deletion of the CTD or mutations that remove acidic residues within this domain improve overall chaperone function, these same changes were found to have very little effect on NA binding affinity (Table 1). Binding to ssDNA is only slightly increased, whereas dsDNA binding affinity is moderately decreased. The net result is that these variants are even better NA-destabilizing agents than WT HTLV-1 NC.
Intramolecular electrostatic interactions between the anionic CTD and cationic NTD of HTLV-1 NC control the binding of this protein to NA by reducing its on rate, as presented schematically in Fig. 11. A previous tryptophan fluorescence quenching study also suggested an intramolecular interaction between the zinc finger domains of HTLV-1 NC and its CTD (56). This interaction is eliminated in the presence of high salt and is absent in an HTLV-1 NC (11–51) construct, which lacks the CTD and 10 residues at the N terminus. These data are in good agreement with our direct measurement of the intermolecular ΔC29/CTD interaction by FA, which was also shown to be highly sensitive to salt (Fig. 5). We estimated that in the presence of physiological salt concentrations, the NTD/CTD interaction free energy is still significant (between 2 and 8 kcal/mol at 100 mm NaCl), and we hypothesize that this interaction contributes to the poor chaperone properties of HTLV-1 NC. Consistent with this hypothesis, addition of the CTD to the NTD domain in trans reduces the chaperone function of the latter at 10 mm NaCl (Fig. 6A), and an HIV-1 NC/HTLV-1 CTD chimera shows significant reductions in chaperone activity relative to WT HIV-1 NC (Fig. 6B). Interestingly, the annealing rate with chimeric NC is not reduced to the level of WT HTLV-1 NC, indicating that other structural features of HTLV-1 NC may also contribute to its poor chaperone activity.
FIGURE 11.

Schematic model of HTLV-1 NC binding to NA based on the data presented in this study. In the unbound state, the anionic CTD interacts with the cationic NTD thereby precluding the latter from effective binding to NA. The requirement for intramolecular NTD/CTD dissociation makes the rate of HTLV-1 NC/NA association slow. In the bound state, the NTD associates with NA, whereas CTD associates with a neighboring NTD, leading to HTLV-1 NC binding cooperativity and slow NA dissociation (see Fig. 9). Assuming that the NTD/CTD interactions are of similar strength in both states, both on and off rates of the protein are decreased by a similar factor upon CTD deletion. This results in similar Kdss values for WT and ΔC29 HTLV NC proteins (see Table 1). High salt concentration is expected to screen the electrostatic NTD/CTD interactions, resulting in more similar chaperone behavior of the WT and ΔC29 proteins, as observed in mini-TAR annealing experiments (see Fig. 4).
Upon binding to NA, intermolecular electrostatic interactions between NA-bound protein neighbors may lead to the apparent salt-dependent binding cooperativity previously observed by Morcock et al. (56). The head-to-tail arrangement shown in Fig. 11 is the simplest model consistent with the available data, but other arrangements can also be envisioned. In contrast, N- and C-terminally truncated HTLV-1 NC (11–51) was shown to bind NA with negligible cooperativity (56). Multimerization on NA inhibits the aggregation ability of HTLV-1 NC WT (Fig. 7) and reduces its NA dissociation kinetics (Figs. 9 and 10), leading to poor overall chaperone function (Figs. 3 and 4) (28). CTD deletion makes NA dissociation (koff) ∼10-fold faster (Figs. 9 and 10) but does not significantly affect its Kdss (see Table 1). Taken together, these results imply that the NA association (kon) of HTLV-1 NC also becomes faster upon reduction (or screening) of the negative charges of the CTD. This supports the notion that the intramolecular electrostatic interaction between the anionic CTD and cationic NTD slows down the NA association rate of this protein. Upon CTD removal (or charge screening by higher salt), both protein association and dissociation from NA are similarly facilitated, such that the Kdss value of the protein remains essentially unaffected.
How does HTLV-1 function with such a weak NA chaperone protein? Regulation of chaperone activity for this NC may be controlled by interaction with other proteins such as reverse transcriptase in vivo. In addition, it is possible that other viral proteins such as matrix perform at least a subset of functions in HTLV-1 and other deltaretroviruses that are typically carried out by NC, such as NA binding and viral RNA selection. For example, matrix has been shown to play a role in viral RNA packaging in another deltaretrovirus, bovine leukemia virus (72).
Interestingly, mature HIV-1 NCp7 is derived from a precursor protein known as NCp15, which has a basic domain known as p1 followed by an acidic extension (p6) at its C terminus (see Fig. 1C). In addition, the high content of proline residues in all CTDs is also conserved (compare Fig. 1, A, B and C). Similar to WT HTLV-1 NC, HIV-1 NCp15 dissociates slowly from ssDNA (68) and polymerizes on ssDNA without facilitating aggregation (73, 74). Furthermore, failure to cleave p6 from NCp15 results in greatly reduced viral infectivity, likely because of defects in the integration step of the life cycle (75). In contrast to HIV-1 NCp7, which facilitates reverse transcription, NCp15 expressed by a mutant HIV-1 was shown to inhibit DNA polymerization by reverse transcriptase (75). Just as truncation of the CTD of HTLV NC greatly improves its NA aggregating and chaperone properties (Figs. 3, 7, 9, and 10), the natural proteolysis of HIV-1 NC from NCp15 to NCp9 and then NCp7 leads to progressive improvement of the NA aggregating (73, 74) and chaperone properties (68)7 of this protein. Whereas the acidic CTD is part of the mature form of HTLV-1 NC, HIV-1 NCp15 is an intermediate and is not present in significant amounts in the mature virus (76). Work to elucidate the biological significance of this difference in NC processing in these two human retroviruses is currently underway.
A question remains as to why HTLV-1 maintains a CTD that is unfavorable to effective chaperone function. Retroviral NCs perform a variety of essential functions throughout the viral life cycle, and there are many differences between HIV-1 and HTLV-1 that may explain why the CTD is retained in one case but not in the other (77–81). In addition, the viruses appear to have evolved different mechanisms to resist cellular restriction factors such as human APOBEC3G. Extensive research in the last several years (for reviews see Refs. 82–84) has shown that the cellular cytidine deaminases of the APOBEC family are co-packaged into retroviruses via an RNA-mediated interaction with viral Gag proteins. In the case of HIV-1, the viral Vif protein binds APOBEC3G specifically, and targets it for degradation. In contrast, HTLV-1 does not appear to express an accessory protein that interferes with APOBEC3G activity (34). Although APOBEC3G can be found in HTLV-1 particles (85, 86), it has been proposed that the virus reduces the amount of APOBEC3G packaged into virions via a mechanism that is mediated by the CTD of NC (34). Interestingly, the mutants studied here that improve chaperone activity and NA dissociation kinetics of HTLV-1 NC also allowed for higher levels of APOBEC3G packaging and increased viral restriction (34). A possible model to explain NC CTD-dependent APOBEC3G exclusion involves the cooperative polymerization of HTLV-1 NC on ssRNA, effectively preventing APOBEC3G binding. It is possible that the slow kinetics of the WT HTLV-1 NC/RNA interaction predicted by the results presented here interfere with efficient APOBEC3G binding to this RNA, thereby leading to its exclusion from virions. Further studies to test this hypothesis are underway.
Acknowledgments
We thank Cathy V. Hixson and Donald G. Johnson for their assistance in preparing the recombinant NC proteins.
This work was supported, in whole or in part, by National Institutes of Health Grants GM065056 (to K. M.-F.) and GM072462 (to M. C. W.), Ruth L. Kirschstein National Service Award GM072396 (to K. M. S.-M.), and Contracts N01-CO-12400 and HHSN261200800001E from the NCI. This work was also supported by National Science Foundation Grant MCB-0744456 (to M. C. W.).
This paper is dedicated to the late David Derse, a friend and colleague whose work on HTLV-1 was the inspiration for many of the studies described in this study. Although we are too late, we thank him for the DNA clones that, in typical fashion, he so graciously provided for generating the various NC proteins used in this study.
I. Rouzina and M. Williams, unpublished results.
C. Jones, R. Gorelick, I. Rouzina, and K. Musier-Forsyth, unpublished data.
- NA
- nucleic acid(s)
- TAR
- trans-activation response element
- NC
- nucleocapsid protein
- nt
- nucleotide(s)
- WT
- wild-type
- HTLV-1
- human T-cell leukemia virus type 1
- HIV-1
- human immunodeficiency virus type 1
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- APOBEC3G
- apolipoprotein B editing catalytic polypeptide 3G
- SM
- single molecule
- FRET
- Förster resonance energy transfer
- FA
- fluorescence anisotropy
- ss
- single-stranded
- ds
- double-stranded
- TCEP-HCl
- tris(2-carboxyethyl) phosphine hydrochloride
- FAM
- 6-carboxyfluorescein
- TEV
- tobacco etch virus
- HPLC
- high pressure liquid chromatography.
REFERENCES
- 1.Rajkowitsch L., Chen D., Stampfl S., Semrad K., Waldsich C., Mayer O., Jantsch M. F., Konrat R., Bläsi U., Schroeder R. (2007) RNA Biol. 4, 118–130 [DOI] [PubMed] [Google Scholar]
- 2.Levin J. G., Guo J., Rouzina I., Musier-Forsyth K. (2005) Prog. Nucleic Acid Res. Mol. Biol. 80, 217–286 [DOI] [PubMed] [Google Scholar]
- 3.Thomas J. A., Gorelick R. J. (2008) Virus Res. 134, 39–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darlix J. L., Garrido J. L., Morellet N., Mély Y., de Rocquigny H. (2007) Adv. Pharmacol. 55, 299–346 [DOI] [PubMed] [Google Scholar]
- 5.Darlix J. L., Gabus C., Nugeyre M. T., Clavel F., Barré-Sinoussi F. (1990) J. Mol. Biol. 216, 689–699 [DOI] [PubMed] [Google Scholar]
- 6.Fu W., Gorelick R. J., Rein A. (1994) J. Virol. 68, 5013–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng Y. X., Copeland T. D., Henderson L. E., Gorelick R. J., Bosche W. J., Levin J. G., Rein A. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 7577–7581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang C., Rong L., Laughrea M., Kleiman L., Wainberg M. A. (1998) J. Virol. 72, 6629–6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laughrea M., Shen N., Jetté L., Darlix J. L., Kleiman L., Wainberg M. A. (2001) Virology 281, 109–116 [DOI] [PubMed] [Google Scholar]
- 10.Kafaie J., Song R., Abrahamyan L., Mouland A. J., Laughrea M. (2008) Virology 375, 592–610 [DOI] [PubMed] [Google Scholar]
- 11.Cristofari G., Darlix J. L. (2002) Prog. Nucleic Acid Res. Mol. Biol. 72, 223–268 [DOI] [PubMed] [Google Scholar]
- 12.Darlix J. L., Lapadat-Tapolsky M., de Rocquigny H., Roques B. P. (1995) J. Mol. Biol. 254, 523–537 [DOI] [PubMed] [Google Scholar]
- 13.Rein A., Henderson L. E., Levin J. G. (1998) Trends Biochem. Sci. 23, 297–301 [DOI] [PubMed] [Google Scholar]
- 14.Tsuchihashi Z., Brown P. O. (1994) J. Virol. 68, 5863–5870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan B., Musier-Forsyth K. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 13530–13535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan B., Weidemaier K., Yip W. T., Barbara P. F., Musier-Forsyth K. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Rocquigny H., Gabus C., Vincent A., Fournié-Zaluski M. C., Roques B., Darlix J. L. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 6472–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hargittai M. R., Gorelick R. J., Rouzina I., Musier-Forsyth K. (2004) J. Mol. Biol. 337, 951–968 [DOI] [PubMed] [Google Scholar]
- 19.Hargittai M. R., Mangla A. T., Gorelick R. J., Musier-Forsyth K. (2001) J. Mol. Biol. 312, 985–997 [DOI] [PubMed] [Google Scholar]
- 20.Prats A. C., Sarih L., Gabus C., Litvak S., Keith G., Darlix J. L. (1988) EMBO J. 7, 1777–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo F., Saadatmand J., Niu M., Kleiman L. (2009) J. Virol. 83, 8099–8107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saadatmand J., Niu M., Kleiman L., Guo F. (2009) Virology 391, 334–341 [DOI] [PubMed] [Google Scholar]
- 23.Thomas J. A., Gagliardi T. D., Alvord W. G., Lubomirski M., Bosche W. J., Gorelick R. J. (2006) Virology 353, 41–51 [DOI] [PubMed] [Google Scholar]
- 24.Carteau S., Gorelick R. J., Bushman F. D. (1999) J. Virol. 73, 6670–6679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao K., Gorelick R. J., Johnson D. G., Bushman F. (2003) J. Virol. 77, 1598–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buckman J. S., Bosche W. J., Gorelick R. J. (2003) J. Virol. 77, 1469–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carteau S., Batson S. C., Poljak L., Mouscadet J. F., de Rocquigny H., Darlix J. L., Roques B. P., Käs E., Auclair C. (1997) J. Virol. 71, 6225–6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart-Maynard K. M., Cruceanu M., Wang F., Vo M. N., Gorelick R. J., Williams M. C., Rouzina I., Musier-Forsyth K. (2008) J. Virol. 82, 10129–10142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darugar Q., Kim H., Gorelick R. J., Landes C. (2008) J. Virol. 82, 12164–12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cruceanu M., Gorelick R. J., Musier-Forsyth K., Rouzina I., Williams M. C. (2006) J. Mol. Biol. 363, 867–877 [DOI] [PubMed] [Google Scholar]
- 31.Narayanan N., Gorelick R. J., DeStefano J. J. (2006) Biochemistry 45, 12617–12628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Record M. T., Jr., Anderson C. F., Lohman T. M. (1978) Q. Rev. Biophys. 11, 103–178 [DOI] [PubMed] [Google Scholar]
- 33.Derse D., Mikovits J., Polianova M., Felber B. K., Ruscetti F. (1995) J. Virol. 69, 1907–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derse D., Hill S. A., Princler G., Lloyd P., Heidecker G. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 2915–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kapust R. B., Tözsér J., Fox J. D., Anderson D. E., Cherry S., Copeland T. D., Waugh D. S. (2001) Protein Eng. 14, 993–1000 [DOI] [PubMed] [Google Scholar]
- 36.Kapust R. B., Tözsér J., Copeland T. D., Waugh D. S. (2002) Biochem. Biophys. Res. Commun. 294, 949–955 [DOI] [PubMed] [Google Scholar]
- 37.Guo J., Wu T., Anderson J., Kane B. F., Johnson D. G., Gorelick R. J., Henderson L. E., Levin J. G. (2000) J. Virol. 74, 8980–8988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vo M. N., Barany G., Rouzina I., Musier-Forsyth K. (2006) J. Mol. Biol. 363, 244–261 [DOI] [PubMed] [Google Scholar]
- 39.Urbaneja M. A., Kane B. P., Johnson D. G., Gorelick R. J., Henderson L. E., Casas-Finet J. R. (1999) J. Mol. Biol. 287, 59–75 [DOI] [PubMed] [Google Scholar]
- 40.Fisher R. J., Fivash M. J., Stephen A. G., Hagan N. A., Shenoy S. R., Medaglia M. V., Smith L. R., Worthy K. M., Simpson J. T., Shoemaker R., McNitt K. L., Johnson D. G., Hixson C. V., Gorelick R. J., Fabris D., Henderson L. E., Rein A. (2006) Nucleic Acids Res. 34, 472–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGhee J. D. (1976) Biopolymers 15, 1345–1375 [DOI] [PubMed] [Google Scholar]
- 42.Hong M. K., Harbron E. J., O'Connor D. B., Guo J., Barbara P. F., Levin J. G., Musier-Forsyth K. (2003) J. Mol. Biol. 325, 1–10 [DOI] [PubMed] [Google Scholar]
- 43.Askjaer P., Kjems J. (1998) J. Biol. Chem. 273, 11463–11471 [DOI] [PubMed] [Google Scholar]
- 44.Toyoshima H., Itoh M., Inoue J., Seiki M., Takaku F., Yoshida M. (1990) J. Virol. 64, 2825–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Golinelli M. P., Hughes S. H. (2001) Virology 285, 278–290 [DOI] [PubMed] [Google Scholar]
- 46.Zuker M. (2003) Nucleic Acids Res. 31, 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vo M. N., Barany G., Rouzina I., Musier-Forsyth K. (2009) J. Mol. Biol. 386, 773–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson P. E., Turner R. B., Wu Z. R., Hairston L., Guo J., Levin J. G., Summers M. F. (2000) Biochemistry 39, 9084–9091 [DOI] [PubMed] [Google Scholar]
- 49.Williams M. C., Rouzina I., Wenner J. R., Gorelick R. J., Musier-Forsyth K., Bloomfield V. A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 6121–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernacchi S., Stoylov S., Piémont E., Ficheux D., Roques B. P., Darlix J. L., Mély Y. (2002) J. Mol. Biol. 317, 385–399 [DOI] [PubMed] [Google Scholar]
- 51.Williams M. C., Gorelick R. J., Musier-Forsyth K. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 8614–8619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heath M. J., Derebail S. S., Gorelick R. J., DeStefano J. J. (2003) J. Biol. Chem. 278, 30755–30763 [DOI] [PubMed] [Google Scholar]
- 53.Beltz H., Clauss C., Piémont E., Ficheux D., Gorelick R. J., Roques B., Gabus C., Darlix J. L., de Rocquigny H., Mély Y. (2005) J. Mol. Biol. 348, 1113–1126 [DOI] [PubMed] [Google Scholar]
- 54.Kankia B. I., Barany G., Musier-Forsyth K. (2005) Nucleic Acids Res. 33, 4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nyborg J. K., Peersen O. B. (2004) Biochem. J. 381, e3–e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morcock D. R., Kane B. P., Casas-Finet J. R. (2000) Biochim. Biophys. Acta 1481, 381–394 [DOI] [PubMed] [Google Scholar]
- 57.McCauley M. J., Williams M. C. (2007) Biopolymers 85, 154–168 [DOI] [PubMed] [Google Scholar]
- 58.Williams M. C., Rouzina I. (2002) Curr. Opin. Struct. Biol. 12, 330–336 [DOI] [PubMed] [Google Scholar]
- 59.Shokri L., Marintcheva B., Eldib M., Hanke A., Rouzina I., Williams M. C. (2008) Nucleic Acids Res. 36, 5668–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pant K., Karpel R. L., Williams M. C. (2003) J. Mol. Biol. 327, 571–578 [DOI] [PubMed] [Google Scholar]
- 61.Rouzina I., Bloomfield V. A. (2001) Biophys. J. 80, 882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rouzina I., Bloomfield V. A. (2001) Biophys. J. 80, 894–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williams M. C., Rouzina I., Bloomfield V. A. (2002) Acc. Chem. Res. 35, 159–166 [DOI] [PubMed] [Google Scholar]
- 64.Pant K., Karpel R. L., Rouzina I., Williams M. C. (2004) J. Mol. Biol. 336, 851–870 [DOI] [PubMed] [Google Scholar]
- 65.Pant K., Karpel R. L., Rouzina I., Williams M. C. (2005) J. Mol. Biol. 349, 317–330 [DOI] [PubMed] [Google Scholar]
- 66.Rouzina I., Pant K., Karpel R. L., Williams M. C. (2005) Biophys. J. 89, 1941–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vo M. N., Barany G., Rouzina I., Musier-Forsyth K. (2009) J. Mol. Biol. 386, 789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cruceanu M., Urbaneja M. A., Hixson C. V., Johnson D. G., Datta S. A., Fivash M. J., Stephen A. G., Fisher R. J., Gorelick R. J., Casas-Finet J. R., Rein A., Rouzina I., Williams M. C. (2006) Nucleic Acids Res. 34, 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin S. L., Cruceanu M., Branciforte D., Wai-Lun Li P., Kwok S. C., Hodges R. S., Williams M. C. (2005) J. Mol. Biol. 348, 549–561 [DOI] [PubMed] [Google Scholar]
- 70.Martin S. L., Bushman D., Wang F., Li P. W., Walker A., Cummiskey J., Branciforte D., Williams M. C. (2008) Nucleic Acids Res. 36, 5845–5854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shokri L., McCauley M. J., Rouzina I., Williams M. C. (2008) Biophys. J. 95, 1248–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang H., Norris K. M., Mansky L. M. (2003) J. Virol. 77, 9431–9438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mirambeau G., Lyonnais S., Coulaud D., Hameau L., Lafosse S., Jeusset J., Justome A., Delain E., Gorelick R. J., Le Cam E. (2006) J. Mol. Biol. 364, 496–511 [DOI] [PubMed] [Google Scholar]
- 74.Mirambeau G., Lyonnais S., Coulaud D., Hameau L., Lafosse S., Jeusset J., Borde I., Reboud-Ravaux M., Restle T., Gorelick R. J., Le Cam E. (2007) PLoS ONE 2, e669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coren L. V., Thomas J. A., Chertova E., Sowder R. C., 2nd, Gagliardi T. D., Gorelick R. J., Ott D. E. (2007) J. Virol. 81, 10047–10054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Henderson L. E., Bowers M. A., Sowder R. C., 2nd, Serabyn S. A., Johnson D. G., Bess J. W., Jr., Arthur L. O., Bryant D. K., Fenselau C. (1992) J. Virol. 66, 1856–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mazurov D., Heidecker G., Derse D. (2007) J. Biol. Chem. 282, 3896–3903 [DOI] [PubMed] [Google Scholar]
- 78.Mazurov D., Heidecker G., Derse D. (2006) Virology 346, 194–204 [DOI] [PubMed] [Google Scholar]
- 79.Derse D., Hill S. A., Lloyd P. A., Chung Hk., Morse B. A. (2001) J. Virol. 75, 8461–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pettit S. C., Sanchez R., Smith T., Wehbie R., Derse D., Swanstrom R. (1998) AIDS Res. Hum. Retroviruses 14, 1007–1014 [DOI] [PubMed] [Google Scholar]
- 81.Derse D., Heidecker G., Mitchell M., Hill S., Lloyd P., Princler G. (2004) Front. Biosci. 9, 2495–2499 [DOI] [PubMed] [Google Scholar]
- 82.Goila-Gaur R., Strebel K. (2008) Retrovirology 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Holmes R. K., Malim M. H., Bishop K. N. (2007) Trends Biochem. Sci. 32, 118–128 [DOI] [PubMed] [Google Scholar]
- 84.Malim M. H. (2009) Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 675–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Navarro F., Bollman B., Chen H., König R., Yu Q., Chiles K., Landau N. R. (2005) Virology 333, 374–386 [DOI] [PubMed] [Google Scholar]
- 86.Sasada A., Takaori-Kondo A., Shirakawa K., Kobayashi M., Abudu A., Hishizawa M., Imada K., Tanaka Y., Uchiyama T. (2005) Retrovirology 2, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]