

Abstract
Prothrombinase converts prothrombin to thrombin via cleavage at Arg320 followed by cleavage at Arg271. Exosite-dependent binding of prothrombin to prothrombinase facilitates active site docking by Arg320 and initial cleavage at this site. Precise positioning of the Arg320 site for cleavage is implied by essentially normal cleavage at Arg320 in recombinant prothrombin variants bearing additional Arg side chains either one or two residues away. However, mutation of Arg320 to Gln reveals that prothrombinase can cleave prothrombin following Arg side chains shifted by as many as two residues N-terminal to the 320 position at near normal rates. Further repositioning leads to a loss in cleavage at this region with an abrupt shift toward slow cleavage at Arg271. In contrast, the binding constant for the active site docking step is strongly dependent on the sequence preceding the scissile bond as well as position. Large effects on binding only yield minor changes in rate until the binding constant passes a threshold value. This behavior is expected for a substrate that can engage the enzyme through mutually exclusive active site docking reactions followed by cleavage to yield different products. Cleavage site specificity as well as the ordered action of prothrombinase on its compound substrate is regulated by the thermodynamics of active site engagement of the individual sites as well as competition between alternate cleavage sites for active site docking.
Introduction
The conversion of the zymogen, prothrombin, to the serine proteinase thrombin is crucial for clot formation following the initiation of the blood coagulation cascade (1–3). The physiologically relevant catalyst for this reaction is considered to be prothrombinase, a membrane-bound enzyme complex containing the serine proteinase, factor Xa, and the cofactor, factor Va (1–3). Accordingly, congenital deficiency of either proteinase or cofactor is associated with comparably catastrophic bleeding (4).
Thrombin formation requires specific proteolysis of the zymogen precursor at two sites (1–3, 5). Although both scissile bonds in prothrombin are available for cleavage, prothrombinase assembled on synthetic phospholipid vesicles acts on its substrate in an apparently ordered fashion. The bulk of flux toward thrombin formation arises from initial cleavage following Arg320 to produce the proteinase meizothrombin (mIIa)3 as an intermediate (5–8). Further processing at Arg271 yields thrombin (IIa) and the activation peptide, fragment 1.2 (F12) (Scheme 1). Cleavage in the opposite order, yielding F12 and the zymogen prethrombin 2 (P2) as intermediates (Scheme 1), contributes in an experimentally undetectable way (<5%) toward thrombin formation (7).
SCHEME 1.

Cleavage products of prothrombin. Line diagrams illustrate the two cleavage sites within intact prothrombin (II). Numbering is according to the 579 residues in the mature protein. Cleavage at Arg320 yields meizothrombin (mIIa), whereas a single cleavage at Arg271 yields F12 and P2. The species obtained following cleavage at both sites are F12 and IIa. A key disulfide bond that covalently links two polypeptide chains in IIa and mIIa is illustrated to aid in the interpretation of the results of SDS-PAGE following disulfide bond reduction.
The kinetic basis for this phenomenon lies in the ability of prothrombinase to act on the Arg320 site with ∼30-fold higher catalytic efficiency than at Arg271 in the otherwise uncleaved zymogen (7). Prior cleavage at Arg320 restores efficient cleavage at Arg271, thereby leading to the essentially ordered action of prothrombinase on the two sites in the substrate (7, 9). These functional effects arise from the predominant role played by exosite interactions involving extended surfaces removed from the catalytic site and substrate structures distant from the cleavage site(s) in mediating the binding of substrate to prothrombinase (10). Equivalent exosite interactions tether all possible substrate forms to the enzyme complex with comparable affinity and regulate the presentation of the individual cleavage sites for active site engagement and catalysis (7, 10–12).
In prothrombin, exosite binding facilitates active site engagement of structures flanking the Arg320 site, whereas elements at the Arg271 site cannot efficiently dock at the active site of prothrombinase (13). Because the two cleavage sites in prothrombin are expected to be ∼36 Åapart (14–16), geometric features of the substrate likely play a major role in the selective presentation of Arg320 for cleavage, thereby explaining the ability of the enzyme complex to preferentially cleave the substrate at this site at the expense of cleavage at Arg271. Initial cleavage at Arg320, which drives the conversion of zymogen to proteinase, has been proposed to alter geometric constraints to facilitate active site engagement by the Arg271 site and its efficient proteolysis in the intermediate, meizothrombin (9, 13).
Exosite-dependent substrate recognition and its attending consequences provide a kinetic and mechanistic framework explaining the action of prothrombinase on prothrombin. A key element of this framework lies in the central role played by substrate geometry in both facilitating and restricting presentation of the individual cleavage sites for active site docking and resultant catalysis. In addition to explaining specificity in terms of the ordered action of prothrombinase on prothrombin, this reliance on precise geometric positioning likely drives specificity in a broader sense. Exosite tethering restricts the action of prothrombinase to prothrombin and prevents cleavage at the numerous other Arg residues within prothrombin despite the established ability of the catalytic site to bind and cleave a variety of Arg-containing sequences (17–19). However, such ideas are not fully consistent with findings made with the Segovia variant of prothrombin (IIRR) purified from a patient and bearing the sequence Asp318-Arg319-Arg320 in place of Asp318-Gly319-Arg320 (20, 21). Cleavage at Arg320-Ile321 in prothrombin, which corresponds to cleavage at the Arg15-Ile16 sequence in chymotrypsinogen, generates a new N terminus, which inserts into the N-terminal binding cleft in a sequence-specific manner to yield a mature serine proteinase (22). Possible cleavage of IIRR at Arg319 rather than at Arg320 is implied by the reduced proteolytic activity of the cleaved product possibly reflecting the inability to generate a new N-terminal sequence beginning at Ile321. These findings cast doubt on the proposed critical role played by precise geometric positioning of the exosite-tethered substrate in determining substrate recognition and specificity.
We have investigated the role played by precise positioning of the substrate in allowing prothrombinase to discriminate between the two cleavage sites in prothrombin. By the use of a series of cleavage site variants of prothrombin, we have assessed the action of prothrombinase on the equivalent of the Arg320 cleavage site incrementally shifted toward the Arg271 cleavage site. Surprisingly, our approach reveals that both preference and rate for cleavage in the Arg320 region in prothrombin are only modestly affected by shifts that produce large changes in the thermodynamics for active site engagement. These new insights into the thermodynamic and kinetic mechanisms at play in regulating ordered prothrombin cleavage have broader implications for the multiple instances in biology wherein proteinases act to cleave at more than one site within their cognate substrates.
EXPERIMENTAL PROCEDURES
Reagents
Human plasma used for protein isolation was a generous gift of the Plasmapheresis Unit of the Hospital of the University of Pennsylvania. H-d-Phenylalanyl-l-pipecolyl-l-arginine-p-nitroanilide (S2238, Chromogenix), d-phenylalanyl-l-proline-l-arginine chloromethyl ketone (FPR-CH2Cl, Calbiochem), 4-aminobenzamidine (pAB, Aldrich), p-amidinophenylmethanesulfonyl fluoride (Sigma), and dansylarginine-N-(3-ethyl-1,5-pentanediyl)amide (DAPA, Hematologic Technologies) were from the indicated suppliers. Stock solutions of S2238, pAB, and DAPA were prepared in water, and concentrations were determined using E342M = 8,270 m−1·cm−1 (S2238), E293M = 15,000 m−1·cm−1 (pAB), and E330M = 4,010 m−1·cm−1 (DAPA) (23–25). Soy bean trypsin inhibitor Sepharose was from Sigma. Small unilamellar phospholipid vesicles (PCPS) composed of 75% (w/w) hen egg l-α-phosphatidylcholine and 25% (w/w) porcine brain l-α-phosphatidylserine (Avanti Polar Lipids) were prepared and quality-controlled as described (26). All measurements were performed in 20 mm Hepes, 150 mm NaCl, 5 mm CaCl2, 0.1% (w/v) polyethylene glycol 8000, pH 7.5 (Assay Buffer) at 25 °C.
Proteins
Prothrombin, factor X, and factor V were isolated from plasma by established procedures (27, 28). Factor Xa and factor Va were purified following preparative activation of factor X by the purified activator from Russell's viper venom or of factor V by thrombin as described previously (5, 28). Both activated species were quality-controlled as described (28). Recombinant factor X with the catalytic serine replaced with alanine (XS195A, recombinant factor X containing Ala in place of the catalytic Ser, numbered according to the homologous residues in chymotrypsinogen (43)) was expressed in HEK293 cells and purified from conditioned media as described in detail (29, 30). Factor XS195A was proteolytically activated and purified by affinity chromatography on soy bean trypsin-Sepharose as described previously (13). Mutagenic primers and the QuikChange mutagenesis kit (Stratagene) were used to introduce the indicated mutations into the coding sequence for the wild type human prothrombin gene contained in an entry cassette in the pENTR vector (7). The resulting cassettes were transferred using λ integrase to an appropriately adapted pcDNA 3.1(+) destination vector using the Gateway cloning system (Invitrogen) (7). The integrity of each expression cassette was verified by DNA sequencing before transfection of HEK293 cells and selection of stable cell lines. Each prothrombin variant was purified from conditioned culture medium by established procedures following large scale expression in serum-free media containing vitamin K (7). Quality control by N-terminal sequencing and quantitative analysis of 4-carboxyglutamate following base hydrolysis (31) established that XS195A and all recombinant variants of prothrombin possessed a correctly processed N terminus with 4-carboxyglutamic acid content that was indistinguishable from that of their counterparts purified from plasma. Recombinant factor V lacking residues 811–1,491 in the B-domain (V810) was expressed on a large scale in baby hamster kidney cells as described previously (32), except that the Dulbecco's modified Eagle's medium/F12 medium lacked phenol red. Conditioned medium collected daily (6 liters) was treated with 10 mm benzamidine, 3 μm p-amidinophenylmethanesulfonyl fluoride and applied at room temperature to a column (4.6 × 10 cm) of SP-Sepharose (Amersham Biosciences) equilibrated in 20 mm Hepes, 0.15 m NaCl, 5 mm CaCl2, 1 mm benzamidine, pH 7.4. Following washing with the same buffer, bound protein was eluted with buffer containing 0.65 m NaCl. Fractions containing factor V activity were stored frozen at −20 °C. Accumulated fractions from 5 days of media collection were pooled and further processed in two halves. Each half was diluted with 5 volumes of 12 mm Hepes, 5 mm CaCl2, pH 7.4, and applied to a 1 × 10-cm Poros HQ/M column (PerkinElmer Life Sciences) equilibrated in 20 mm Hepes, 0.15 m NaCl, 5 mm CaCl2, pH 7.4. Following washing with the same buffer, bound protein was eluted with a linear gradient of increasing NaCl (0.15-1.0 m, 20 ml/min, 19 min). Fractions containing V810 from both runs were pooled, treated with 6 μm p-amidinophenylmethanesulfonyl fluoride, dialyzed for 2.75 h against 50 volumes of 20 mm Hepes, 5 mm CaCl2, pH 7.4, concentrated by centrifugal ultrafiltration to ∼3 mg/ml, and stored frozen at −80 °C (yield = 60 mg). Preparative activation and purification of Va from the recombinant species were done by modifications to published procedures (32, 33). Purified V810 (1 mg/ml, 30 ml) diluted into 20 mm Hepes, 0.15 m NaCl, 5 mm CaCl2, pH 7.4, was preparatively activated with 20 nm thrombin for 10 min at 37 °C. The reaction mixture was quenched with 50 μm FPR-CH2Cl and applied to a 1 × 10-cm Poros HQ/M column equilibrated in the same buffer. Following washing, bound protein was eluted with a linear gradient of increasing NaCl (0.15–0.66 m, 5 ml/min, 7 min). Fractions containing Va were pooled, treated with 15 μm FPR-CH2Cl, diluted with 3 volumes of 20 mm MES, 5 mm CaCl2, pH 6.8, and applied to a 1 × 5-cm Poros HS-50 column (PerkinElmer Life Sciences) equilibrated in the same buffer containing 0.15 m NaCl. Following washing, bound protein was eluted with a linear gradient of increasing NaCl (0.15–0.66 m, 10 ml/min, 12 min). Fractions containing factor Va were pooled, exchanged into 20 mm Hepes, 0.15 m NaCl, 5 mm CaCl2, pH 7.4, by repeated centrifugal ultrafiltration, concentrated to ∼3.5 mg/ml, and stored at −80 °C in single use aliquots (yield = 16.5 mg). Previous studies have established that Va produced from recombinant V810 by the action of thrombin possesses the same polypeptide structure as the activated product from factor V purified from plasma. Fluorescence binding measurements and measurements of prothrombin activation established that Va derived from recombinant V810 was indistinguishable from Va derived from plasma V in its ability to bind factor Xa and to function as a cofactor within prothrombinase (not shown). The two types of Va have been used interchangeably in this study. Protein concentrations were determined using the following molecular weights and extinction coefficients (E2800.1%): Xa and XaS195A (recombinant factor Xa containing Ala in place of the catalytic Ser), 45,300, 1.16 (34); all prothrombin variants, 72,000, 1.47 (35); thrombin, 37,500, 1.89 (35); V810, 216,000, 1.54 (32); and Va, 168,000, 1.78 (33).
Analysis of Prothrombin Cleavage by SDS-PAGE
All prothrombin variants were exchanged into Assay Buffer by centrifugal gel filtration using Sephadex G-25 before use. Reaction mixtures (800 μl) maintained at 25 °C and containing 1.4 μm IIVariant (recombinant prothrombin variants as defined in Scheme 2), 30 μm PCPS, 60 μm DAPA, and 30 nm Va in Assay Buffer were initiated by the addition of 0.2 nm Xa. Aliquots (40 μl) were withdrawn at the indicated times and quenched by mixing with 16 μl of 0.2 m Tris, 6.4% (w/v) SDS, 32% (v/v) glycerol, 0.04% (w/v) bromphenol blue, 50 mm EDTA, 50 mm dithiothreitol, pH 6.8, in a 96-well PCR plate (Eppendorf). Proteolysis by thrombin in the quenched samples was further limited by the addition of 36 μm FPR-CH2Cl. Samples (40 μl) were heated at 85 °C for 5 min, subject to electrophoresis using 10% precast Tris-Glycine gels (Invitrogen), and protein bands were visualized using the Colloidal Blue stain (Invitrogen) and following the manufacturer's instructions. Stained gels were imaged using a high resolution infrared fluorescence scanner (Odyssey, Li-Cor) using λEX = 685 nm and λEM = 720 nm. Control experiments established the linear dependence of the integrated signal on loaded prothrombin and fragments extending well beyond the amounts expected in these gels.
SCHEME 2.

Cleavage site variants of prothrombin. The sequence surrounding the two cleavage sites in prothrombin are listed for the variants used in this study. Vertical arrows denote the position of the scissile bonds in wild type prothrombin. Mutated residues are shown in red, and residues in blue denote the new N-terminal sequence following cleavage required for the expression of proteolytic activity in the product. All variants possessed the same sequence flanking the Arg271 site as the wild type species.
N-Terminal Sequence Analysis of Prothrombin Cleavage Products
Reaction mixtures containing 5 μm IIVariant, 30 μm PCPS, 60 μm DAPA, and 30 nm Va in Assay Buffer were incubated at 25 °C for 45 min following the addition of 1 nm Xa. Following quenching as described above, samples were subject to SDS-PAGE, electrophoretically transferred to polyvinylidene fluoride membranes, and visualized by staining as described previously (27). The N-terminal sequence of excised bands was determined by automated Edman degradation by Dr. Jan Pohl at the Emory University Microchemical Facility or at the Biotechnology Branch of the Centers for Disease Control.
Activity Measurements
Reaction mixtures (200 μl) in Assay Buffer and maintained at 25 °C contained 1.4 μm IIVariant, 30 μm PCPS, 6 μm DAPA, and 30 nm Va. Prothrombin cleavage was initiated by the addition of 0.2 nm Xa, and aliquots (10 μl) withdrawn at various times were quenched by mixing with Assay Buffer containing 50 mm EDTA in place of 5 mm Ca2+. Quenched samples were further diluted in the same buffer, and initial velocities of S2238 hydrolysis were determined following the addition of 100 μm peptidyl substrate. Concentrations of proteinase formed were determined by interpolation from the linear dependence of the rate of S2238 hydrolysis on known concentrations of thrombin determined in the presence of the same concentration of DAPA as in the quenched samples.
Substrate Binding Studies
The ability of substrate variants to engage the active site of Xa within prothrombinase in the absence of catalysis was assessed by fluorescence studies with pAB as described previously (13). As before, each substrate titration employed three reaction mixtures to provide for appropriate controls (13). Reaction mixtures (0.6 ml each) in 0.5 × 0.5-cm quartz cuvettes and prepared in Assay Buffer contained: A, no probe (1 μm XaS195A, 1.2 μm Va, 200 μm PCPS, and 10 μm FPR-CH2Cl); B, no enzyme (1.2 μm Va, 200 μm PCPS, 10 μm FPR-CH2Cl, and 25 μm pAB); and C, experimental (1 μm XaS195A, 1.2 μm Va, 200 μm PCPS, 10 μm FPR-CH2Cl, and 25 μm pAB). Steady state fluorescence intensity was measured in a PTI QuantaMaster fluorescence spectrophotometer (Photon Technology International) using λEX = 320 nm and λEM = 374 nm with a long pass filter (LP 345, CVI Laser) in the emission beam. Intensities were obtained by integration of the signal for 30 s following incremental additions of titrant, previously exchanged into Assay Buffer. Fluorescence intensities measured for reactions B and C were corrected for scattering effects (from reaction A) and processed as described previously to derive FOBS/FP,Free representing the ratio of observed fluorescence to that observed with free pAB (13).
Data Analysis
Concentrations of Va and PCPS were chosen based on published equilibrium constants and stoichiometries to ensure that all added Xa was saturably incorporated into the Xa-Va-PCPS ternary complex (36). Thus, the concentration of prothrombinase was considered equal to the concentration of the limiting concentration of Xa. Some figures illustrate representative findings from 2 to 5 experiments performed at an equivalent level of detail and frequently with at least one different protein preparation. All replicates were incorporated into the quantitative analyses of prothrombin consumption or fluorescence measurements wherein the data points denote means ± 1 S.D. Steady state rate expressions were verified by symbolic derivation using the applet King-Altman, publicly available through BioKin Inc., as well as by their ready reduction to the independently derived rapid equilibrium form.
Quantitative densitometry was performed using ImageQuant (Amersham Biosciences) and by volume integration of the bands in unscaled images obtained from the fluorescent scanner. The fate of prothrombin was determined by normalizing the integrated volume from this band to the total stained material in each lane to correct for variations in loaded material and by setting the staining intensity observed in the t = 0 lane to 1. This focus on prothrombin avoids the complexities associated with correcting for the differential fractional staining of the product bands in which both product species as well as their fractional staining are dependent on the variant being analyzed. Progress curves of the fate of prothrombin as a function of time were fitted to the logarithmic approximation previously described to yield initial rates (37). Confidence limits (95%) were determined by propagating errors in the fitted coefficients used to compute initial rates.
The ability of substrate variants to engage the active site of prothrombinase was inferred from pAB displacement studies using the model, assumptions, and considerations detailed previously (13). The program Dynafit, provided as a generous gift by Petr Kuzmic (BioKin Inc.), was used to combine the numerical solution of equilibria with non-linear least squares analysis (38). In the present work, the fluorescence enhancement of pAB upon binding XaS195A within prothrombinase was fixed at 30-fold, and the equilibrium dissociation constant for exosite-dependent interaction of substrate variant was fixed at 100 nm, in accordance with measured values (7). We continue to employ the unverified assumption that the fluorescence yield of pAB bound to the active site within prothrombinase is unperturbed when the substrate initially engages the enzyme through exosite binding. Analysis with these constraints yielded fitted values for the equilibrium constants (Ks* and KE,P) describing the mutually exclusive interactions between the active site of Xa within prothrombinase and either the substrate or pAB as well as the stoichiometry for the interaction of the prothrombin variant with prothrombinase.
RESULTS
Experimental Strategy
The postulated role for precise substrate geometry in the preferential presentation of the Arg320 site for active site docking and cleavage in prothrombinase was examined using a series of prothrombin variants wherein the Arg residue at this site was systematically repositioned toward the NH2 terminus and the Arg271 site (Scheme 2). The first variant, IIRR, was prepared with the intent of testing the previous implication that the vicinal arginines offer an alternate P1 4 residue for efficient recognition and cleavage by prothrombinase (20, 21). In the second variant (IIRGR, Scheme 2), the Asp-Gly-Arg sequence preceding the cleavage site in IIWT (where IIWT denotes wild type prothrombin) was replaced with Arg-Gly-Arg to test for possible cleavage at Arg318 as well as Arg320. The following four register shift variants (II-1δ through II-4δ) contained Gln in place of Arg320 to render this site uncleavable with the Arg-Gly-Arg motif shifted in one-residue increments toward the N terminus, culminating with IIQ320 that lacked any Arg residues in the immediate vicinity (Scheme 2). We pursued the use of the Arg-Gly-Arg motif in this register shift strategy because of the added advantage of obtaining pairs of variants with Arg residues at the same sequence position to provide additional opportunities for the verification of unexpected findings. Because perturbed active site docking by the substrate is expected to have pronounced effects on Vmax (10), we also pursued studies at the physiological concentration (1.4 μm) of prothrombin and its variants, which is well above saturation (7). In agreement with this design, an ∼3.5-fold increase in the concentration of prothrombin did not change the rate of prothrombin cleavage (not shown).
Discriminate Action of Prothrombinase on Vicinal Arginines in the Substrate
The action of prothrombinase on IIWT was associated with the consumption of prothrombin, the transient appearance of a band (F12-A) uniquely associated with the formation of mIIa as an intermediate, followed by bands arising from the production of IIa (Fig. 1A). These results are comparable with a series of published findings, including those made with prothrombin isolated from plasma, which can be interpreted in terms of the initial action of prothrombinase at Arg320 to yield mIIa as an intermediate followed by its cleavage at Arg271 to yield IIa (7). Comparable results, both qualitatively and quantitatively, were obtained for the action of prothrombinase on IIRR (Fig. 1B). N-terminal sequencing of the band corresponding to the B-chain of thrombin yielded the same sequence as was found in the product of IIWT, denoting cleavage at Arg320 with no evidence for cleavage at the alternate Arg319 site in IIRR (below). Accordingly, the action of prothrombinase on either IIWT or IIRR yielded similar progress curves for proteinase product formation when assessed with a peptidyl substrate yielding initial rates that only differed by ∼15% (Fig. 2). Cleavage at Arg319 rather than Arg320 is not expected to yield the appropriate N-terminal sequence necessary for proteinase formation (22). Thus, comparable progress curves obtained by measuring peptidyl substrate hydrolysis by the products produced confirm near quantitative cleavage at the authentic Arg320 site by prothrombinase despite the presence of an alternate P1 residue at Arg319. In agreement, initial velocity studies yielded steady state kinetic constants, albeit not interpretable in the usual sense, for the action of prothrombinase on IIWT and IIRR that only differed by ∼20% (not shown). These results lend support for a major role of precise substrate geometry in facilitating the action of prothrombinase at Arg320 in prothrombin. It remains a formal possibility that it is actually Arg319 that is initially cleaved by prothrombinase, which then facilitates rapid cleavage at Arg320. However, this possibility is not consistent with the ∼1,000-fold lower catalytic efficiencies observed in the action of factor Xa (or prothrombinase) on substrates containing only Arg followed by a leaving group, relative to tri- or tetrapeptidyl substrates containing additional residues N-terminal to the P1 Arg (39).
FIGURE 1.

Cleavage of IIWT and IIRR by prothrombinase. Reaction mixtures containing 1.4 μm IIWT (A) or IIRR (B), 30 μm PCPS, 60 μm DAPA, and 30 nm Va in Assay Buffer were initiated by the addition of 0.2 nm Xa. Samples were quenched, analyzed by SDS-PAGE following disulfide bond reduction, stained with Colloidal Blue, and visualized as described under “Experimental Procedures.” Lanes 1–18 correspond to reaction times of 0, 0.33, 0.66, 1.0, 1.33, 1.66, 2, 2.33, 2.66, 3, 4, 6, 8, 12, 20, 30, 45, and 65 min. Identities of the relevant species are indicated in the margin, and two gels, aligned using molecular weight markers, are presented in each panel. The IIaA polypeptide species (Scheme 1) runs off the gel with the dye front in this gel system.
FIGURE 2.

Formation of proteinase products from prothrombin variants. Reaction mixtures contained 1.4 μm IIVariant, 30 μm PCPS, 6 μm DAPA, 30 nm Va, and 0.2 nm Xa. The initial velocity of S2238 hydrolysis was determined on aliquots withdrawn and quenched at the indicated times and converted to the concentration of proteinase formed as described under “Experimental Procedures.” Data obtained with IIWT (●), IIRR (○), IIRGR (▴), or II-1δ (▵) are illustrated. The line was arbitrarily drawn.
Cleavage of Register Shift Variants of Prothrombin by Prothrombinase
The action of prothrombinase on IIRGR, assessed by peptidyl substrate hydrolysis by products, also yielded a progress curve that was similar to that obtained with IIWT but with a modestly decreased initial velocity (Fig. 2). These findings also imply efficient processing of the zymogen at Arg320 by prothrombinase despite the presence of an alternate Arg at position 318. The findings with IIRGR further support a role for substrate geometry in determining the Arg residue selected for cleavage. Comparable rates of product formation observed with IIWT, IIRR, and IIRGR, all resulting from cleavage at Arg320, further highlight the ability of the active site of Xa within prothrombinase to productively accommodate a variety of peptidyl sequences (17). The action of prothrombinase on II-1δ and the remaining register shift variants failed to yield products with detectable ability to hydrolyze the peptidyl substrate (Fig. 2 and not shown). Because these variants all contain Gln at position 320 and are not expected to develop the appropriate N terminus required for the formation of functional proteinase, further analyses were performed by SDS-PAGE (Fig. 3). The action of prothrombinase on IIRGR yielded a time course for cleavage comparable with that observed with IIWT and comparable interpretations in terms of ordered cleavage at Arg320 followed by cleavage at Arg271 (Fig. 3A). N-terminal sequencing of the species tentatively identified as thrombin B-chain yielded the same sequence as seen following cleavage of IIWT (Table 1). Thus, as with IIRR, IIRGR was also cleaved at Arg320 with no overt evidence for cleavage at the adjacent Arg318 by prothrombinase.
FIGURE 3.

Cleavage of the register shift prothrombin variants by prothrombinase. Experimental conditions, reactant concentrations, and reaction times were identical to those presented in the legend for Fig. 1 except that the variants analyzed were different. The stained gels illustrate the fate of IIRGR (A), II-1δ (B), II-2δ (C), II-3δ (D), II-4δ (E), and IIQ320 (F) following the addition of prothrombinase. Identities of the polypeptide species are denoted in the margins.
TABLE 1.
NH2-terminal sequence for the IIaB or P2 species produced upon cleavage of the prothrombin variants
| Substrate variant | Sequence |
|---|---|
| IIWT | IVEGSD |
| IIRR | IVEGSD |
| IIRGR | IVEGSD |
| II-1δ | Q IVEGSD |
| II-2δ | GQ IVEGS |
| II-3δ | TATSEY |
| II-4δ | TATSEY |
| IIQ320 | TATSEY |
The action of prothrombinase on II-1δ or II-2δ yielded cleavage profiles resembling that observed with IIRGR (Fig. 3, B and C). For these register shift variants, prothrombin consumption was similar to that seen with IIRGR. The band (F12-A) uniquely reflecting the intermediate, mIIa, persisted with a delayed appearance of the band (F12) reflecting thrombin formation (Fig. 3, B and C). Traces of product (P2) arising from initial cleavage at Arg271, evident with II-1δ, were somewhat more pronounced in the case of IIδ-2 (Fig. 3, B and C). These findings suggest near normal action of prothrombinase at one of the Arg residues in the 320 region but impaired subsequent cleavage at Arg271. The observations are in line with the lack of measurable proteolytic activity in the products and the central role previously established for some aspect of the zymogen to proteinase transition in facilitating subsequent cleavage at Arg271 in the mIIa intermediate (9). N-terminal sequence analysis of the band co-migrating with the thrombin B-chain revealed that both II-1δ and II-2δ were quantitatively cleaved at the Arg closest to the 320 site (Table 1). In contrast to the conclusions developed with IIRR and IIRGR, these findings instead indicate that prothrombinase is not fastidious with respect to the position of the Arg residue in this region and question the proposed role for precise geometry in facilitating active site docking by the substrate and its cleavage.
Analysis of the action of prothrombinase on II-3δ and II-4δ produced evidence for a reduction in the rate of prothrombin consumption with a change in the products obtained (Fig. 3, D and E). The principal products were P2 and F12, arising from cleavage at Arg271. F12-A, reflecting the formation of mIIa as an intermediate, which featured prominently in cleavage studies with other variants, was not observed. Traces of thrombin B chain, seen only after a long time in the cleavage of II-3δ, was further reduced in the case of II-4δ (Fig. 3, D and E). The results with these variants closely resembled those obtained for the cleavage of IIQ320, which is cleaved only at Arg271 (Fig. 3F). Accordingly, N-terminal sequencing confirmed that cleavage at Arg271 accounted for the majority of the products obtained with II-3δ and II-4δ (Table 1). We were unable to identify the site of cleavage responsible for the formation of trace amounts of material migrating in the position expected for thrombin B chain. Thus, the expected geometric effects on the rate of cleavage are evident but only in a qualified way. It also remains unclear why IIRR or IIRGR appear to be exclusively cleaved at Arg320, whereas findings with II-1δ and II-2δ indicate that prothrombinase can readily act on an Arg positioned at 319 and 318.
Kinetics of Prothrombin Consumption
Progress curves describing the fate of the variant prothrombin species following the addition of prothrombinase were constructed by quantitative densitometry (Fig. 4) and yielded initial rates for prothrombin consumption (Table 2). In tolerable agreement with previous findings (7), the rate of consumption of IIWT was ∼24-fold greater than that of IIQ320 (Table 2). This finding reflects the fact that prothrombinase acts on the Arg271 site in intact prothrombin with an ∼30-fold lower kcat than at the Arg320 site (7). Consequently, the inferred initial rate for IIWT consumption was consistent with the published kinetic constants for the action of prothrombinase on the Arg320 site in intact prothrombin (7). Progress curves and initial rates for IIWT, IIRR, IIRGR, and II-1δ were similar, and the rate observed with II-2δ was only modestly lower (Fig. 4 and Table 2). In contrast, the consumption rates for II-3δ and II-4δ were significantly decreased and yielded progress curves more in line with that obtained for IIQ320 (Fig. 4 and Table 2). Taken together with the cleavage patterns observed with these variants (Figs. 2 and 3), these data yield the conclusion that although an N-terminal shift by 2 residues of the P1 Arg in the 320 region can readily be tolerated with modest effects on the rate, further shifts lead to a substantial loss in the ability of prothrombinase to act on the substrate in this region. This loss of function is evident as an abrupt switch in the pathway for cleavage because prothrombin consumption is then presumably determined by the slower rate of cleavage at Arg271.
FIGURE 4.

Fate of prothrombin variants following cleavage by prothrombinase. Progress curves for prothrombin consumption were constructed by quantitative densitometry following fluorescence scanning of gels as illustrated in Figs. 1 and 3 and normalized as described. The data sets correspond to those describing the fate of IIWT (●), IIRR (○), IIRGR (▴), II-1δ (▵), II-2δ (▾), II-3δ (▿), II-4δ (■), and IIQ320 (□). Data points and error bars denote means ± 1 S.D. from 2–5 experiments. The lines were arbitrarily drawn. Initial rates determined from these progress curves are presented in Table 2.
TABLE 2.
Parameters governing the action of prothrombinase on prothrombin variants
All fitted parameters are listed ± 95% confidence limits.
| Substrate variant | Initial rate ± S.D.a | Normalized rate | Ks* ± S.D.b | KE,P ± S.D. | n ± S.D. | kcat320c |
|---|---|---|---|---|---|---|
| μm/min/nm E | μm | mol of S/mol of E | s−1 | |||
| IIWT | 5.73 ± 0.66 | 1.0 | 0.018 ± 0.002 | 58.4 ± 5.1 | 1.04 ± 0.03 | 96 |
| IIRR | 5.10 ± 1.15 | 0.89 | 0.13 ± 0.01 | 62.5 ± 2.5 | 0.98 ± 0.02 | 97 |
| IIRGR | 4.65 ± 0.71 | 0.81 | 0.25 ± 0.01 | 58.8 ± 3.0 | 1.02 ± 0.04 | 100 |
| II-1δ | 3.93 ± 0.47 | 0.69 | 0.31 ± 0.01 | 45.9 ± 0.9 | 1.20 ± 0.02 | 89 |
| II-2δ | 2.65 ± 0.61 | 0.46 | 0.41 ± 0.01 | 57.6 ± 1.6 | 1.03 ± 0.02 | 66 |
| II-3δ | 0.80 ± 0.25 | 0.14 | 0.88 ± 0.03 | 54.5 ± 2.0 | 1.08 ± 0.03 | 0 |
| II-4δ | 0.69 ± 0.24 | 0.12 | 3.4 ± 0.12 | 69.4 ± 2.5 | 0.90 ± 0.03 | 0 |
| IIQ320 | 0.24 ± 0.08 | 0.04 | 4.3 ± 0.11 | 69.7 ± 1.7 | 0.90 ± 0.02 | 0 |
a Initial rates represent those for the total rate of prothrombin consumption determined from progress curves illustrated in Fig. 4.
b Fitted parameters determined from fluorescence binding measurements with pAB represent the unimolecular equilibrium constant for active site docking (wherein Ks* = k−/k+), the equilibrium dissociation constant for the binding of the probe to prothrombinase assembled with XaS195A (KE,P), and the stoichiometry reflecting moles of substrate engaging the active site at saturation.
c kcat320 reflects the intrinsic kcat for the action of prothrombinase in the 320 region and was calculated from initial rate using Equation 1.
Active Site Docking by Prothrombin Variants
A thermodynamic explanation for the findings was sought from binding studies examining the ability of the substrate variants to engage the active site of Xa within prothrombinase. We employed fluorescence measurements of pAB displacement from the active site of XaS195A within prothrombinase to infer the unimolecular equilibrium constant (Ks*) and stoichiometry for active site docking following exosite tethering of the substrate but in the absence of catalysis (13). In agreement with published results (13), IIWT could readily displace the probe from the active site of prothrombinase, whereas IIQ320 did so poorly (Fig. 5). Based on previous studies with variants containing Gln substitutions at position 271 or both 320 and 271, this result arises from the ability of Arg320 but not Arg271 in prothrombin to engage the active site in a highly favorable reaction (13). Analysis according to a previously established system of equilibria (13) yielded fitted values of Ks* and stoichiometry that were consistent with reported values, indicating a large difference in the unimolecular equilibrium constant for active site engagement by Arg320 in IIWT relative to Arg271 in IIQ320 (Table 2). The fitted values for KE,P were in agreement with the directly measured value of 50 ± 6 μm, whereas the smaller value for Ks* obtained for IIQ320 in the present work most likely reflects the fact that it represents a lower limit estimate due to the uncertainty arising from the small extent of probe displacement (13).
FIGURE 5.

Active site docking by prothrombin variants. Fluorescence measurements at equilibrium were determined using λEX = 320 nm, λEM = 374 nm, and reaction mixtures containing 1 μm XaS195A, 1.2 μm Va, 200 μm PCPS, 10 μm FPR-CH2Cl, 25 μm pAB, and increasing concentrations of the indicated variant. Data are presented as FOBS/FP,Free, reflecting the ratio of measured fluorescence intensity to that for pAB in solution following corrections for scatter and normalization. Titration curves are presented for IIWT (●), IIRR (○), IIRGR (▴), II-1δ (▵), II-2δ (▾), II-3δ (▿), II-4δ (■), and IIQ320 (□). With the exception of II-4δ and IIQ320 for which only one representative experiment is shown, data points and error bars denote means ± 1 S.D. from 3–5 different experiments. The lines are drawn following analysis as described under “Experimental Procedures” using the fitted constants listed in Table 2.
Studies with the other prothrombin variants yielded a family of displacement curves distributed between those obtained for IIWT and IIQ320 (Fig. 5). Altered extents of probe displacement are expected in a system wherein the signal arises from a unimolecular binding of exosite-tethered substrate to the active site with systematically increased Ks*. Individual data sets could adequately be described by the same system of equilibria (Fig. 5), with the fitted parameters listed in Table 2. Each variant bound to prothrombinase with the expected stoichiometry of ∼1 and yielded fitted values of KE,P that were consistent with the directly measured value (Table 2). However, fitted values of Ks* for each of the variants were very significantly increased relative to Ks* for Arg320 in IIWT (Table 2). Surprisingly, as much as a 28-fold increase in Ks*, comparing IIWT with II-2δ, was only associated with a relatively modest ∼2-fold change in the rate of prothrombin consumption and without effect on the essentially ordered action of prothrombinase at Arg320 followed by Arg271 (Table 2 and Fig. 3). Loss in cleavage at the Arg320 region seen with II-3δ and II-4δ was associated with larger increases in Ks* (Table 2). Thus, stringent requirements for effective active site docking, both in terms of the sequence preceding the scissile bond as well as in terms of the position of the P1 Arg, are evident from measurement of Ks*. This stringency is obscured in measurements of the initial rate of cleavage and is only apparent when Ks* becomes very large. Discordance between the thermodynamics of active site docking by the substrate and the rate of its cleavage yields the unexpected conclusion that large changes in Ks* are not always associated with detectable changes in function.
Competitive Active Site Interactions within Prothrombin
Substrate recognition by prothrombinase through an initial active site-independent exosite binding step followed by active site docking and cleavage has been established in numerous studies and applies to both cleavage reactions (7, 11). The simplest model describing the kinetics of prothrombin consumption requires consideration of a bifurcating pathway that follows exosite-dependent tethering of the substrate to the enzyme (Scheme 3). Exosite-tethered substrate (determined by KEXO) must engage the active site of the catalyst in one of two configurations to permit substrate cleavage at the two sites. Active site docking by the Arg320 site (Ks*320) followed by catalysis (kcat320) yields mIIa, whereas active site docking by the Arg271 site (Ks*271) followed by catalysis (kcat271) yields P2 and F12 (Scheme 3). For IIWT, predominant flux through the initial formation of mIIa and not the formation of P2 arises because Ks*320 ≪ Ks*271 (13). Based on these ideas, we define prothrombin as a compound substrate, one that is tethered to the enzyme in a particular way to permit active site docking in two mutually exclusive modes that lead to catalysis at the individual sites and the formation of different products (Scheme 3).
SCHEME 3.

Kinetic pathways for prothrombin binding and cleavage by prothrombinase. The annotated scheme illustrates the initial binding of prothrombin through exosite interactions with prothrombinase determined by KEXO. Exosite-bound substrate then engages the active site through one of two mutually exclusive active site docking steps. Active site engagement by Arg320, determined by Ks*320, leads to the cleavage at the 320 site and the formation of mIIa. Active site engagement by Arg271, determined by Ks*271, leads to the cleavage at the 271 site and the formation of P2 plus F12. Definition of KEXO, Ks*320, and Ks*271 in terms of rate constants is shown, and the intrinsic kcat for cleavage at either the 320 site or the 271 site is listed as kcat320 and kcat271.
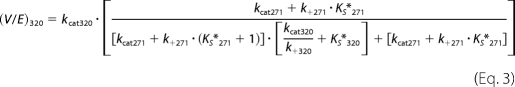
As the concentration of substrate and variants was well above KEXO in our experiments (7), flux through either arm of the bifurcating pathway proceeds at Vmax, which is related in a complex way to the kcat for the individual steps as well as the rate and/or equilibrium constants for the mutually exclusive active site docking steps (Scheme 3). This is illustrated by the expressions for Vmax derived employing the initial velocity and rapid equilibrium assumptions (Equations 1 and 2) or the more general steady state case (Equations 3 and 4). The initial rate of prothrombin consumption, also at Vmax, is determined by the sum of contributions from both possible pathways (Equation 5).
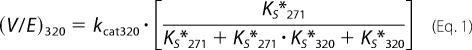 |
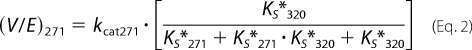 |
 |
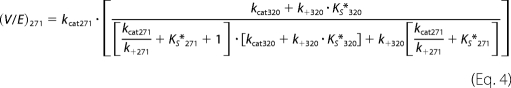 |
These expressions provide unexpected insights into the kinetic behavior arising from reciprocal regulation of the two cleavage reactions (Scheme 3). Because the steady state expressions require knowledge of rate constants (k+320 and k+271) that are presently unavailable, we have utilized the rapid equilibrium approach along with measured values for Ks*320, Ks*271, kcat320, and kcat271 for illustrative purposes, although similar effects are also evident with the steady state expressions and a range of values for the two unknown rate constants. Simulations using measured values for kcat320, kcat271, and Ks*271 revealed that the rate of prothrombin consumption was surprisingly insensitive to changes in Ks*320 over several orders of magnitude. Although these calculations partly replicate the trend observed experimentally where large changes in measured Ks*320 only yield modest changes in rate (Table 2), they are unrealistic because they presuppose that the intrinsic kcat for cleavage of all register shift mutants are identical and equal to kcat320.
This possibility was addressed by calculating the intrinsic kcat for cleavage of the variants in the 320 region using the measured Ks*, the rate of prothrombin consumption, and Equation 1 (Table 2). Values calculated for IIWT, IIRR, IIRGR, and II-1δ were identical and equal to the previously measured value of kcat320 = 94 ± 2 s−1 (7). Thus, the intrinsic kcat appears identical for these variants and is unchanged even when the cleavage site is shifted one residue toward the N terminus. The calculated kcat was modestly lower for the II-2δ variant and was set to 0 for the remainder in which no cleavage in the 320 region in intact prothrombin could be detected. Application of these values to Equations 1, 2, and 5 yielded a reasonable description of the dependence of the initial rate of prothrombin consumption on Ks*320 measured for the range of variants (Fig. 6). The total rate of prothrombin consumption was maximal and essentially independent of Ks*320 at values below ∼0.2 but decreased precipitously at higher values of Ks*320. The fractional contribution from cleavage at Arg320 toward prothrombin consumption remained near 1 and only decreased significantly at Ks*320>0.25. Conversely, the fractional contribution from cleavage at Arg271 to prothrombin consumption remained near zero and only became significant at Ks*320>0.25. Thus, for the cleavage of prothrombin by prothrombinase, small changes in rate with continued predominant flux through the formation of the intermediate mIIa actually mask large perturbations in Ks*320. However, increases in Ks*320 beyond a threshold value lead to significant decreases in the rate of prothrombin consumption, which is accompanied by predominant flux through initial cleavage at Arg271, yielding P2 as an intermediate. These ideas, based on the expected kinetic behavior of this bifurcating reaction pathway, provide an explanation for the present observations.
FIGURE 6.

Kinetic accounting of the action of prothrombinase on prothrombin variants. Normalized rates ± 2 S.D. for the consumption of all prothrombin variants with the exception of IIQ320 are plotted versus measured values for Ks*320. The solid black line was calculated using Equations 1, 2, and 5 using the values in Table 2 (see “Results”), Ks*271 = 4.3, kcat320 = 94 s−1, and kcat271 = 100 s−1. The red and blue lines denote the fractional contributions of cleavage at the 320 region or the 271 site to the rate of prothrombin cleavage.
DISCUSSION
Multiple lines of evidence point to a predominant role for exosite-dependent substrate recognition in driving the affinity of prothrombinase for prothrombin, restricting substrate specificity, and directing the essentially ordered action of the enzyme complex on the two scissile bonds in the biological substrate (10). Implicit in these ideas is the presumed importance of precise positioning of elements flanking the Arg320 site for preferential active site docking in exosite-tethered prothrombin, thereby leading to initial cleavage at Arg320 followed by cleavage at Arg271 (10).
In this work, we provide a platform to test for the consequences of systematic repositioning of the P1 Arg within the 320 region on the action of prothrombinase on prothrombin. On the one hand, functional studies with these variants suggest that the enzyme is surprisingly tolerant to N-terminal shifts of the cleavage site by one or two residues. Further shifts, however, lead to an abrupt loss in cleavage at this region. In contrast, thermodynamic constants for active site binding show large increases with any perturbation either in the sequence preceding the scissile bond or in the position of that sequence. The latter findings are in line with an essential role played by precise positioning of the scissile bond for preferential active site docking by the Arg320 site. The discrepant observations can be reconciled by the unique behavior of prothrombin as a compound substrate wherein competitive active site docking steps between alternate cleavage sites regulate the kinetics of action of prothrombinase at any one site.
The kinetic explanation for our findings reflects the fact that large increases in Ks*320 result in only minor effects on the rate for cleavage at Arg320 within prothrombin, which otherwise engages the active site in a highly favorable way. Thus, the predominant pathway for prothrombin consumption via initial cleavage at the 320 region and its rate of consumption remain unchanged, masking the major role played by precise positioning of this region for preferential active site engagement in prothrombin tethered by exosite interactions to prothrombinase. Such positioning effects are only functionally evident when Ks* for active site docking of elements in the 320 region exceed a threshold value whereupon the rate of prothrombin consumption is decreased greatly and proceeds via cleavage at Arg271. Thus, the previously presumed effects of substrate geometry and precise positioning indeed play an essential role in regulating the action of prothrombinase on prothrombin except that they are only observed in a peculiarly qualified way in kinetic studies. This provides another striking example illustrating the foregone dangers of establishing structure-function relationships from kinetic studies without full appreciation of mechanistic details of the system.
The impetus for our approach was the inconsistency between prevailing ideas and the implications of findings made with the naturally occurring Segovia variant of prothrombin (20, 21). However, studies with IIRR now indicate that it functions indistinguishably from IIWT (or prothrombin purified from plasma) as a substrate for prothrombinase and quantitatively yields thrombin. We speculate that the use of factors Xa and V of bovine origin in the previous studies could represent one basis for this discrepancy (21). The prior studies also described irregularities between F12 formation, clotting activity, and peptidyl substrate hydrolytic activity of the activation products that are difficult to reconcile with the established properties of the products of prothrombin cleavage. Nevertheless, as the patient was described with a bleeding problem (20), it remains possible that cleavage at the alternate vicinal Arg residue becomes significant under circumstances not properly approximated in studies with synthetic phospholipid vesicles. An alternate possibility is that another functionally relevant clotting factor mutation, co-segregating with an otherwise silent Segovia mutation, was missed in the characterization of the patient.
The essentially normal action of prothrombinase on the IIRR and IIRGR variants with quantitative cleavage at Arg320 supports the previous contention that the active site of factor Xa within prothrombinase can effectively engage and cleave a wide range of peptidyl sequences provided that they are properly presented for active site docking by exosite-bound substrate (17). The much larger values of Ks*320 measured for these variants (Table 2) instead point to a clear ability of the active site of proteinase within prothrombinase to discriminate between different sequences preceding the P1 Arg. However, the two-step binding strategy employed for substrate recognition and cleavage ensures that a very significant weakening of the active site docking step will be functionally inconsequential. Although the findings are contradictory, they nevertheless highlight the predominant role played by exosite interactions in enforcing specificity and assisting the action of prothrombinase on its biological substrate. It also suggests that numerous mutations N-terminal to the Arg320 cleavage site will be tolerated without major or even detectable changes in function. Subsite mapping studies with peptidyl substrates and phage peptides indicate that the Asp/Glu-Gly-Arg sequence preceding either cleavage site is not optimal for binding to the active site of factor Xa (18, 19). If, as previously postulated (10), exosite-dependent substrate recognition is only relevant to the function of prothrombinase and not free Xa, we speculate that our apparently contradictory observations may reflect the evolutionary pressure required to maintain a biological substrate that is poorly cleaved by free Xa but acted on efficiently by prothrombinase.
Although functional measurements indicate the efficient action of prothrombinase at Arg320 in IIRR or IIRGR, they provide little insight as to why the alternate Arg residues in these variants are not cleaved when the enzyme complex can act on the II-1δ and II-2δ variants at Arg319 or Arg318. The explanation for these findings likely lies in the more favorable Ks* observed for presumed active site docking of the Arg320 residue in IIRR or IIRGR relative to Ks* for active site engagement by II-1δ and II-2δ. We propose that preferential cleavage at Arg320 in IIRR or IIRGR reflects the fact that active site engagement by the Arg residues at 319 or 318 is substantially weaker and effectively competed for. Cleavage at the alternate sites is only revealed when Arg320 is mutated to Gln to eliminate its ability to engage the active site. Thus, it is not only the position-dependent value for Ks* that determines the rate of cleavage at any one site but also internal competition between alternate sites present in the substrate. Geometric effects on Ks* as well as internal competition between alternate cleavage sites, uncovered in the present work, probably represent major contributors to the selective action of prothrombinase on just two sites within the substrate despite the presence of multiple Arg residues following sequences that can be accommodated at the active site of the proteinase within the enzyme complex.
An ∼30-fold lower Vmax for cleavage at Arg271 relative to cleavage at Arg320 in otherwise intact prothrombin has been inferred from studies with variants in which the individual sites were rendered uncleavable by substitution with Gln, which cannot effectively engage the primary specificity site in the proteinase (7). We realize in retrospect that this approach biases measurements by eliminating the potential contribution of competitive effects within the substrate to the rate of cleavage at the individual sites. For the IIQ271 variant that is only cleaved at Arg320, elimination of the already much weaker active site docking of the 271 site is not expected to affect rate (Fig. 6). However, this is not the case for cleavage at Arg271 in the IIQ320 variant. It follows that the Vmax for cleavage at Arg271, as well as flux toward thrombin formation via the formation of P2, is likely to be even less than that estimated with the IIQ320 variant. Based on the position-dependent effects we describe, it is surprising that the putatively distant Arg271 site in intact prothrombin is even capable of engaging the active site of the catalyst, however weakly, accounting for slow cleavage at this site. We offer the reasonable speculation that predicted disorder in the 271–320 region allows for elements flanking the Arg271 site to be captured by the active site of Xa within prothrombinase with low but finite probability. It also follows that alterations in substrate or enzyme complex that influence exosite tethering and/or the constrained presentation of substrate for active site docking will impact discrimination between the two cleavage sites. This may explain why P2 is readily seen as an intermediate of prothrombin cleavage in the absence of membranes or Va and to variable extents with all components of prothrombinase even in different studies from collaborating laboratories (5, 40–42).
Although the intent of studies with the register shift variants was to probe the contribution of geometric effects to substrate recognition, we have no structural basis to presuppose that N-terminal shifts of the cleavage site indeed have predictable effects on substrate geometry. Another potential weakness in our strategy is that the P′ sequences differ for each of the variants. Thus, changes in Ks* observed for the variants could reflect unanticipated perturbations in the mutated region of the substrate as well as from effects of the P′ sequence in affecting active site docking. Regardless of the physical basis by which Ks* is systematically affected for the variants, the combination of rate measurements along with thermodynamic studies of active site engagement reveal surprising and previously unconsidered aspects of the action of prothrombinase on its biological substrate that would probably not have been evident by any other approach. In this vein, it is likely that a complete accounting of the kinetics of action of prothrombinase on prothrombin can be developed using the more realistic steady state rate expressions with knowledge of the key rate constants for the two active site docking steps.
We define prothrombin as a compound substrate for prothrombinase, one that is tethered to the enzyme complex through exosite interactions, which permits two mutually exclusive active site docking steps followed by catalysis to yield different products. The nature of the product formed is dictated by the thermodynamics of the individual active site docking steps as well as by reciprocal kinetic effects reflecting the ability of the two sites to compete internally for active site docking. These ideas encompass a refined framework for the further consideration of how specificity for the individual cleavage sites is achieved as well as how perturbed interactions at one cleavage site can affect the action of prothrombinase at the second. These ideas also potentially provide insights into enzymic specificity at the level of explaining the restricted action of prothrombinase on only two sites in the substrate when other Arg-containing sequences can be accommodated at the active site of the catalyst.
Exosite-dependent substrate recognition is prevalent among the serine proteinases of coagulation and in numerous other systems in which enzymes act on macromolecular substrates (10). The proteolytic conversion of fibrinogen to fibrin, the activation of factor IX, the activation of factors V and VIII, and the proteolytic inactivation of Va and VIIIa by activated protein C represent examples of coagulation reactions requiring multiple cleavages in the substrate with at least some evidence for cleavage in a particular order. The ideas outlined in the present study may partly apply to explain function in these cases as well as in other reaction systems in which enzymes act on macromolecular substrates in an apparently ordered fashion.
Supplementary Material
Acknowledgments
The technical assistance of Steven Orcutt and Martin Lehr is gratefully acknowledged. Baby hamster kidney cells stably expressing V810 and human embryonic kidney cells stably expressing XS195A were generously provided by Rodney Camire. We are also grateful to Rodney Camire and Bill Church for critical review of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL-74124 and HL-47465 (to S. K.).
This article was selected as a Paper of the Week.
Substrate residues are denoted using the nomenclature of Schecter and Berger (44).
- IIa
- thrombin
- mIIa
- meizothrombin
- DAPA
- dansylarginine-N-(3-ethyl-1,5-pentanediyl)amide
- F12
- fragment 1.2
- FPR-CH2Cl
- d-phenylalanyl-l-proline-l-arginine chloromethyl ketone
- P2
- prethrombin 2
- pAB
- 4-aminobenzamidine
- S2238
- H-d-phenylalanyl-l-pipecolyl-l-arginine-p-nitroanilide
- MES
- 2-(N-morpholino)ethanesulfonic acid.
REFERENCES
- 1.Mann K. G., Jenny R. J., Krishnaswamy S. (1988) Annu. Rev. Biochem. 57, 915–956 [DOI] [PubMed] [Google Scholar]
- 2.Mann K. G., Nesheim M. E., Church W. R., Haley P., Krishnaswamy S. (1990) Blood 76, 1–16 [PubMed] [Google Scholar]
- 3.Mann K. G. (2003) Chest 124, 4S–10S [DOI] [PubMed] [Google Scholar]
- 4.Camire R. M., Pollack E. S. (2006) in Hemostasis and Thrombosis. Basic Principles and Clinical Practice (Colman R. W., Marder V. J., Clowes A. J., George J. N., Goldhaber S. Z. eds) pp. 59–89, Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 5.Krishnaswamy S., Church W. R., Nesheim M. E., Mann K. G. (1987) J. Biol. Chem. 262, 3291–3299 [PubMed] [Google Scholar]
- 6.Krishnaswamy S., Mann K. G., Nesheim M. E. (1986) J. Biol. Chem. 261, 8977–8984 [PubMed] [Google Scholar]
- 7.Orcutt S. J., Krishnaswamy S. (2004) J. Biol. Chem. 279, 54927–54936 [DOI] [PubMed] [Google Scholar]
- 8.Rosing J., Tans G. (1988) Thromb. Haemost. 60, 355–360 [PubMed] [Google Scholar]
- 9.Bianchini E. P., Orcutt S. J., Panizzi P., Bock P. E., Krishnaswamy S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 10099–10104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krishnaswamy S. (2005) J. Thromb. Haemost. 3, 54–67 [DOI] [PubMed] [Google Scholar]
- 11.Boskovic D. S., Krishnaswamy S. (2000) J. Biol. Chem. 275, 38561–38570 [DOI] [PubMed] [Google Scholar]
- 12.Boskovic D. S., Troxler T., Krishnaswamy S. (2004) J. Biol. Chem. 279, 20786–20793 [DOI] [PubMed] [Google Scholar]
- 13.Hacisalihoglu A., Panizzi P., Bock P. E., Camire R. M., Krishnaswamy S. (2007) J. Biol. Chem. 282, 32974–32982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vijayalakshmi J., Padmanabhan K. P., Mann K. G., Tulinsky A. (1994) Protein Sci. 3, 2254–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin P. D., Malkowski M. G., Box J., Esmon C. T., Edwards B. F. (1997) Structure 5, 1681–1693 [DOI] [PubMed] [Google Scholar]
- 16.Lee C. J., Lin P., Chandrasekaran V., Duke R. E., Everse S. J., Perera L., Pedersen L. G. (2008) J. Thromb. Haemost. 6, 83–89 [DOI] [PubMed] [Google Scholar]
- 17.Orcutt S. J., Pietropaolo C., Krishnaswamy S. (2002) J. Biol. Chem. 277, 46191–46196 [DOI] [PubMed] [Google Scholar]
- 18.Bianchini E. P., Louvain V. B., Marque P. E., Juliano M. A., Juliano L., Le Bonniec B. F. (2002) J. Biol. Chem. 277, 20527–20534 [DOI] [PubMed] [Google Scholar]
- 19.Hsu H. J., Tsai K. C., Sun Y. K., Chang H. J., Huang Y. J., Yu H. M., Lin C. H., Mao S. S., Yang A. S. (2008) J. Biol. Chem. 283, 12343–12353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akhavan S., Rocha E., Zeinali S., Mannucci P. M. (1999) Br. J. Haematol. 105, 667–669 [DOI] [PubMed] [Google Scholar]
- 21.Collados M. T., Fernández J., Páramo J. A., Montes R., Borbolla J. R., Montaño L. F., Rocha E. (1997) Thromb Res. 85, 465–477 [DOI] [PubMed] [Google Scholar]
- 22.Khan A. R., James M. N. G. (1998) Protein Sci. 7, 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lottenberg R., Jackson C. M. (1983) Biochim. Biophys. Acta 742, 558–564 [DOI] [PubMed] [Google Scholar]
- 24.Evans S. A., Olson S. T., Shore J. D. (1982) J. Biol. Chem. 257, 3014–3017 [PubMed] [Google Scholar]
- 25.Nesheim M. E., Prendergast F. G., Mann K. G. (1979) Biochemistry 18, 996–1003 [DOI] [PubMed] [Google Scholar]
- 26.Walker R. K., Krishnaswamy S. (1994) J. Biol. Chem. 269, 27441–27450 [PubMed] [Google Scholar]
- 27.Baugh R. J., Krishnaswamy S. (1996) J. Biol. Chem. 271, 16126–16134 [DOI] [PubMed] [Google Scholar]
- 28.Buddai S. K., Toulokhonova L., Bergum P. W., Vlasuk G. P., Krishnaswamy S. (2002) J. Biol. Chem. 277, 26689–26698 [DOI] [PubMed] [Google Scholar]
- 29.Camire R. M. (2002) J. Biol. Chem. 277, 37863–37870 [DOI] [PubMed] [Google Scholar]
- 30.Camire R. M., Larson P. J., Stafford D. W., High K. A. (2000) Biochemistry 39, 14322–14329 [DOI] [PubMed] [Google Scholar]
- 31.Price P. A. (1983) Methods Enzymol. 91, 13–17 [DOI] [PubMed] [Google Scholar]
- 32.Toso R., Camire R. M. (2004) J. Biol. Chem. 279, 21643–21650 [DOI] [PubMed] [Google Scholar]
- 33.Toso R., Camire R. M. (2006) J. Biol. Chem. 281, 8773–8779 [DOI] [PubMed] [Google Scholar]
- 34.Di Scipio R. G., Hermodson M. A., Davie E. W. (1977) Biochemistry 16, 5253–5260 [DOI] [PubMed] [Google Scholar]
- 35.Mann K. G., Elion J., Butkowski R. J., Downing M., Nesheim M. E. (1981) Methods Enzymol. 80, 286–302 [DOI] [PubMed] [Google Scholar]
- 36.Krishnaswamy S. (1990) J. Biol. Chem. 265, 3708–3718 [PubMed] [Google Scholar]
- 37.Lu W. P., Fei L. (2003) Anal. Biochem. 316, 58–65 [DOI] [PubMed] [Google Scholar]
- 38.Kuzmic P. (1996) Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 39.Lottenberg R., Hall J. A., Pautler E., Zupan A., Christensen U., Jackson C. M. (1986) Biochim. Biophys. Acta 874, 326–336 [DOI] [PubMed] [Google Scholar]
- 40.Boskovic D. S., Giles A. R., Nesheim M. E. (1990) J. Biol. Chem. 265, 10497–10505 [PubMed] [Google Scholar]
- 41.Kim P. Y., Nesheim M. E. (2007) J. Biol. Chem. 282, 32568–32581 [DOI] [PubMed] [Google Scholar]
- 42.Bukys M. A., Kim P. Y., Nesheim M. E., Kalafatis M. (2006) J. Biol. Chem. 281, 39194–39204 [DOI] [PubMed] [Google Scholar]
- 43.Padmanabhan K., Padmanabhan K. P., Tulinsky A., Park C. H., Bode W., Huber R., Blankenship D. T., Cardin A. D., Kisiel W. (1993) J. Mol. Biol. 232, 947–966 [DOI] [PubMed] [Google Scholar]
- 44.Schechter I., Berger A. (1967) Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.