

Abstract
UDP-galactopyranose mutases (UGM) are the enzymes responsible for the synthesis of UDP-galactofuranose (UDP-Galf) from UDP-galactopyranose (UDP-Galp). The enzyme, encoded by the glf gene, is present in bacteria, parasites, and fungi that express Galf in their glycoconjugates. Recently, a UGM homologue encoded by the cj1439 gene has been identified in Campylobacter jejuni 11168, an organism possessing no Galf-containing glycoconjugates. However, the capsular polysaccharide from this strain contains a 2-acetamido-2-deoxy-d-galactofuranose (GalfNAc) moiety. Using an in vitro high performance liquid chromatography assay and complementation studies, we characterized the activity of this UGM homologue. The enzyme, which we have renamed UDP-N-acetylgalactopyranose mutase (UNGM), has relaxed specificity and can use either UDP-Gal or UDP-GalNAc as a substrate. Complementation studies of mutase knock-outs in C. jejuni 11168 and Escherichia coli W3110, the latter containing Galf residues in its lipopolysaccharide, demonstrated that the enzyme recognizes both UDP-Gal and UDP-GalNAc in vivo. A homology model of UNGM and site-directed mutagenesis led to the identification of two active site amino acid residues involved in the recognition of the UDP-GalNAc substrate. The specificity of UNGM was characterized using a two-substrate co-incubation assay, which demonstrated, surprisingly, that UDP-Gal is a better substrate than UDP-GalNAc.
Introduction
In nature, hexose sugars are found predominantly in the thermodynamically favored pyranose ring form; however, hexose sugars in the furanose ring form are found in bacteria, fungi, and parasites (1, 2). For example, d-galactofuranose (Galf)5 is a component in many microbial cell surface oligosaccharides (3, 4) and is a major structural component of the mycobacterial cell wall (5). In many pathogenic microorganisms, these Galf residues are essential for cell viability or play a crucial role in cell physiology (6, 7). For this reason, and because hexofuranose sugars are absent in mammalian cell saccharide structures (1), there has been a surge of interest in studying and identifying inhibitors of Galf biosynthesis (8).
The sugar nucleotide UDP-Galf is the precursor of Galf and is incorporated into growing oligosaccharides via galactofuranosyltransferase-mediated reactions (9). First identified in Escherichia coli (10), the enzyme UDP-d-galactopyranose mutase (UGM) is responsible for the biosynthesis of UDP-Galf via the ring contraction of UDP-galactopyranose (UDP-Galp). UGM is encoded by the glf gene for which homologues have since been identified in Klebsiella pneumoniae (11), mycobacterial species (12), and in various eukaryotic pathogens (13, 14). Since the advent of rapid genome sequencing, a number of putative UGMs have been identified throughout the microbial species; however, very few of the gene products have been confirmed by biochemical analysis.
UGM is a flavoprotein and catalyzes the reversible ring contraction of UDP-Galp to UDP-Galf via a unique mechanism (Fig. 1) (15). The noncovalently bound FAD co-factor is directly involved in catalysis and must be in the reduced form for the enzyme to be active (16). Because of the interest in UGM as a drug target (8), significant work has been done to study its mechanism, and it has been shown that the reduced FADH− acts as a nucleophile and displaces the anomeric UDP to form a covalent intermediate (17). Formation of an iminium ion breaks the O5–C1 bond of the galactose moiety leading to a covalently bound acyclic intermediate. This species can then cyclize to the furanose ring form.
FIGURE 1.

UGM reaction mechanism.
Although the enzyme mechanism is generally understood, there are still many unanswered questions about the enzyme-substrate interactions. The UGM protein structure contains a mobile loop region, which adopts either an open or closed form in the crystal structures that have been determined to date (15, 18) with the closed structure being the catalytically active form. This loop has been shown to close upon substrate binding (19), and a conserved arginine (Arg-174 in K. pneumoniae, Arg-170 in E. coli, and Arg-180 in Mycobacterium tuberculosis) has been found to be essential for UGM activity (20). This arginine appears to stabilize the negatively charged diphosphate backbone of the sugar nucleotide substrate. Many synthetic analogues (21–26) have been used to probe the mechanism of UGM and investigate substrate binding, but until recently, no ligand-bound crystal structures have been available. Tryptophan fluorescence (15) and molecular modeling have predicted that the uridine of the UDP-Gal base stacks with Trp-160 (numbering for K. pneumoniae) (27); in contrast, recent crystal structures of the K. pneumoniae UGM with bound UDP-Glcp (28) and UDP-Galp (29) show that the uridine stacks against tyrosine 155 in the active site. This discrepancy demonstrates that many of the key binding interactions responsible for the substrate specificity of UGM still remain to be elucidated.
Although Galf is the most common naturally occurring hexofuranose, it is not unique. 6-Deoxy-d-galactofuranose (30), 6-deoxy-l-altrofuranose (31), and 2-acetamido-2-deoxy-d-galactofuranose (32, 33), among others, have also been identified in bacterial saccharide structures. However, little is known about the biosynthesis of these other hexofuranose sugars. Previous work has established that the UGM from K. pneumoniae, which has been the most studied, is unable to catalyze the synthesis of either UDP-Fucf or UDP-GalfNAc (26, 34). Recently it has been shown that a homologue of the glf gene, fcf2 in E. coli O52, acts as a Fucf mutase enzyme for the biosynthesis of TDP-Fucf (35). This protein has 60% identity to the K. pneumoniae UGM, but the origin of the difference in substrate tolerance is unknown.
The bacterium Campylobacter jejuni is a foodborne pathogen that is a leading cause of diarrheal disease worldwide (36). Infections by this organism have also been linked to the development of the neurological disorder Guillain-Barré syndrome (37). Previous work showed that the capsular polysaccharide (CPS) from the 11168 strain contains a GalfNAc residue (Fig. 2) (33). C. jejuni 11168 also contains a homologue of the glf gene cj1439. Because no Galf residues have been found in C. jejuni 11168 glycoconjugates, it has been proposed that the cj1439 gene product is responsible for the biosynthesis of UDP-GalfNAc from UDP-GalpNAc (33). Herein, we report studies on the protein produced by expression of cj1439 and demonstrate its activity as a UDP-N-acetylgalactopyranose mutase (UNGM). We also have demonstrated that the enzyme can use both UDP-Gal and UDP-GalNAc as substrates and have investigated the origins of this substrate selectivity using site-directed mutagenesis to identify key residues that allow for the turnover of UDP-GalNAc.
FIGURE 2.

Capsular polysaccharide structure of C. jejuni 11168. Shown is the tetrasaccharide repeat unit of C. jejuni 11168 (HS:2 serotype) with the GalfNAc residue shown in boldface.
EXPERIMENTAL PROCEDURES
Preparation of UDP-sugars
UDP-Galp and UDP-GalpNAc were obtained from Aldrich and used without further purification. UDP-Galf was prepared from synthetic Galf-1-phosphate (38) using galactose-1-phosphate uridyltransferase and UDP-glucose pyrophosphorylase as described by Errey et al. (39). All stock solutions were prepared by dissolving the appropriate quantity of UDP-sugar in 100 mm potassium phosphate (pH 7.4). Before use, stock solutions were calibrated by HPLC co-injection with a known concentration of UDP.
Cloning, Expression, and Purification of Glf Proteins
For MFF1 (Ec-glf mutant) and Cj1439c (Cj-glf mutant) complementation and in vivo Glf analyses, glf alleles were put under the control of the constitutive C. jejuni-E. coli shuttle promoter replacing the gfp gene on plasmid pWM1007 (40). Cj-glf alleles were amplified by PCR using oligonucleotides CS261 and CS262 that introduce restriction sites for EcoRI and BsrGI, whereas Ec-glf was amplified with oligonucleotides CS362 and CS363 introducing an EcoRI site in the 5′ end of Ec-glf. For both species, a C-terminal His tag was introduced via PCR, where the plasmid pJHCV32 (41) and chromosomal DNA of C. jejuni 11168-V26 (42) served as template DNA, respectively. The EcoRI-BsrGI-digested Cj-glf PCR product was ligated with the purified 8643-bp pWM1007 vector DNA fragment obtained after digestion with the same enzymes. The Ec-glf PCR product, subsequently treated with T4 DNA polymerase and EcoRI, was inserted into the purified 8297-bp pWM1007 vector subsequently treated with SfuI, T4 DNA polymerase, and EcoRI. For expression in C. jejuni, the kan (kanamycin) cassette within the pWM1007-Cj-glf construct was replaced by the cat (chloramphenicol) cassette after EcoRV digestion of the vector and ligation with an 842-bp DNA fragment containing the cat cassette isolated from plasmid pRY109 (43) after SmaI digestion. A similar strategy was carried out for the pWM1007 Ec-glf constructs, except that the 8428-bp vector fragment was purified after partial digestion with EcoRV. Orientation of the cat gene on the resulting plasmids (same orientation as the nonpolar kan cassette) was verified by restriction analyses. For high yield expression of Ec-glf, the corresponding gene was amplified by PCR using oligonucleotides CS372 and CS373 to introduce NdeI and XhoI restriction sites, respectively. Plasmid pJHCV32 (41) served as template DNA for the PCR. After restriction digestion, the purified DNA fragment was ligated into plasmid pET22b cut with the same enzymes.
Expression of soluble C-terminal hexahistidine-tagged E. coli UGM protein from plasmid pET-Ec-glf in BL21 was observed after induction with 0.01 mm isopropyl β-d-1-thiogalactopyranoside for 2 h at room temperature (22 °C). C-terminal hexahistidine-tagged C. jejuni UNGM was expressed in E. coli DH5α from plasmid pWM1007 (kan)-Cj-glf after growth for 18 h at 28 °C. Soluble UNGM-His6 proteins were purified by nickel-nitrilotriacetic acid affinity chromatography as described previously.
UNGM Activity Assay
The activity of purified wild-type and mutant proteins was assayed by incubating a mixture of sugar nucleotide (UDP-Galp, UDP-Galf, UDP-GalpNAc, or UDP-GalfNAc, 1 mm) and mutase protein (3.9 μm) in 30 μl of 100 mm potassium phosphate buffer (pH 7.4) containing freshly prepared sodium dithionite (20 mm) for 2-, 5-, 10-, and 20-min periods at 37 °C. Reactions were monitored by HPLC (Varian Prostar 210) following conditions similar to those previously reported by Zhang and Liu (16). A C18 column (Microsorb-MV, Varian, 4.6 × 250 mm) was used, and elution was done with 50 mm triethylammonium acetate buffer (pH 6.5) containing 1.5% acetonitrile. A flow rate lower than that described earlier (0.6 ml/min) (16) was used to increase separation, and the UV detector was set to a wavelength of 262 nm. Base-line resolution for all substrates was achieved, and the retention times for UDP-Galp, UDP-Galf, UDP-GalpNAc, and UDP-GalfNAc were found to be 8.8, 10.6, 10.0, and 12.5 min, respectively. The amount of conversion was determined by integration of the product and starting material peaks.
Enzymatic Synthesis of UDP-GalfNAc (Compound 2)
To confirm the identity of the product of the UNGM incubation with UDP-GalpNAc, a milligram scale reaction of UDP-GalfNAc was carried out. UDP-GalpNAc (1 mm, 25 mg) was incubated with 4.7 μm UNGM in 2 ml of 100 mm potassium phosphate buffer (pH 7.4) containing freshly prepared sodium dithionite (20 mm) for 1 h at 37 °C. Compound 2 was purified by HPLC using a C18 column (Microsorb, Varian, 21.4 × 250 mm). The eluent used was 50 mm triethylammonium acetate (pH 6.5) containing 1.5% acetonitrile at a flow rate of 7.0 ml/min. The retention time of compound 2 was 22.3 min under these conditions. The product was further purified by chromatography on a Sephadex G-15 column (21.4 × 250 mm) eluting with Milli Q H2O. The product was lyophilized to obtain a white powder (1 mg, 4% yield). The 1H NMR data for the product can be found in the supplemental Table S2. Negative ion high resolution electrospray ionization mass spectrometry m/z calculated for C17H25N3O17P2 [M − 2H]2− was 302.5335 and found was 302.5339.
NMR Spectroscopy
NMR spectra were obtained at 27 °C using a Varian DirectDrive two-channel spectrometer operating at 499.822 MHz for 1H. Samples were prepared at concentrations of ∼1.5 mm in D2O. Chemical shifts were externally referenced to 0.1% acetone signal (2.225 ppm). A sweep width of 6010 Hz was used for measurement of the one-dimensional 1H NMR spectrum with a 3.0 s relaxation time. The free induction decay signal gave a final digital resolution of 0.1 Hz/point. All coupling constants are reported in hertz (see supplemental Table S2). A two-dimensional 1H–1H COSY spectrum of compound 2 (supplemental Fig. S1) was obtained using a 6010 Hz sweep width with a 1.0-s acquisition time, a 0.1 relaxation delay, and 426 t1 increments.
Determination of the Pyranose-Furanose Distribution at Equilibrium
The distribution of pyranose to furanose ring forms was measured at equilibrium and determined by integration of the appropriate peaks from the HPLC. The ratio was determined for both UDP-Gal and UDP-GalNAc substrates in both the forward and reverse directions. For the forward reaction, UDP-Galp or UDP-GalpNAc (1 mm) was incubated with the appropriate mutase protein (3.9 μm) in 30 μl of 100 mm potassium phosphate buffer (pH 7.4) with freshly prepared sodium dithionite (20 mm) until equilibrium was reached as indicated by a constant product/substrate ratio by HPLC. The same procedure was used for the reverse reaction but instead using UDP-Galf or UDP-GalfNAc (1 mm) as the starting substrate.
Complementation of C. jejuni 11168 glf Knock-out
Shuttle plasmids (pWM1007/cat derivatives) expressing the E. coli glf and C. jejuni glf genes were mobilized into C. jejuni 11168 wild-type and the glf mutant as described (44). Capsular polysaccharides of the resulting strains were prepared as described (45) and visualized after 16.5% deoxycholate PAGE by silver staining using the protocol of Tsai and Frasch (46) with the modification that fixing was performed for only 2 h.
UGM Activity in E. coli
The E. coli glf mutant strain MFF1 (47) was transformed with plasmids containing E. coli wbbL (pMF19 (47)) and either empty vector pWM1007 or pWM1007 containing glf from E. coli or from C. jejuni. Strains were grown overnight after induction with 0.5 mm isopropyl β-d-1-thiogalactopyranoside. Lipopolysaccharides (LPS) were extracted following a modified protocol of Marolda et al. (48). Briefly, cells were adjusted to an absorbance of 3.0 (600 nm wavelength), resuspended in 150 μl of lysis buffer (2% SDS, 4% β-mercaptoethanol, 10% glycerol, 0.5 m Tris (pH 6.8)), and incubated at 100 °C for 10 min. After addition of 2 μl of proteinase K (20 mg/ml), samples were incubated for 2 h at 60 °C. To this solution was added 150 μl of hot phenol, and samples were incubated at 70 °C for 15 min, followed by 10 min on ice. After centrifugation, the aqueous phase was mixed with 250 μl of EtOH and centrifuged, and the precipitated LPS were dried at room temperature. LPS corresponding to an absorbance (A600) of 0.45 were separated by SDS-PAGE, and LPS silver staining was performed according to Tsai and Frasch (46).
Homology Model of C. jejuni UNGM with Bound UDP-GalfNAc
A homology model of C. jejuni 11168 UNGM was generated based on the crystal structure of the E. coli UGM in the closed ring form (Protein Data Bank code 1I8T, chain A) (15) using ESyPred3D (49). Chain A was used as it has the mobile loop region in the active “closed” conformation. Using Autodock 4.0 (50), the UDP-GalpNAc and UDP-GalfNAc substrates were modeled into the active site. For each substrate, the low energy conformation, in which the nucleotide conformation agreed with the published crystal structure data of UGM bound to UDP-Glcp (28) and UDP-Galp (29), was chosen to represent a plausible binding mode.
Mutagenesis of C. jejuni UNGM
The R59H, R168K, and R59H/R168K mutants were prepared using the QuikChange XL II protocol by Stratagene (51). In the case of the R59H and R168K, mutants were prepared from the pWM1007 plasmid containing the glf gene isolated from E. coli DH5α cells using R59H_F and R59H_R or R168K_F and R168K_R (supplemental Table S1) as primer pairs, respectively. In the case of R59H/R168K, the double mutant was prepared using the R59H mutant pWM1007 plasmid and the R168K_F and R168K_R primer pair. After mutagenesis, the plasmid DNA was isolated from the XL10-gold cells and sequenced before being transformed into DH5α cells for protein expression.
Competitive Substrate Specificity Assay
A mixture of UDP-Galf (0.5 mm) and UDP-GalfNAc (0.5 mm) was incubated with the appropriate mutase protein (3.9 μm) in 100 mm potassium phosphate buffer (pH 7.4) containing freshly prepared sodium dithionite (20 mm) for 5 min. The reactions were again monitored using the same modified HPLC conditions of Zhang and Liu (16). In this case, the use of a lower concentration of acetonitrile (1.25%) and a flow rate of 0.6 ml/min allowed for better resolution of all four product and substrate peaks (UDP-Galp, UDP-Galf, UDP-GalpNAc, and UDP-GalfNAc). Using these conditions, these compounds were found to have retention times of 10.0, 12.4, 11.6, and 15.4 min, respectively. The relative specificity was determined by integration of the UDP-Galp and UDP-GalpNAc peaks.
Determination of UNGM and Mutant Kinetic Parameters
Kinetic parameters for wild-type C. jejuni UNGM and each of the mutants were determined following a kinetic assay modified from the procedure reported by Zhang and Liu (16). Reactions were prepared containing an appropriate concentration of the desired protein with UDP-Galf or UDP-GalfNAc (10, 12.5, 25, 50, 100, 250, and 500 μm) in a final volume of 60 μl of 100 mm potassium phosphate (pH 7.4) containing 20 mm of freshly prepared sodium dithionite. Incubations were carried out for 5 min at 37 °C and then promptly quenched by heating to 90 °C for 5 min. The sugar nucleotides do not decompose by loss of UDP under these quenching conditions, as evidenced by the lack of a peak in these HPLCs for UDP, which has a retention time of 11.2 min. The incubation mixtures were monitored by HPLC as described above. In all cases, a concentration of protein was used so that less than 40% conversion to the pyranose product was observed. The concentrations of UDP-Galp or UDP-GalpNAc were determined by integration of the appropriate peaks on the HPLC trace, and these were used to determine the initial velocities. Each assay was performed in duplicate for all proteins for each of the furanose substrates. The calculated enzyme specificity was determined using a modification of the Michaelis-Menten Equation 1 for two competing substrates (52),
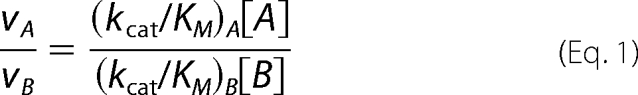 |
RESULTS
C. jejuni UNGM Functions as a UNGM in Vitro
When annotated, the glf gene product from C. jejuni 11168 was proposed to be a UGM based on homology (53), despite the lack of Galf residues its glycoconjugates. Although it was later suggested that Glf may function in biosynthesis of GalfNAc in the CPS (33), the function of the protein has never been examined. In this study, the purified C. jejuni UNGM was examined by incubation with UDP-GalpNAc (compound 1) under the reducing conditions modified from Liu and Zhang (16), and conversion to products was monitored by reverse phase HPLC. Formation of a longer retention time product (12.5 min) was observed, and at equilibrium, a ratio of 7:93 with respect to the UDP-GalpNAc peak resulted, as seen in Fig. 3A (traces i and ii). This reflects the equilibrium ratios observed for the K. pneumoniae (26) and E. coli (16) UGM with UDP-Gal. Scaling up the reaction gave access to sufficient quantities of the reaction product to characterize by 1H NMR spectroscopy (Fig. 4B) and mass spectrometry. The observed mass was consistent with the product being UDP-GalfNAc (compound 2), and the resonances in the 1H NMR spectrum closely matched those previously reported for GalfNAc residues in the CPS of C. jejuni 11168 (33) and the O-specific polysaccharide of Proteus penneri strain 22 (32). In particular, the characteristic coupling pattern for the GalfNAc H-4 proton and the upfield shift of the H-5 proton confirmed a galactofuranose ring configuration (11). Incubation of the isolated product 2 with UNGM under the same reducing conditions gave, as expected, an equilibrium ratio of 7:93 UDP-GalfNAc to UDP-GalpNAc (Fig. 3B).
FIGURE 3.

Functional characterization of C. jejuni UNGM. A, with UDP-GalNAc as the substrate. Trace i, retention time for a standard solution of UDP-GalpNAc. Trace ii, incubation of UDP-GalpNAc with C. jejuni UNGM at equilibrium. Trace iii, standard solution of the purified product (UDP-GalfNAc) from the reaction of UDP-GalpNAc with C. jejuni UNGM. Trace iv, incubation of UDP-GalfNAc with C. jejuni UNGM at equilibrium. The same ratio of 93:7 UDP-GalpNAc to UDP-GalfNAc is seen as in trace ii. B, with UDP-Gal as the substrate. Trace i, retention time for a standard solution of UDP-Galp. Trace ii, incubation of UDP-Galp with UNGM at equilibrium. Trace iii, standard solution of UDP-Galf produced enzymatically from Galf-1-phosphate. Trace iv, incubation of UDP-Galf with C. jejuni UNGM at equilibrium. The same ratio of 93:7 UDP-Galp to UDP-Galf seen in trace ii is also observed. The peak at ∼8 min corresponds to UMP.
FIGURE 4.

1H NMR analysis of the product of C. jejuni UNGM reaction with UDP-GalpNAc. A, region of the 1H NMR spectrum of the starting material UDP-GalpNAc. B, same region of the 1H NMR spectrum for the UNGM reaction product. The observed chemical shifts and coupling constants are consistent for UDP-GalNAc in the furanose ring form.
E. coli UGM Does Not Interconvert UDP-GalpNAc and UDP-GalfNAc
Earlier work established that the UGM of K. pneumoniae does not interconvert UDP-GalpNAc and UDP-GalfNAc (26), but no published data were available for the E. coli UGM. The E. coli UGM has a higher sequence identity, 60%, with the C. jejuni 11168 UNGM than with the K. pneumoniae UGM (38% identity) with which it shares a function. Therefore, we wanted to determine whether the E. coli UGM recognized UDP-GalpNAc as a substrate. We found that no conversion of UDP-GalpNAc was detected using the E. coli UGM (Table 1), even upon prolonged incubations (60 min). A similar experiment, using UDP-GalfNAc as the substrate, also demonstrated no conversion. To ensure that the protein was active, both UDP-Galp and UDP-Galf were used in the assay, and activity levels consistent with previously published results (16) were observed.
TABLE 1.
Kinetic parameters for the C. jejuni UNGM and mutants
| Enzyme | Substrate | Km | kcat | kcat/Km | UDP-Gal:UDP-GalNAc |
|---|---|---|---|---|---|
| μm | min−1 | μm−1 min−1 | |||
| Wild type | UDP-Galf | 45 ± 3 | 178 ± 4 | 4.1 | 1.5 |
| UNGM | UDP-GalfNAc | 40 ± 6 | 114 ± 5 | 2.8 | |
| R59H | UDP-Galf | 92 ± 20 | 590 ± 50 | 6.4 | 15 |
| UDP-GalfNAc | 61 ± 7 | 26 ± 1 | 0.43 | ||
| R168K | UDP-Galf | 65 ± 9 | 96 ± 4 | 1.5 | 3.8 |
| UDP-GalfNAc | 231 ± 40 | 89 ± 7 | 0.39 | ||
| R59H/R168K | UDP-Galf | 77 ± 9 | 380 ± 20 | 4.9 | 21 |
| UDP-GalfNAc | 59 ± 6 | 13.8 ± 0.5 | 0.23 | ||
| E. coli UGM | UDP-GalfNAc | NDa | NDa | NDa | NDa |
a ND means none detected.
C. jejuni CPS Production Is Restored by Expressing the cj1439c Allele in Trans
To test the activity of the glf-His allele in vivo, the corresponding proteins were expressed in trans in C. jejuni wild-type and the glf mutant strain (Fig. 5A). CPS formation was followed by silver staining of crude CPS preparations. Expression of the Cj-glf-His allele resulted in restoration of CPS in the cj1439c(glf) mutant, whereas no CPS formation was observed upon expression of Ec-glf-His. The expression of an additional copy of Cj-glf-His, Ec-glf-His, or the presence of the empty plasmid (pWM1007/cat, negative control) in C. jejuni 11168 cells did not affect CPS formation.
FIGURE 5.

C. jejuni glf gene complements CPS in C. jejuni Δglf strain and LPS in E. coli Δglf strain. A, separation of C. jejuni 11168 crude CPS preparations by 16.5% deoxycholate PAGE. Equivalent amounts of sample were loaded in each lane that originated from bacterial cell cultures adjusted to an absorbance (A600) of 3.0. Silver staining showed formation of CPS in the Cj-wt strain as well as Cj-wt with pMW1007/cat, pMW1007/Cj-glf, or pMW1007/Ec-glf plasmids. wt is wild type The C. jejuni 1439c (glf) knock-out strain (Cj-Δ) and the Cj-Δ strain with pMW1007/cat plasmid showed no CPS formation. Cj-Δ strain complemented with pMW1007/Cj-glf showed restoration of CPS production, and no CPS was observed in Cj-Δ complemented with pMW1007/Ec-glf. In all strains lipooligosaccharide (LOS) formation was not affected. B, LPS of E. coli strains derived from MFF1 were extracted after overnight growth and separated by SDS-PAGE. Equivalent amounts of sample were loaded in each lane that originated from bacterial cell cultures adjusted to an A600 of 0.45. Silver staining shows the production of a fast migrating band composed of lipid A core plus a GlcNAc residue (wbbL −, glf −); a band of higher molecular weight due to addition of an incomplete O antigen subunit to the lipid A core, which is only deficient of the Galf residue (wbbL +, glf −); and smooth LPS when both wbbL and either the E. coli or the C. jejuni glf are present (wbbL +, Ec-glf and wbbL +, Cj-glf), respectively.
C. jejuni UNGM Interconverts UDP-Galf and UDP-Galp in Vitro
The natural CPS of C. jejuni 11168 contains only GalfNAc residues and no Galf residues (33). To probe whether this observation is due to the specificity of the C. jejuni UNGM, the protein was incubated with both UDP-Galp (compound 3) and UDP-Galf (compound 4) (39). Interconversion between compounds 3 and 4 was observed in both cases (Fig. 3B). As was observed for compound 2, the equilibrium ratio was consistent with the 7:93 UDP-Galf to UDP-Galp ratio reported previously (16).
In a second experiment, co-incubation of UNGM with a 50:50 mixture of UDP-Galf and UDP-GalfNAc was carried out. This experiment was intended to provide an estimate of the relative specificity of the enzyme for these two substrates. At equilibrium, the ratio of the corresponding pyranose product was 2.48:1 in favor of UDP-Galp as shown in Fig. 7.
FIGURE 7.

Co-incubation assay of C. jejuni UNGM with 50:50 UDP-GalfNAc and UDP-Gaf. After co-incubation, the relative amounts of UDP-Galp and UDP-GalpNAc were determined based on the total amount of products observed. Shown are the average of three co-incubation assays for each of the C. jejuni UNGM and the R59H, R168K, and R59H/R168K UNGM mutants. Also shown is the specificity of the protein represented by the ratio of UDP-Galp to UDP-GalpNAc.
C. jejuni UNGM Has UGM Activity in E. coli
Because the specificity of UNGM allows it to act as a UGM in vitro, we desired a method to test this activity in a cellular environment similar to the Δglf complementation experiment in C. jejuni. Unfortunately, no C. jejuni serotypes containing Galf residues have been identified. Conveniently, common E. coli laboratory strains such as W3110 are derived from serogroup O16, which contains Galf as a component of the LPS O-antigen. In this species, the O-antigen is composed of a pentasaccharide repeating unit of (1→2)-β-d-Galf-(1→6)-α-d-Glcp-(l→3)-α-l-Rhap-(1→3)-[(1→6)-α-d-Glcp]-α-d-GlcpNAc (47). The W3110 strain does not display a smooth LPS phenotype, due to an insertion sequence disrupting a gene encoding the rhamnosyltransferase (wbbL), which is required for the attachment of the second sugar of the O-antigen subunits. Re-introduction of the wbbL gene on a plasmid restores smooth LPS biosynthesis (54, 55). By mutating the E. coli O16 glf gene in a wbbL-complemented strain, only one incomplete O chain subunit, devoid of Galf, is attached to the lipid A core (Fig. 5B, lane 2) (47). Wild-type LPS production is restored when E. coli glf is reintroduced (Fig. 5B, lane 3) (47). To prove in vivo UGM activity of the C. jejuni UNGM, we repeated the complementation of the E. coli Δglf mutation using a plasmid carrying C. jejuni glf. As shown in the 4th lane of Fig. 5B, the resulting strain produced full-length LPS, indicating that intact O chain subunits containing Galf were synthesized and polymerized. Incorporation of GalfNAc into the O chain can be ruled out because subunits of the O16 LPS are connected via an α-(1→2) linkage to Galf, which would be prevented by the presence of an acetamido group at position 2, as in GalfNAc. Furthermore, to the best of our knowledge, serogroup O16 E. coli strains do not produce UDP-GalpNAc. Thus, the E. coli complementation studies indicate that C. jejuni UNGM can interconvert UDP-Galf and UDP-Galp in vivo.
Modeling the Active Site of UNGM Suggests the Origin of UDP-GalNAc Recognition
To investigate the origin of the increased substrate scope of C. jejuni UNGM compared with the highly homologous E. coli UGM, we examined differences in the active site residues. No crystal structure for the C. jejuni UNGM has been determined; therefore, a homology model was generated based on the E. coli UGM for which a structure is available (15). After building the protein model, UDP-GalpNAc (Fig. 6A) and UDP-GalfNAc (Fig. 6B) were docked into the active site. Inspection of the resulting structure showed that the nucleotide portion was bound in a similar conformation to that observed in the crystal structure of K. pneumoniae UGM bound to UDP-Gal (28), which was recently reported. As was seen in the K. pneumoniae structure, a key base stacking interaction is predicted to occur with a tyrosine residue (Tyr-150).
FIGURE 6.

Homology model of the active site of C. jejuni UNGM. A, homology model of C. jejuni UNGM with the UDP-GalpNAc substrate docked. The UDP-GalpNAc appears to be bound in an active conformation with the GalpNAc C-1 located in proximity to the FADH− cofactor. UDP is positioned similar to that in the crystal structure of K. pneumoniae bound to UDP-Glc by Kiessling and co-workers (28). Nonconserved active site residues Arg-59 and Arg-168 and their distance from the acetamido carbonyl oxygen are highlighted. B, homology model of C. jejuni UNGM with the UDP-GalfNAc substrate bound. The UDP-GalfNAc appear bound in an inactive conformation (no possible interaction between GalfNAc C-1 and FADH− cofactor). Nonconserved active site residues Arg-59 and Arg-168 are highlighted.
Active site residues are highly conserved between the three bacterial UGM for which x-ray structures have been determined (E. coli, K. pneumonia, and M. tuberculosis) and the C. jejuni UNGM. Only one residue in the UNGM active site, arginine 59, differed from the conserved histidine found in all three bacterial UGMs (supplemental Fig. S2). A second residue, arginine 168 in UNGM, differs from the conserved lysine found in the E. coli and K. pneumoniae UGM. As seen in Fig. 6A, Arg-59 appears to be in close proximity to the carbonyl oxygen of the acetamido moiety of UDP-GalpNAc. The other residue, Arg-168, is located adjacent to the conserved arginine, in this case Arg-169, in the mobile loop region that has previously been shown to be essential for UGM activity (20).
Mutagenesis of Arginines 59 and 168 Reduces the Ability of UNGM to Catalyze Interconversion of UDP-GalfNAc to UDP-GalpNAc
The two active site arginine residues, Arg-59 and -168, were examined for their role in the interconversion of UDP-GalfNAc and UDP-GalpNAc by UNGM. Site-directed mutagenesis was used to convert these residues to the corresponding amino acid from the E. coli/K. pneumoniae UGMs, i.e. arginine 59 to histidine and arginine 168 to lysine. The double mutant was also constructed with both R59H and R168K mutations. The specificity of the mutant UNGM was determined using a co-incubation assay with the same 50:50 mixture of UDP-Galf and UDP-GalfNAc. Again, the ratio of the corresponding pyranose products was used to determine the relative selectivity of each mutant for the two substrates (Fig. 7). The R59H mutation resulted in a decrease in the conversion of UDP-GalfNAc and the selectivity for the corresponding substrate, whereas UDP-Galf conversion increased by greater than 10-fold over the wild-type UNGM. The R168K mutation had a less pronounced effect causing only an approximate 2-fold increase in the selectivity for UDP-Galf compared with the wild-type enzyme. Mutation of both residues R59H and R168K resulted in the largest decrease in the conversion of UDP-GalfNAc to UDP-GalpNAc and a selectivity increase for UDP-Galf of greater than 12-fold in comparison to the wild-type UNGM.
Analysis of C. jejuni UNGM and Mutant Kinetics with UDP-Galf and UDP-GalfNAc Supports Results of the Co-incubation Assay
At equilibrium, the pyranose ring form is favored in the reaction of pyranose-furanose mutases. Therefore, substrate kinetics are often measured starting with the furanose form and monitoring the formation of the thermodynamically favored pyranose isomer (56). Thus, the kinetic parameters were determined for UNGM and each of the mutants using UDP-GalfNAc and UDP-Galf as substrates. As seen in Table 1, the Km value of wild-type UNGM is approximately the same for both UDP-Galf and UDP-GalfNAc; however, the kcat value is larger for UDP-Galf. The R59H mutant showed only small changes in the Km value for both substrates as compared with the wild-type UNGM. At the same time, there is a significant decrease in kcat values observed for UDP-GalfNAc in addition to an increase in the kcat values for UDP-Galf. Conversely, the R168K mutant displayed approximately the same kcat value for both substrates, but the Km value was significantly larger for UDP-GalfNAc as the substrate. For each protein, the first-order rate constants were approximated by calculating kcat/Km values for each substrate (Table 1). The ratio of the first-order rate constants for UDP-Galf over UDP-GalfNAc could be used to approximate the specificity of each protein for UDP-Galf as the incubation times for each substrate remained constant (52). The calculated specificities in each case mirror those determined using the co-incubation assay (Fig. 7), but the former approach underestimates these differences.
DISCUSSION
In this paper, we have tested a previously proposed hypothesis (57) that the C. jejuni 11168 gene cj1439 encodes a protein responsible for the biosynthesis of the UDP-GalfNAc. Our investigations have shown that, unlike the highly homologous UGM from E. coli and K. pneumoniae, the C. jejuni enzyme is able to convert the sugar nucleotide UDP-GalpNAc to UDP-GalfNAc, the precursor of GalfNAc in the CPS. Thus, the protein is a UNGM. In addition, we have demonstrated that the C. jejuni UNGM also converts UDP-Galp to UDP-Galf both in vitro and in vivo in E. coli. We also identified two amino acids in the active site of UNGM that play a role in the interconversion of UDP-GalfNAc and UDP-GalpNAc. Our co-incubation assay allowed us to examine, in a single reaction, the substrate specificity of wild-type UNGM protein and each of the UNGM mutants.
Cj1439 Encodes for a Protein with Both UGM and UNGM Activity
This study represents the first demonstration of an enzyme involved in the interconversion of UDP-GalpNAc and UDP-GalfNAc. This enzyme, which bears a high sequence similarity to known UGMs, produces UDP-GalfNAc from UDP-GalpNAc presumably via a similar ring contraction mechanism. Both in vitro investigations and an in vivo complementation experiment support the role of UNGM in the biosynthesis of the GalfNAc in C. jejuni 11168. In addition, we have demonstrated that the C. jejuni UNGM has dual substrate specificity and can interconvert the furanose and pyranose isomers of both UDP-Gal and UDP-GalNAc. Furthermore, the UGM activity of the C. jejuni enzyme has been demonstrated in vivo where it is able to complement the activity of the E. coli UGM in a glf gene knock-out.
It is not unusual that bacteria with compact genomes express enzymes that exhibit more than one activity. These bifunctional enzymes are widespread among bacteria and allow for the synthesis of many complex structures advantageous to the survival of the organism while still maintaining a small genome size. An example is the wbbO gene product from K. pneumoniae, a galactosyltransferase that catalyzes the transfer of both Galp and Galf residues in the biosynthesis of the lipopolysaccharide O1 antigen (58). Bifunctional enzymes have also been characterized in C. jejuni. For example, a single UDP-GlcNAc/Glc 4-epimerase was shown to be involved in the biosynthesis of three cell surface glycoconjugates in strain 11168 (59). Because the CPS structures in C. jejuni are highly variable between serotypes, it is reasonable to hypothesize that a bifunctional UNGM would be advantageous. However, no Galf residues have been identified in any glycoconjugate from this organism. Thus, the inclusion of GalfNAc, instead of Galf, into the CPS appears to be due to the specificity of the cognate glycosyltransferase.
Comparing Calculated Specificity of C. jejuni UNGM to the Experimentally Determined Specificity
In this study, two methods were used to determine the substrate specificity of the C. jejuni UNGM. The first was the direct determination from the ratio of product conversion observed in the co-incubation assay (Fig. 7). The second was calculated using the observed kinetic parameters for each protein with UDP-Galf or UDP-GalfNAc (Table 1). The observed trend of substrate specificity is the same by either method; however, the calculated specificity in each case is lower than that determined using the co-incubation assay. The calculated substrate specificity may not accurately represent the observed specificity as the combined rate of the two competing reactions may be greater than, equal to, or lower than the rate of each individual reaction (60). Because of this, it appears that the co-incubation assay more accurately represents the substrate specificity of the UNGM for its two competing substrates UDP-Galf and UDP-GalfNAc.
Effect of Two Active Site Arginines on UDP-GalNAc Recognition by UNGM
We believed that residues in, or in proximity to, the active site would play a role in the different substrate specificity of the C. jejuni UNGM compared with other known UGM enzymes. We also rationalized that these key residues would be conserved in the other UGM but not in the C. jejuni enzyme. The only two residues that fit these criteria were Arg-59 and Arg-168. In both cases, these residues were found to be other basic amino acids, histidine and lysine, respectively, in the E. coli and K. pneumoniae UGM. In M. tuberculosis, Arg-59 was also replaced by histidine; however, Arg-168 was found to be a threonine rather than the lysine observed in E. coli and K. pneumoniae.
Mutagenesis of R168K only resulted in a 2-fold increase in selectivity for UDP-Galf over UDP-GalfNAc in the co-incubation assay; however, there were more significant changes observed for the kinetic parameters. This amino acid change resulted in a decrease in the catalytic activity of the protein for both substrates compared with the wild-type UNGM, but most interesting was that this substitution resulted in a significant increase in the Km value with UDP-GalfNAc but not with UDP-Galf. As the Km value approximates the dissociation constant for the enzyme-substrate complex, this supports the notion that Arg-168 has a role in binding and stabilizing UDP-GalNAc in the active site. It has previously been proposed that amino acid residues in the mobile loop of UGMs are involved in substrate recognition (19, 20). As Arg-168 is located in the mobile loop, its ability to stabilize the UDP-GalNAc enzyme-substrate complex is consistent with the role of the mobile loop in substrate recognition. This arginine residue is located 4.2 Åfrom the acetamido carbonyl oxygen in our homology model with docked UDP-GalpNAc (Fig. 6A), indicating a possible hydrogen bonding interaction.
Mutation of the other active site arginine (Arg-59) indicates that it also plays an important role in the catalytic activity of UNGM. Mutagenesis of R59H results in a greater than 4-fold decrease in kcat for UDP-GalfNAc, while simultaneously resulting in a 3-fold increase in kcat for UDP-Galf. The Km value is also changed for both substrates, but this appears minor in comparison with the observed changes in kcat. Considering our homology model, Arg-59 is located within 4.5 Åof the carbonyl oxygen of the acetamido group of UDP-GalpNAc (Fig. 6A). In the E. coli UGM crystal structure, the equivalent residue His-59 sits below the active site (data not shown) and does not protrude into the active site as does Arg-59 in UNGM. Therefore, it appears that Arg-59 is able to interact with the UDP-GalNAc substrate to stabilize the intermediate. This may occur by preventing the formation of nonproductive oxazoline-like intermediates, which could be formed by an intramolecular reaction of the acetamido group (Fig. 8). When the substrate is UDP-Gal, then there is no possibility of forming such intermediates. Therefore, Arg-59 does not aid in catalysis. Instead, because arginine is more bulky than histidine, it could lower the catalytic rate due to steric interactions in the active site during catalysis.
FIGURE 8.

Formation of oxazoline intermediate. A, acetamido group of GalNAc can undergo an unproductive intramolecular reaction to form an FAD-bound oxazoline intermediate that prevents conversion to GalfNAc. B, possible interaction between Arg-59 and the GalNAc acetamido group that would prevent formation of an oxazoline intermediate and promote conversion to GalfNAc.
The mutagenesis of both R59H and R168K results in a larger decrease in turnover of UDP-GalfNAc substrate while simultaneously increasing turnover of UDP-Galf. This results in an increased selectivity for UDP-Galf than observed for either of the single mutants (Fig. 7; Table 1). Somewhat surprisingly, the increase in the Km value for UDP-GalfNAc seen for the R168K mutant was not observed in the case of the double mutant. This suggests that the amino acids play a synergistic role in allowing the enzyme to interconvert UDP-GalfNAc and UDP-GalpNAc, rather than an additive role in which Arg-59 is the major determinant.
Subtle Amino Acid Substitutions Result in Changes in C. jejuni UNGM Substrate Tolerance
Despite the high sequence identity of C. jejuni UNGM with the E. coli, M. tuberculosis, and K. pneumoniae UGM, the UNGM possesses the ability to recognize UDP-GalNAc as a substrate. Most of the sequence variability occurs in solvent-exposed residues, and the amino acids making up the active site are nearly identical in all four mutase enzymes (supplemental Fig. S2, boxed residues). The two residues identified here as playing an important role in allowing recognition of UDP-GalNAc are relatively conservative replacements of the residues found in the other mutase enzymes; however, they nevertheless have a significant effect on the substrate selectivity of the enzyme. It should be appreciated that small variations in amino acids leading to changes in substrate specificity are well known in carbohydrate-active enzymes. For example, the blood group GTA and GTB glycosyltransferases, which use UDP-GalpNAc versus UDP-Galp, respectively, as the donor substrate, differ in only four amino acids (61). Similarly, in Neisseria meningitidis, a single amino acid change in the capsule polymerase determines the substrate specificity for either Glcp or Galp transferase activity (62).
Although the two arginine residues identified in this study influence the substrate specificity, the mutagenesis of either residue or both failed to result in a complete loss in UNGM activity. It is therefore clear that other amino acids further removed from the active site also contribute to the specificity of the enzyme, and these remain to be elucidated. As well, we have demonstrated that C. jejuni UNGM can function as a UGM in vivo; however, there have been no Galf residues reported in C. jejuni glycoconjugates. In this context, another unresolved issue is if other C. jejuni strains containing the glf gene possess Galf-containing glycoconjugates, and investigations into the specificity of the transferases accepting donors from bifunctional enzymes are warranted. This study further demonstrates the intricacies in bacterial glycoconjugate biosynthesis. Detailed understanding of these systems will allow for the development of novel antimicrobials targeting these pathogen-specific pathways.
Supplementary Material
This work was supported in part by the Alberta Ingenuity Centre for Carbohydrate Science and National Sciences and Engineering Research Council of Canada.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S2 and Tables S1–S2.
- Galf
- d-galactofuranose
- Cj
- Campylobacter jejuni
- CPS
- capsular polysaccharide
- Ec
- Escherichia coli
- LPS
- lipopolysaccharide
- UGM
- UDP-galactopyranose mutase
- UNGM
- UDP-N-acetylgalactopyranose mutase
- Galp
- galactopyranose
- HPLC
- high pressure liquid chromatography.
REFERENCES
- 1.Peltier P., Euzen R., Daniellou R., Nugier-Chauvin C., Ferrières V. (2008) Carbohydr. Res. 343, 1897–1923 [DOI] [PubMed] [Google Scholar]
- 2.Richards M. R., Lowary T. L. (2009) ChemBioChem 10, 1920–1938 [DOI] [PubMed] [Google Scholar]
- 3.de Lederkremer R. M., Colli W. (1995) Glycobiology 5, 547–552 [DOI] [PubMed] [Google Scholar]
- 4.Whitfield C. (1995) Trends Microbiol. 3, 178–185 [DOI] [PubMed] [Google Scholar]
- 5.Brennan P. J., Nikaido H. (1995) Annu. Rev. Biochem. 64, 29–63 [DOI] [PubMed] [Google Scholar]
- 6.Lee R. E., Smith M. D., Nash R. J., Griffiths R. C., McNeil M., Grewal R. K., Yan W. X., Besra G. S., Brennan P. J., Fleet G. W. (1997) Tetrahedron Lett. 38, 6733–6736 [Google Scholar]
- 7.Pan F., Jackson M., Ma Y., McNeil M. (2001) J. Bacteriol. 183, 3991–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pedersen L. L., Turco S. J. (2003) Cell. Mol. Life Sci. 60, 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belánová M., Dianisková P., Brennan P. J., Completo G. C., Rose N. L., Lowary T. L., Mikusová K. (2008) J. Bacteriol. 190, 1141–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nassau P. M., Martin S. L., Brown R. E., Weston A., Monsey D., McNeil M. R., Duncan K. (1996) J. Bacteriol. 178, 1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Köplin R., Brisson J. R., Whitfield C. (1997) J. Biol. Chem. 272, 4121–4128 [DOI] [PubMed] [Google Scholar]
- 12.Weston A., Stern R. J., Lee R. E., Nassau P. M., Monsey D., Martin S. L., Scherman M. S., Besra G. S., Duncan K., McNeil M. R. (1997) Tuber Lung Dis. 78, 123–131 [DOI] [PubMed] [Google Scholar]
- 13.Beverley S. M., Owens K. L., Showalter M., Griffith C. L., Doering T. L., Jones V. C., McNeil M. R. (2005) Eukaryotic Cell 4, 1147–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakker H., Kleczka B., Gerardy-Schahn R., Routier F. H. (2005) Biol. Chem. 386, 657–661 [DOI] [PubMed] [Google Scholar]
- 15.Sanders D. A., Staines A. G., McMahon S. A., McNeil M. R., Whitfield C., Naismith J. H. (2001) Nat. Struct. Biol. 8, 858–863 [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q., Liu H. (2000) J. Am. Chem. Soc. 122, 9065–9070 [Google Scholar]
- 17.Soltero-Higgin M., Carlson E. E., Gruber T. D., Kiessling L. L. (2004) Nat. Struct. Mol. Biol. 11, 539–543 [DOI] [PubMed] [Google Scholar]
- 18.Beis K., Srikannathasan V., Liu H., Fullerton S. W., Bamford V. A., Sanders D. A., Whitfield C., McNeil M. R., Naismith J. H. (2005) J. Mol. Biol. 348, 971–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao X. H., Bleile D. W., Yuan Y., Chao J., Sarathy K. P., Sanders D. A., Pinto B. M., O'Neill M. A. (2009) Proteins Struct. Funct. Bioinformat. 74, 972–979 [DOI] [PubMed] [Google Scholar]
- 20.Chad J. M., Sarathy K. P., Gruber T. D., Addala E., Kiessling L. L., Sanders D. A. R. (2007) Biochemistry 46, 6723–6732 [DOI] [PubMed] [Google Scholar]
- 21.Caravano A., Sinaÿ P., Vincent S. P. (2006) Bioorg. Med. Chem. Lett. 16, 1123–1125 [DOI] [PubMed] [Google Scholar]
- 22.Itoh K., Huang Z., Liu H. W. (2007) Org. Lett. 9, 879–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlson E. E., May J. F., Kiessling L. L. (2006) Chem. Biol. 13, 825–837 [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q., Liu H. (2001) J. Am. Chem. Soc. 123, 6756–6766 [DOI] [PubMed] [Google Scholar]
- 25.Yuan Y., Bleile D. W., Wen X., Sanders D. A., Itoh K., Liu H. W., Pinto B. M. (2008) J. Am. Chem. Soc. 130, 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Errey J. C., Mann M. C., Fairhurst S. A., Hill L., McNeil M. R., Naismith J. H., Percy J. M., Whitfield C., Field R. A. (2009) Org. Biomol. Chem. 7, 1009–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan Y., Wen X., Sanders D. A., Pinto B. M. (2005) Biochemistry 44, 14080–14089 [DOI] [PubMed] [Google Scholar]
- 28.Gruber T. D., Borrok M. J., Westler W. M., Forest K. T., Kiessling L. L. (2009) J. Mol. Biol. 391, 327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gruber T. D., Westler W. M., Kiessling L. L., Forest K. T. (2009) Biochemistry 48, 9171–9173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng L., Senchenkova S. N., Yang J., Shashkov A. S., Tao J., Guo H., Cheng J., Ren Y., Knirel Y. A., Reeves P. R., Wang L. (2004) J. Bacteriol. 186, 4510–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanniffy O. M., Shashkov A. S., Moran A. P., Prendergast M. M., Senchenkova S. N., Knirel Y. A., Savage A. V. (1999) Carbohydr. Res. 319, 124–132 [DOI] [PubMed] [Google Scholar]
- 32.Arbatsky N. P., Shashkov A. S., Mamyan S. S., Knirel Y. A., Zych K., Sidorczyk Z. (1998) Carbohydr. Res. 310, 85–90 [DOI] [PubMed] [Google Scholar]
- 33.St Michael F., Szymanski C. M., Li J., Chan K. H., Khieu N. H., Larocque S., Wakarchuk W. W., Brisson J. R., Monteiro M. A. (2002) Eur. J. Biochem. 269, 5119–5136 [DOI] [PubMed] [Google Scholar]
- 34.Eppe G., Peltier P., Daniellou R., Nugier-Chauvin C., Ferrières V., Vincent S. P. (2009) Bioorg. Med. Chem. Lett. 19, 814–816 [DOI] [PubMed] [Google Scholar]
- 35.Wang Q., Ding P., Perepelov A. V., Xu Y., Wang Y., Knirel Y. A., Wang L., Feng L. (2008) Mol. Microbiol. 70, 1358–1367 [DOI] [PubMed] [Google Scholar]
- 36.Allos B. M. (2001) Clin. Infect. Dis. 32, 1201–1206 [DOI] [PubMed] [Google Scholar]
- 37.Kaldor J., Speed B. R. (1984) Br. Med. J. 288, 1867–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delederkremer R. M., Nahmad V. B., Varela O. (1994) J. Org. Chem. 59, 690–692 [Google Scholar]
- 39.Errey J. C., Mukhopadhyay B., Kartha K. P., Field R. A. (2004) Chem. Commun. 2706–2707 [DOI] [PubMed] [Google Scholar]
- 40.Miller W. G., Bates A. H., Horn S. T., Brandl M. T., Wachtel M. R., Mandrell R. E. (2000) Appl. Environ. Microbiol. 66, 5426–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marolda C. L., Welsh J., Dafoe L., Valvano M. A. (1990) J. Bacteriol. 172, 3590–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carrillo C. D., Taboada E., Nash J. H., Lanthier P., Kelly J., Lau P. C., Verhulp R., Mykytczuk O., Sy J., Findlay W. A., Amoako K., Gomis S., Willson P., Austin J. W., Potter A., Babiuk L., Allan B., Szymanski C. M. (2004) J. Biol. Chem. 279, 20327–20338 [DOI] [PubMed] [Google Scholar]
- 43.Yao R., Alm R. A., Trust T. J., Guerry P. (1993) Gene 130, 127–130 [DOI] [PubMed] [Google Scholar]
- 44.Nothaft H., Liu X., McNally D. J., Li J., Szymanski C. M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 15019–15024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bacon D. J., Szymanski C. M., Burr D. H., Silver R. P., Alm R. A., Guerry P. (2001) Mol. Microbiol. 40, 769–777 [DOI] [PubMed] [Google Scholar]
- 46.Tsai C. M., Frasch C. E. (1982) Anal. Biochem. 119, 115–119 [DOI] [PubMed] [Google Scholar]
- 47.Feldman M. F., Marolda C. L., Monteiro M. A., Perry M. B., Parodi A. J., Valvano M. A. (1999) J. Biol. Chem. 274, 35129–35138 [DOI] [PubMed] [Google Scholar]
- 48.Marolda C. L., Lahiry P., Vinés E., Saldías S., Valvano M. A. (2006) Methods Mol. Biol. 347, 237–252 [DOI] [PubMed] [Google Scholar]
- 49.Lambert C., Léonard N., De Bolle X., Depiereux E. (2002) Bioinformatics 18, 1250–1256 [DOI] [PubMed] [Google Scholar]
- 50.Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. (1998) J. Comput. Chem. 19, 1639–1662 [Google Scholar]
- 51.Bian X. L., Rosas-Acosta G., Wu Y. C., Wilson V. G. (2007) J. Virol. 81, 2899–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fersht A. (1999) in Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Julet M. R. ed) pp. 116–117, W. H. Freeman & Co., New York [Google Scholar]
- 53.Parkhill J., Wren B. W., Mungall K., Ketley J. M., Churcher C., Basham D., Chillingworth T., Davies R. M., Feltwell T., Holroyd S., Jagels K., Karlyshev A. V., Moule S., Pallen M. J., Penn C. W., Quail M. A., Rajandream M. A., Rutherford K. M., van Vliet A. H., Whitehead S., Barrell B. G. (2000) Nature 403, 665–668 [DOI] [PubMed] [Google Scholar]
- 54.Liu D., Reeves P. R. (1994) Microbiology 140, 49–57 [DOI] [PubMed] [Google Scholar]
- 55.Stevenson G., Neal B., Liu D., Hobbs M., Packer N. H., Batley M., Redmond J. W., Lindquist L., Reeves P. (1994) J. Bacteriol. 176, 4144–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee R., Monsey D., Weston A., Duncan K., Rithner C., McNeil M. (1996) Anal. Biochem. 242, 1–7 [DOI] [PubMed] [Google Scholar]
- 57.Karlyshev A. V., Champion O. L., Churcher C., Brisson J. R., Jarrell H. C., Gilbert M., Brochu D., St Michael F., Li J., Wakarchuk W. W., Goodhead I., Sanders M., Stevens K., White B., Parkhill J., Wren B. W., Szymanski C. M. (2005) Mol. Microbiol. 55, 90–103 [DOI] [PubMed] [Google Scholar]
- 58.Guan S., Clarke A. J., Whitfield C. (2001) J. Bacteriol. 183, 3318–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bernatchez S., Szymanski C. M., Ishiyama N., Li J., Jarrell H. C., Lau P. C., Berghuis A. M., Young N. M., Wakarchuk W. W. (2005) J. Biol. Chem. 280, 4792–4802 [DOI] [PubMed] [Google Scholar]
- 60.Cha S. (1968) Mol. Pharmacol. 4, 621–629 [PubMed] [Google Scholar]
- 61.Rose N. L., Palcic M. M., Evans S. V. (2005) J. Chem. Educ. 82, 1846–1852 [Google Scholar]
- 62.Claus H., Stummeyer K., Batzilla J., Mühlenhoff M., Vogel U. (2009) Mol. Microbiol. 71, 960–971 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.