

Abstract
Pteridine reductase (PTR1) is a target for drug development against Trypanosoma and Leishmania species, parasites that cause serious tropical diseases and for which therapies are inadequate. We adopted a structure-based approach to the design of novel PTR1 inhibitors based on three molecular scaffolds. A series of compounds, most newly synthesized, were identified as inhibitors with PTR1-species specific properties explained by structural differences between the T. brucei and L. major enzymes. The most potent inhibitors target T. brucei PTR1, and two compounds displayed antiparasite activity against the bloodstream form of the parasite. PTR1 contributes to antifolate drug resistance by providing a molecular bypass of dihydrofolate reductase (DHFR) inhibition. Therefore, combining PTR1 and DHFR inhibitors might improve therapeutic efficacy. We tested two new compounds with known DHFR inhibitors. A synergistic effect was observed for one particular combination highlighting the potential of such an approach for treatment of African sleeping sickness.
Introduction
Antifolates are exploited to treat malaria, bacterial infections, various cancers, rheumatoid arthritis, and psoriasis.1,2 However, despite such widespread applications, they are ineffective against the protozoan parasites Trypanosoma and Leishmania species, the causal agents of neglected diseases such as human African trypanosomiasis (HAT,a Sleeping Sickness) and the different forms of leishmaniasis. This is surprising because these parasites are folate and pterin auxotrophs, totally reliant on pteridine salvage from their hosts.3,4
In mammals, biopterin and reduced derivatives are cofactors for aromatic amino acid hydroxylations, the biosynthesis of neurotransmitters and nitric oxide signaling,(5) and oxidation of glycerol ethers.(6) Although a role in trypanosomatids is less clear, biopterins are essential for metacyclogenesis and implicated in resistance to reactive oxygen and nitrogen species in Leishmania.7−10 More is known about folates in trypanosomatid biology, where they contribute to DNA and protein synthesis and cellular methylation (Figure 1a).(11) In mammals, folate cofactors contribute to the same processes but in addition also to purine biosynthesis, an aspect of metabolism lost in trypanosomatids who acquire purines from the host.(12) A key enzyme of folate metabolism is dihydrofolate reductase (DHFR, EC 1.5.1.3, Figure 1), a target for the antifolates methotrexate (MTX), pyrimethamine (PYR), and trimethoprim (TMP).(13) A mechanism that contributes to trypanosomatid resistance to typical antifolates is amplification of the gene encoding the NADPH-dependent pteridine reductase 1 (PTR1, EC 1.5.1.33), an enzyme unique to these parasites.(14) PTR1 catalyzes reduction of biopterin to dihydrobiopterin (H2B), H2B to tetrahydrobiopterin (H4B, Figure 1b), and since it can reduce other pterins/folates, it provides a bypass for DHFR inhibition.7,14 PTR1 overexpression promotes antifolate resistance in T. cruzi,(15) and a gene knockout in L. major is lethal unless a supplement of reduced biopterin is provided.(16) Even in the presence of reduced biopterin the modified parasites display increased susceptibility to antifolates.14,16 These observations suggest that dual DHFR-PTR1 inhibition may provide a successful treatment for trypanosomatid infections. Potent DHFR inhibitors are already known, and we worked on design of novel PTR1 inhibitors concentrating on the enzyme from T. brucei (TbPTR1), since this organism causes the disease with greatest unmet medical need, HAT.
Figure 1.

(a) Folate cycle and cellular processes supported in trypanosomatids. Enzymes are shown next to the reactions that they catalyze. The dimeric bifunctional dihydrofolate reductase−thymidylate synthase (DHFR-TS, PDB code 2H2Q) used by trypanosomatids is shown in the center of the cycle with DHFR and TS domains colored blue and red, respectively. The PTR1 tetramer is shown bottom left next to the DHFR catalyzed reactions, which PTR1 can also catalyze. T. brucei, unlike L. major, lacks serine hydroxymethyl transferase (SHMT), and the glycine cleavage complex (GCC) accomplishes synthesis of CH2−H4F.10,41 (b) Two-stage reduction of biopterin to H2B and H4B catalyzed by PTR1.
We identified three scaffolds to support the design of new PTR1 inhibitors based on structural data for TbPTR1 and the L. major enzyme (LmPTR1).17−20 With rounds of molecular modeling and design, chemical synthesis, enzyme assays, and structure determination of 14 TbPTR1−ligand complexes we developed potent inhibitors for both LmPTR1 and TbPTR1. The inhibitors display selectivity with respect to the orthologues, and the structural basis of such discrimination is described. The inhibitors are cytotoxic to cultured bloodstream form (BSF) T. brucei with micromolar potency. Strikingly, potency is improved when one of the new PTR1 inhibitors is used in combination with MTX.
Results and Discussion
PTR1 Structure and Organization of the Active Site
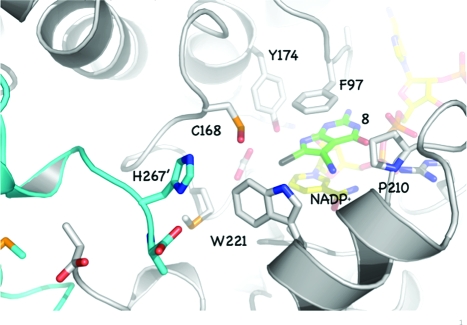
PTR1 is a tetrameric short-chain oxidoreductase with a single α/β-domain subunit constructed around a seven-stranded parallel β-sheet sandwiched between two sets of α-helices, a Rossmann fold repeat (Figure 2).(19) An elongated active site is formed primarily by a single subunit but with one end created by the C-terminus of a partner subunit. A feature of the short-chain oxidoreductase family is the presence of a flexible substrate-binding loop which links β6 to α6, positioned on one side of the active site (Figure 2). NADPH contributes to the formation of the catalytic center between the nicotinamide and Phe97. Here, the ribose and a phosphate of the cofactor, Ser95, and two catalytically important residues, Asp161 and Tyr174, are positioned to interact with ligands (Figure 3a).(18)
Figure 2.

PTR1 subunit architecture and position of the active site. (a) Side view of the subunit of the ternary complex with cofactor and folate. α6, β6, and the substrate binding loop are colored red. The cofactor and folate are depicted as blue and black sticks, respectively. (b) Orthogonal view to (a) in the orientation used for all other molecular images. Trp221 is represented as stick model on α6.
Figure 3.

(a) TbPTR1 in complex with MTX.(17) Cys168 is modified by addition of dimethylarsinoyl. Atoms are colored as follows: N, blue; O, red; As, purple; P, orange; S, yellow; C of PTR1 and NADPH, gray; C of ligands, pale-yellow. An exception is made for the side chain of Phe97, which for the purpose of clarity, since it is directly over the ligand binding position, is shown in thin dark gray lines. Hydrogen bonds are depicted as dashed lines, and water molecules are shown as red spheres. (b) TbPTR1 with folate. In several of the new structures, as seen here, Cys168 is oxidized to sulfenic acid and a number of others are modified by dithiothreitol (DTT, Supporting Information). (c) MTX and folate in the orientation adopted when bound to PTR1. Hydrogen bond donor and acceptor groups are designated D and A, respectively. For comparative purposes the structure of pyrimethamine (PYR), a potent DHFR inhibitor, is also shown. (d) Three scaffolds based on the pterins of MTX and folate. For scaffold II, X = O or S. For scaffold III, X = CH2 or S.
The structures of substrate/product complexes of PTR1 are highly similar. The pterin N8 accepts a hydrogen bond from Tyr174 OH, and O4 accepts hydrogen bonds from Arg14 and water, which bridges to the cofactor pyrophosphate (Figure 3b). The orientation of substrate/product pteridines is distinct from that of MTX, which is flipped 180° such that MTX N8 interacts with the cofactor pyrophosphate via a water bridge and N4 donates a hydrogen bond to Tyr174 OH (Figure 3a). The pteridines of MTX and substrates/products have the potential to donate or accept eight hydrogen bonds, and the difference in the orientations of the folate and MTX pterins appears driven to maximize hydrogen-bonding capacity. The position of hydrogen bond donors/acceptors at the 4- and 8-positions determines the orientation such that an acceptor is placed to interact with a water molecule that in turn interacts with pyrophosphate. The orientation of the MTX pteridine matches five of these acceptor/donor groups with those of substrates and products (Figure 3c). N8 of biopterin/folate is an acceptor, but in H2B and H2F it is a donor. Since the partner for interaction is the hydroxyl of Tyr174, able to function as donor or acceptor, then N8 can match with MTX N4. The two remaining hydrogen bonds that might be formed involve groups accessible to solvent (O4 and N4 for substrates; N4 and N5 for MTX), and therefore, whether they are donor or acceptor is a moot point. The proximity of the phosphate to MTX N1 suggests that, as observed when MTX binds DHFR,(21) the inhibitor is protonated (Figure 3c). For both ligands the aminobenzoate group is directed toward Trp221 with the glutamate extruding from the active site, exposed to solvent (Figure 3a,b).
Residues 207−215 in TbPTR1 form the substrate-binding loop (Figure 2). In LmPTR1 this loop is conformationally labile.(20) The rms fit for all atoms for this stretch of residues is 3.0 Å when comparing substrate and MTX complexes. In TbPTR1 the loop appears more rigid, the rms fit for the folate and MTX complexes is 0.6 Å over these residues, yet it retains enough flexibility to accommodate ligands in either the substrate-like or MTX-like orientation (Figure 3c). In TbPTR1, the position of α6 puts Trp221 near the pABA group on one side of the substrate, with Pro210 on the other side (Figure 3a). The presence of the tryptophan/proline combination in TbPTR1 reduces the size of the pABA binding region and introduces a significant chemical change in this area of the active site compared to LmPTR1 which presents a histidine/aspartate combination at the corresponding positions.(17)
Scaffold Identification and Molecular Design
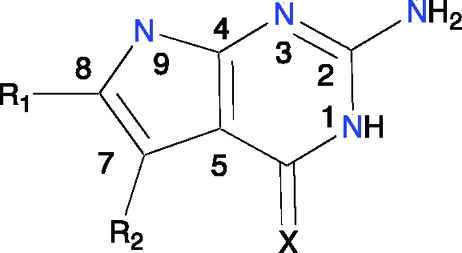
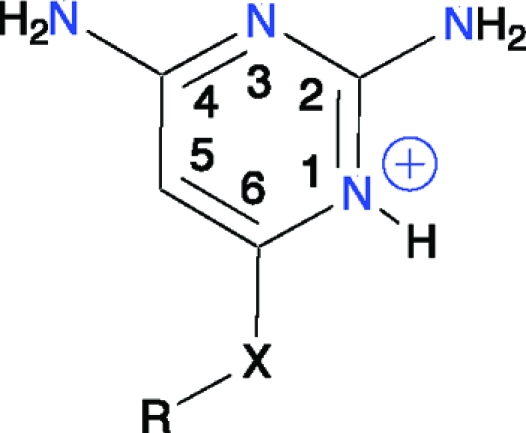
The pterin orientations of MTX and folate, with their distinctive hydrogen-bonding and π-stacking interactions with PTR1, provide frameworks from which to derive novel inhibitors, and three scaffolds for ligand design were identified. The MTX-like framework provided scaffold I (Figure 3d, Table 1) with modification at C6 and C7 providing capacity to generate additional interactions and improve affinity for the target. A substrate-like pyrrolo[2,3-d]pyrimidine framework was selected as scaffold II (Figure 3d, Table 2), with C7 and C8 as suitable branch points. The simplest framework that should retain extensive interactions with the enzyme is 2,4-diaminopyrimidine with a hydrogen bond acceptor group at C6. This provided scaffold III (Figure 3d, Table 3).
Table 1. Scaffold Ia.

|
Ki (μM) |
||||
|---|---|---|---|---|
| compd | R1 | R2 | TbPTR1 | LmPTR1 |
| MTX | CH2N(CH3)C6H5CONHCH(COOH)CH2CH2COOH | H | 0.152 | 0.039 |
| 1 | CH (CH3)2 | CH(CH3)2 | 3.3 | 0. 24 |
| 2 | C6H5 | NH2 | 1.2 | 3.4 |
| 3 | CH3 | (CH3)2 | >35 | 12 |
The Ki values of MTX for TbPTR1 and LmPTR1 are from ref (15).
Table 2. Scaffold II.
|
Ki (μM) |
|||||
|---|---|---|---|---|---|
| compd | R1 | R2 | X | TbPTR1 | LmPTR1 |
| 4a | H | H | O | >35 | >27 |
| 5 | H | H | S | >35 | >27 |
| 6a | H | CN | O | 5.8 | >27 |
| 7 | H | CH2CH2C6H5 | O | 0.96 | >27 |
| 8 | Br | CN | O | 3.9 | >27 |
| 9 | C6H5 | CN | O | 0.71 | >27 |
| 10a | C6H4CH2CH2 | CN | O | 0.50 | 16.4 |
| 11 | C6H4OCH3 | CN | O | 0.36 | 3.4 |
| 12a | C6H4CHO (meta) | CN | O | 0.29 | 4.2 |
| 13 | C7H5O2 | CN | O | 0.40 | 2.6 |
Crystal structures of these compounds complexed to PTR1 were not determined.
Table 3. Scaffold III.
|
Ki (μM) |
||||
|---|---|---|---|---|
| compd | R | X | TbPTR1 | LmPTR1 |
| 14 | NH2 | >35 | >27 | |
| 15 | c-C3H5 | NH | >35 | >27 |
| 16 | C6H4CH3 | S | 5.4 | ∼27 |
| 17 | CH2C6H5 | S | 3.2 | 0.60 |
| 18 | CH2C6H4OCH3 (para) | S | 18 | 2.7 |
The scaffolds formed the basis for molecular modeling. By varying substituents at the branch points, we sought to enhance interactions with PTR1 or to generate ideas for compounds that might allow us to investigate the relative importance of certain active site features with respect to ligand affinity. Consideration was given to the types of substituents commonly exploited in medicinal chemistry and to synthetic tractability.22,23 The majority of sought compounds required the development of new synthetic protocols.(23) The synthesis of two compounds are reported here. Potential inhibitors were assayed to determine inhibition properties against PTR1 and the most potent prioritized for structural characterization. On the basis of this first round of results, further modeling, syntheses, enzymatic assays, and structural characterization were carried out to investigate the structure−activity relationship around each scaffold and to improve affinity for the target. A series of high-resolution crystal structures of TbPTR1 with the new inhibitors and folate were determined. Supporting Information carries experimental details, crystallographic results (Tables S1−S3), and figures (S1−S3) depicting ligands and interactions within the PTR1 active site.
Scaffold I Is More Effective against LmPTR1 Than TbPTR1
Scaffold I compounds 1 and 2 are LmPTR1 inhibitors.(17)1 displays Ki = 0.24 μM, but it is not as effective as MTX (Ki = 0.039 μM, Table 1). By comparison, 2 is a relatively poor inhibitor of LmPTR1 (Ki = 3.4 μM). Scaffold I appears less effective for inhibition of TbPTR1 (Table 1). The complex structures of 1 and 2 with TbPTR1 reveal that they both adopt the MTX orientation (Figures 4a and S1). The pteridine of 2 binds with the phenyl substituent placed in a hydrophobic section of the active site, forming an edge-to-face contact with Trp221. However, there is a potential clash of the amine substituent at C7 with Pro210. The structure of the complex reveals that in three of the subunits that constitute the asymmetric unit, the β6−α6 loop adopts a conformation in which the polypeptide is further away from the catalytic center thereby accommodating bulkier substituents on this scaffold. In the fourth subunit of the asymmetric unit the loop is completely disordered. The isopropyl substituents at C6 and C7 of 1 emphasize this point with the β6−α6 loop positioned even further from the active site (∼4 Å), leaving the compound more exposed to solvent. This, together with a degree of steric clash, likely contributes to reduced affinity for TbPTR1. C7 is therefore a poor branch point for modification of scaffold I. A major factor in the different affinities of the scaffold I inhibitors against LmPTR1 and TbPTR1 is likely due to the different conformations of the β6−α6 loop. In LmPTR1, the flexible β6−α6 loop is more able to accommodate inhibitors extending from C7 with TbPTR1 less adaptable in this respect.
Figure 4.

Scaffold representatives in the TbPTR1 active site as revealed by crystallographic analyses: (a) 2; (b) 7; (c) 13; (d) 17. In the PTR1−17 complex, Cys168 displays two rotamers. The inhibitor complex structures were determined between 2.4 and 1.6 Å resolution (Tables S1−S3).
In compound 3 N8 is a hydrogen bond donor, and this has a dramatic effect on the mode of binding. The TbPTR1−3 complex reveals the inhibitor in a substrate-like orientation in two of the subunits of the asymmetric unit but displaying static disorder involving both substrate-like and MTX-like conformations in the other two subunits. A similar observation has been made for MTX-based inhibitors of LmPTR1.(24) When the MTX orientation is adopted, the β6−α6 loop is pushed away from the active site by the dimethyl substituents in a similar manner as when 2 binds. In the substrate-like orientation the dimethyl substituents at C7 of 3 would clash with the nicotinamide and Phe97, therefore restricting access to the active site (data not shown). As a result, N2 can only interact with Ser95 and the cofactor pyrophosphate via a water bridge in a fashion similar to that of TMP when it binds LmPTR1.(19)
Scaffold II Is More Effective against TbPTR1 Than LmPTR1
We first investigated if there was scope for modification at the 2-amino group and at N9 of this scaffold. Modeling suggested that inhibition would only result if the compounds adopted a new orientation in the active site (data not shown). The addition of a tertiary butylcarbonyl at the 2-amino group resulted in a complete loss of inhibition likely due to steric clash with the nicotinamide and Ser95. In similar fashion, the placement of an allyl group at N9 abrogated binding probably because of steric clash with Tyr174. These positions are inappropriate for further development.
Scaffold II compounds adopt the substrate-like orientation when they bind TbPTR1 (Figure 4b,c and Figure S2A−G). Compound 4 is a poor inhibitor (Ki > 27 μM, Table 2), and the substitution of O4 by S4 (5) had no effect (Table 2). The addition of a nitrile at C7 (6) improved potency against TbPTR1 (Ki = 5.8 μM) through additional van der Waals interactions with the β6−α6 loop of TbPTR1. The addition of 7-phenylethyl (7) dramatically improved inhibition against TbPTR1 (Ki = 0.96 μM). Although the phenyl group is placed near Trp221 (Figure 4b), the two-carbon link is too short to allow formation of π−π interactions between these aromatic side chains.
Modifications of scaffold II at C8, designed to occupy a solvent filled cavity between the catalytic center and Met163, Cys168 together with His267 of the neighboring subunit, proved highly significant (Table 2). While the 7-carbonitrile is maintained, addition of 8-bromo (8) improved potency against TbPTR1 slightly (Ki = 3.9 μM) and was useful from a synthetic chemistry perspective. Replacement of the bromine with a phenyl group (9) significantly improved inhibitor potency (Ki = 0.71 μM) and provided additional points from which to branch out. The addition of a 4-ethylphenyl group (10) improved inhibition further against TbPTR1 (Ki = 0.50 μM). A 4-methoxyphenyl group (11) improved inhibition again (Ki = 0.36 μM) with the oxygen forming an additional hydrogen bond to solvent within the active site. The strongest inhibition of TbPTR1 was achieved with the addition of a 3-formylphenyl (12, Ki = 0.29 μM). We were unable to derive a structure of the PTR1−12 complex, but modeling suggests that the carbonyl may interact with Asp161 if the latter were protonated. Increasing the size of the substituent at C8 from a phenyl (9) to 1,3-methylenedioxy (13) improved inhibitor potency slightly against TbPTR1 (Ki = 0.40 μM, Table 2), likely because of additional van der Waals interactions formed between the ligand with Met163 and Cys168 and perhaps with a small contribution from a weak hydrogen bond formed by the side chain of Asp161 and the ligand (Figure 4c, the average O···O separation is 3.8 Å over the four copies per asymmetric unit). Scaffold II proved more effective against TbPTR1 than LmPTR1 (Table 2). An explanation is that the more flexible β6−α6 loop of LmPTR1 and relatively open binding site are unable to interact closely with these inhibitors, leaving them exposed to solvent. In TbPTR1 a less flexible β6−α6 loop together with the hydrophobic Met163 and Cys168 may provide less competition with solvent and more van der Waals interactions to stabilize the inhibitor complexes.
Scaffold III
The simplest of the scaffold III series, compound 14, was a weak inhibitor of LmPTR1 and TbPTR1 (Table 3), forming similar interactions with TbPTR1 and the cofactor as the pterin of folate and MTX (Figures 4d and S2). The addition of cyclopropyl at N6 (15) had little effect, and the structure indicates that the substituent was unable to form hydrophobic interactions with the protein. Increasing the size of the substituent, to methylphenyl, coupled with replacement of N6 by S6 (16) improved activity against TbPTR1 and LmPTR1 (Table 3). This may have been partly due to the substitution of the hydrogen-bond-donating N6 to a hydrogen-bond-accepting S6, adding an interaction with Arg14, and partly to the size of the hydrophobic substituent, which provided favorable interactions with the β6−α6 loop. Increasing the chain length by one carbon (17) increased flexibility, facilitating interactions with a hydrophobic region of the β6−α6 loop, and inhibition of TbPTR1 was moderately improved (Ki = 3.2 μM). Inhibition of LmPTR1 was, however, increased dramatically (Ki = 0.60 μM) likely because of contacts that the inhibitor forms with His241 in LmPTR1 (data not shown). The methylphenyl substituent does not appear to be a suitable position for further modification, since 18 reduces inhibition against both TbPTR1 and LmPTR1 (Table 3).
Testing Inhibitors against BSF T. brucei
Having derived new, potent inhibitors of PTR1, we sought to determine their effect on cultured parasites. We tested two of the most potent TbPTR1 inhibitors (11 and 13) together with MTX and PYR against BSF T. brucei (Figure 5a). MTX and PYR (Figure 3c) were selected for this purpose because they are both potent inhibitors of DHFR. No effort was made to reduce the high levels of folate commonly used in media (HML9 + 10% fetal calf serum) to culture T. brucei, and our results therefore represent a highly stringent test of compound efficacy against the parasites.
Figure 5.

Trypanocidal activity of PTR1 and DHFR inhibitors. (a) Dose−response plots for cultured T. brucei parasites subjected to increasing concentration of inhibitor. Points are mean values of three separate determinations conducted in quadruplicate (n = 12), std dev ≤ 5%. (b) Changes in 13 ED50 values in combination with varying concentrations (0, 0.5, 1.0, 1.5, 2.0, 2.5 μM) of MTX. Values are the mean ± std dev (n = 4).
MTX displayed an ED50 of 2.7 ± 0.1 μM (Figure 5a), a value approximately 10-fold higher than the Ki against PTR1,(18) 1000-fold higher than the Ki against DHFR.(25) The DHFR inhibitor PYR was less effective with an ED50 of 26.6 ± 0.7 μM, greater than 100-fold higher than the Ki against a trypanosomatid DHFR.(26) The inefficiency of PYR against T. brucei may reflect poor uptake, inability to compete with high folate levels in the culture media, and/or the ability of T. brucei to use PTR1 as a bypass of DHFR inhibition. The combination of MTX and PYR does not act synergistically (Figure S4A), and these compounds are likely competing with respect to binding DHFR. The PTR1 inhibitors 11 and 13 had limited efficacy against BSF T. brucei with ED50 values of 274 ± 7.5 and 123 ± 3.3 μM, respectively (Figures 5a and S4B). The degree of uptake and off-target effects may contribute to the concentrations required to produce lethal doses. However, 13 acts synergistically with MTX (the area under the slope is less than half the product of the two ED50 values (Figure 5b)).
In conclusion, we identified and exploited three molecular scaffolds for the generation of novel inhibitors of PTR1 targeting two species of important human pathogens. The inhibitors display PTR1-species specific properties, which can be explained by differences, in particular of the β6−α6 substrate-binding loops, between the active sites of LmPTR1 and TbPTR1. Scaffolds I and III are the most effective against LmPTR1, while scaffold II is the most effective against TbPTR1. Testing against BSF, T. brucei indicates that two of the scaffold II compounds, 11 and 13, are lethal to the parasites although with modest ED50 values. However, when 13 is tested in combination with MTX, there is an improvement in efficacy, which highlights the potential of therapy for African sleeping sickness by combination of such DHFR and PTR1 inhibitors.
Methods
Protein Purification and Storage
Recombinant LmPTR1 and TbPTR1were expressed and purified by established methods.17,18 The enzymes for assay were prepared at 15 mg mL−1 in 50 mM Tris-HCl, 250 mM NaCl, 20% (v/v) glycerol, pH 7.5. For crystallization, TbPTR1 was at 22 mg mL−1 in 20 mM Tris-HCl, pH 7.5. Aliquots were flash frozen in liquid N2 and stored at −20 °C.
Inhibitor Sourcing and Assay
MTX, PYR, and compounds 1, 2, 3, 4, and 14 were purchased from Sigma Aldrich, and 15 was a gift from Novartis. Compounds 16 and 17 were prepared by the method of Davies et al.(27) Synthetic routes to new compounds except 10 and 18 have been described;(21) details of the preparation of 10 and 18 follow. 1H NMR and mass spectrometry of inhibitors that were assayed confirmed purity at >95%.
6-[(4′-Methoxybenzyl)sulfanyl]-2,4-pyrimidinediamine (18)
To a suspension of 2,6-diamino-4-pyrimidinethiol (0.6 g, 2.5 mmol) and sodium hydroxide (0.16 g, 4.0 mmol) in ethanol (15 mL) and water (10 mL) was added 1-(bromomethyl)-4-methoxybenzene (0.6 g, 2.55 mmol). The reaction mixture was stirred at room temperature for 46 h. The precipitate was collected by filtration and washed with water (10 mL) and n-hexane (10 mL) to afford the required product as a white solid (0.44 g, 1.68 mmol, 67%; mp 189−191 °C). 1H NMR (400 MHz, DMSO-d6, 25 °C, TMS): δ 3.72 (3H, s; OCH3), 4.25 (2H, s; SCH2), 5.81 (1H, s; C5−H), 6.86 (4H, d + s, 3J(H,H) = 8.6 Hz, C′3-H and NH2), 7.11 (2H, brs, NH2), 7.32 (2H, d, 3J(H,H) = 8.6 Hz, C2′-H). 13C NMR (100 MHz, DMSO-d6, 25 °C, TMS): δ 32.63 (SCH2), 55.06 (OCH3), 90.89 (C5), 113.9 (2C, ArCH), 128.54 (ArC), 130.16 (2C, ArCH), 158.16 (C6), 158.48 (ArC), 161.94 (C2), 163.16 (C4). IR (KBr) 3335, 3146, 1656, 1512, 1303, 1249, 1102, 972, 754, 614 cm−1. HREIMS found m/z 262.0889, C12H14N4OS requires 262.0888 (M+).
[(3E)-4-Nitro-3-butenyl]benzene
To a solution of 1-nitro-4-phenyl-2-butanol(28) (2.94 g, 15 mmol) in dichloromethane (20 mL) at 0 °C was added methanesulfonyl chloride (1.7 g, 15 mmol) followed triethylamine (3.02 g, 30 mmol). The mixture was warmed to room temperature and stirred for 20 min, then poured into water (15 mL) and extracted with dichloromethane (20 mL). The organic extract was washed with aqueous saturated sodium bicarbonate (20 mL × 3) and then dried with anhydrous magnesium sulfate and concentrated. The residue was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:9) to afford the product as a yellow oil which was stored at −20 °C (1.45 g, 8.2 mmol, 55%). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 2.65 (2H, dq, 3J(H,H) = 7.7, 1.4 Hz, PhCH2CH2), 2.90 (2H, t, 3J(H,H) = 7.7 Hz, PhCH2CH2), 7.02 (1H, dt, 3J(H,H) = 13.4, 1.4 Hz, CH = CHNO2), 7.25−7.42 (6H, m, CH=CHNO2 and Ph). 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ 30.23 (CH2CH2Ph), 34.08 (CH2CH2Ph), 126.77 (1C, ArCH), 128.44, 128.87 (4C, ArCH), 139.75 (CH=CHNO2), 140.16 (1C, ArC), 141.55 (CH=CHNO2). IR (neat) 3028, 2929, 1953, 1734, 1648, 1523 (CNO2), 1454, 1351, 1089, 953 (CH=CH), 840, 751, 700 cm−1.
2,6-Diamino-5-[1′-(nitromethyl)-3′-phenylpropyl]-4(3H)-pyrimidinone
It was necessary to synthesize this as an intermediate en route to some targeted compounds. To a mixture of [(3E)-4-nitro-3-butenyl]benzene (1.27 g, 7.2 mmol) in a mixture of water (15 mL) and ethyl acetate (15 mL) at room temperature was added 2,6-diamino-4(3H)-pyrimidinone (0.9 g, 7.2 mmol). The resulting mixture was stirred in an oil bath at 90 °C for 24 h. The organic layer was separated, washed with brine (30 mL), dried, and concentrated. The resulting solid was dried in vacuo (120 mmHg) to give the product as a yellow solid (2.06 g, 6.8 mmol, 94%; mp 143−145 °C). 1H NMR (400 MHz, DMSO-d6, 25 °C, TMS): δ 1.67−1.76 (1H, m, CH2CH2Ph), 2.08−2.18 (1H, m, CH2CH2Ph), 2.43−2.64 (2H, m, CH2CH2Ph), 3.42 (1H, brs, CHCH2CH2Ph), 4.77−5.07 (2H, m, CH2NO2), 5.95 (2H, brs, C6-NH2), 6.08 (2H, brs, C2-NH2), 7.13−7.41 (5H, m, ArH), 9.85 (1H, brs, NH). 13C NMR (100 MHz, DMSO-d6, 25 °C, TMS): δ 31.83 (CHCH2CH2), 32.98 (CHCH2CH2), 35.09 (CHCH2CH2), 77.65 (CH2NO2), 84.08 (C5), 125.55 (1C, ArC), 127.99, 128.22 (4C, ArCH), 142.34 (1C, ArC), 153.55 (C6), 161.93 (C2), 162.81 (C4). IR (KBr) 3471, 3401, 3183, 2859, 1625, 1595, 1536 (CNO2), 1493, 1450, 1378, 697 cm−1. HRFABMS found m/z 301.1410, C14H17N5O3 requires 302.1413 (MH+).
(1E/Z)-2-(2′,4′-Diamino-6′-oxo-1′,6′-dihydro-5′-pyrimidinyl)-4-phenylbutanaloxime
To a suspension of 2,6-diamino-5-[1′-(nitromethyl)-3′-phenylpropyl]-4(3H)-pyrimidinone (0.82 g, 2.7 mmol) and tin(II) chloride (0.77 g, 4.0 mmol) in tetrahydrofuran (70 mL) was added thiophenol (1.2 mL) and triethylamine (1.8 mL). The reaction mixture was stirred at room temperature for 1 h. The remaining tin(II) chloride was removed by filtration, and the liquid portion was concentrated under reduced pressure (400 mmHg). The residue was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1:1 to ethyl acetate/methanol = 1:1) to afford a white solid (0.45 g, 1.56 mmol, 58%; mp >250 °C) as an E/Z mixture (3:1 by 1H NMR). The E-isomer was purified for characterization. 1H NMR (400 MHz, DMSO-d6, 25 °C, TMS): δ 1.94−2.01 (2H, m, CH2CH2Ph), 2.42−2.48 (2H, m, CH2CH2Ph), 3.45 (1H, q, J = 7.4 Hz, CHCH2CH2Ph), 5.77 (2H, brs, C6-NH2), 6.03 (2H, brs, C2-NH2), 7.13−7.27 (5H, m, ArH), 7.58 (1H, d, 3J(H,H) = 6.9 Hz, CH=NOH), 9.83 (1H, brs, OCNH), 10.60 (1H, s, CH=NOH). 13C NMR (100 MHz, DMSO-d6, 25 °C, TMS): δ (E-isomer) 35.47 (CHCH2CH2), 32.45 (CHCH2CH2), 33.37 (CHCH2CH2), 152.10 (CHNO), 86.45 (C5), 125.45 (1C, ArC), 128.07, 128.17 (4C, ArC), 153.51 (C6), 161.81 (C2), 161.99 (C4). IR (KBr) 3371, 3104, 1621, 1596, 1503, 1430, 1374, 1020, 987, 787, 694, 559 cm−1. HRFABMS found 288.1462, C14H17N4O2 requires 288.1460 (MH+).
2-Amino-5-(2′-phenylethyl)-3,7-dihydro-4H-pyrrolo[2,3-d]pyrimidin-4-one (10)
(a) For cyclization with Dowex-50, H+(1E/Z)-2-(2′,4′-diamino-6′-oxo-1′,6′-dihydro-5′-pyrimidinyl)-4-phenylbutanaloxime (0.2 g, 0.7 mmol) was heated for 15 h at reflux with Dowex-50 (H+ form, 0.33 g) in water (25 mL). The reaction mixture was then diluted with methanol (50 mL) and the Dowex resin filtered. The methanol was evaporated under vacuum and the precipitated was filtered and washed with water (15 mL) to give the product as a brown solid (0.16 g, 0.63 mmol, 90%). Spectroscopic data are detailed below.
(b) For the Nef reaction method, to an aqueous solution of sodium hydroxide (0.2 g, 5.0 mmol) in water (5 mL) was added 2,6-diamino-5-[1′-(nitromethyl)-3′-phenylpropyl]-4(3H)-pyrimidinone (0.25 g, 0.82 mmol) at room temperature. The mixture was stirred for 2 h, and then it was slowly added to an aqueous solution of sulfuric acid (97%, 0.69 g, 7.0 mmol) in water (5 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h and at room temperature for 1 h. The color of the mixture changed to gray. Concentrated ammonium acetate was added at 0 °C to adjust the pH to 7. The precipitated solid was collected and purified by column chromatography (silica gel, ethyl acetate/methanol = 9:1) to give the product as a dark-brown solid (0.09 g, 0.35 mmol, 43%; mp 155−157 °C). 1H NMR (400 MHz, DMSO-d6, 25 °C, TMS): δ 2.81−2.85 (2H, m, CH2CH2Ph), 2.88−2.92 (2H, m, CH2CH2Ph), 5.99 (2H, s, NH2), 6.31 (1H, d, 3J(H,H) = 1.5 Hz, C6−H), 7.13−7.28 (5H, m, ArH), 10.16 (1H, brs, NH), 10.59 (1H, s, OCNH). 13C NMR (100 MHz, DMSO-d6, 25 °C, TMS): δ 28.34 (ArCH2CH2), 36.37 (ArCH2CH2), 98.92 (C4a), 113.50(C5), 118.07 (C6), 125.60 (1C, ArCH), 128.20, 128.37 (4C, ArCH), 142.51 (1C, ArC), 151.37 (C7a), 152.27 (C2), 159.48 (C4). IR (KBr) 3340, 3925, 1631, 1433, 1343, 1137, 786, 698, 620 cm−1. HREIMS found 254.1168, C14H14N4O requires 254.1169 (M+).
Inhibition Assay
Compounds were dissolved at 100 mM in DMSO and screened against LmPTR1 and TbPTR1 with H2B (10 mM in 0.1 M NaOH) as the substrate. The IC50 values were determined from dose−response plots using SigmaPlot(29) adjusted with Morrison’s equation for tight-binding inhibition.(30)Ki values were derived from the IC50 values using the equation for competitive inhibition as published.(17)
Cytotoxicity Studies
BSF T. brucei brucei strain Lister 427, were cultured at 37 °C and 5% CO2 in HMI-9 medium supplemented to maintain neomycin drug pressure to express T7 polymerase and the tetracycline repressor.(31) The Alamar blue viability test(32) established ED50 values against BSF T. brucei (strain 427) for MTX, PYR, 11, and 13. For combination exposures, ED50 values were determined for PYR (Figure S4A), 11 (Figure S4B), or 13 in the presence of sub-ED50 concentrations of MTX (2.5, 2.0, 1.5, 1.0, and 0.5 μM).
TbPTR1-Ligand Cocrystallization
Crystals were grown by vapor diffusion in hanging drops consisting of 1.5 μL of protein solution (TbPTR1 at 6−10 mg mL−1, 1 mM NADPH, 1 mM substrate or inhibitor, 1% (v/v) DMSO, and 20 mM DTT) and an equal volume of the reservoir (1.5−3 M sodium acetate, 10−50 mM sodium citrate in the pH range 4.5−6.0). Crystals grew to 0.5 mm × 0.3 mm × 0.1 mm over a few days. Crystals grown in excess of 2.6 M sodium acetate were flash-cooled in a stream of N2 to −173 °C directly from the mother liquor. Those obtained in less than 2.6 M sodium acetate were cryoprotected with either 3 M sodium acetate or 30% (v/v) glycerol.
X-ray Data Collection and Structure Determination
X-ray data were collected in-house using a Rigaku Micromax 007 X-ray generator equipped with an Raxis IV++ detector, at station 14.1 of the Synchrotron Radiation Source (SRS), Daresbury, U.K., and beamlines BM14, ID23-1, ID23-2, and ID29 of the European Synchrotron Radiation Facility (ESRF), Grenoble, France (Supporting Information Tables S1−S3). Data were processed with the CCP4 software suite.(33) X-ray images were integrated and scaled with MOSFLM(34) and SCALA(35) or XDS and XSCALE.(36) Structures were solved by molecular replacement(37) using TbPTR1 as the starting model.(17) Several rounds of restrained refined were carried out using REFMAC5(38) together with inspection of electron and difference density Fourier maps, model manipulation, and identification of solvent ions and ligands with COOT.(39) Figures were generated with PyMol.(40)
Acknowledgments
We thank Novartis Animal Health Inc, Basel, Switzerland, for the gift of 15. Supported by awards were from BBSRC (Structural Proteomics of Rational Targets, BBS/B/14434) and The Wellcome Trust (Grant Nos. 082596 and 083481, Senior Fellowship Grant 064771 to T.K.S.). We thank the SRS Daresbury and ESRF Grenoble for synchrotron beam time and support. J.I. and V.P.M. thank CAPES for a postdoctoral fellowship BEX 2000/04-0 and M.Sc. fellowship, respectively. The University of Strathclyde provided studentship support for J.K.H. and J.H.L.
Supporting Information Available
Figures of all ligands in a TbPTR1 active site that have been characterized; tables of crystallographic details; graphs detailing the trypanocidal activity of pyrimethamine or compound 11 in combination with MTX. This material is available free of charge via the Internet at http://pubs.acs.org.
Coordinates and diffraction data have been deposited with the Protein Data Bank under accession codes 3BMC, 3BMN, 3BMO, 3BMQ, 3JQ6, 3JQ7, 3JQ8, 3JQ9, 3JQA, 3JQB, 3JQC, 3JQD, 3JQE, 3JQF, 3JQG.
Footnotes
Abbreviations: BSF, bloodstream form; DHFR, dihydrofolate reductase; HAT, human African trypanosomiasis; Lm, Leishmania major; MTX, methotrexate; PTR1, pteridine reductase; PYR, pyrimethamine; TMP, trimethoprim; Tb, Trypanosoma brucei.
Supplementary Material
References
- McGuire J. J. Anticancer antifolates: current status and future directions. Curr. Pharm. Des. 2003, 9, 2593–2613. [DOI] [PubMed] [Google Scholar]
- Gangjee A.; Jain H. D.; Kurup S. Recent advances in classical and non-classical antifolates as antitumor and antiopportunistic infection agents: part I. Anti-Cancer Agents Med. Chem. 2007, 7, 524–542. [DOI] [PubMed] [Google Scholar]
- Barrett M. P.; Gilbert I. H. Targeting of toxic compounds to the trypanosome′s interior. Adv. Parasitol. 2006, 63, 125–183. [DOI] [PubMed] [Google Scholar]
- Kidder G. W.; Dutta B. N. The growth and nutrition of Crithidia fasciculata. J. Gen. Microbiol. 1958, 18, 621–638. [DOI] [PubMed] [Google Scholar]
- Thöny B.; Auerbach G.; Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347, 1–16. [PMC free article] [PubMed] [Google Scholar]
- Snyder F.; Malone B.; Piantadosi C. Tetrahydropteridine-dependent cleavage enzyme for O-alkyl lipids:substrate specificity. Biochim. Biophys. Acta 1973, 316, 259–265. [DOI] [PubMed] [Google Scholar]
- Nare B.; Hardy L. W.; Beverley S. M. The roles of pteridine reductase 1 and dihydrofolate reductase−thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J. Biol. Chem. 1997, 272, 13883–13891. [DOI] [PubMed] [Google Scholar]
- Cunningham M. L.; Titus R. G.; Turco S. J.; Beverley S. M. Regulation of differentiation to the infective stage of the protozoan parasite Leishmania major by tetrahydrobiopterin. Science 2001, 292, 285–287. [DOI] [PubMed] [Google Scholar]
- Moreira W.; Leblanc E.; Ouellette M. The role of reduced pterins in resistance to reactive oxygen and nitrogen intermediates in the protozoan parasite Leishmania. Free Radical Biol. Med. 2009, 46, 367–375. [DOI] [PubMed] [Google Scholar]
- Nare B.; Garraway L. A.; Vickers T. J.; Beverley S. M. PTR1-dependent synthesis of tetrahydrobiopterin contributes to oxidant susceptibility in the trypanosomatid protozoan parasite Leishmania major. Curr. Genet. 2009, 55, 287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C.Biochemical Role of Folate in Cellular Metabolism. In Folate in Health and Disease; Bailey L. B., Ed.; Marcel Dekker: New York, 1995; pp 23−42. [Google Scholar]
- Opperdoes F. R.; Coombs G. H. Metabolism of Leishmania: proven and predicted. Trends Parasitol. 2007, 23, 149–158. [DOI] [PubMed] [Google Scholar]
- Blakley R. L. Eukaryotic dihydrofolate reductase. Adv. Enzymol. Relat. Areas Mol. Biol. 1995, 70, 23–102. [DOI] [PubMed] [Google Scholar]
- Bello A. R.; Nare B.; Freedman D.; Hardy L. W.; Beverley S. M. PTR1: a reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 11442–11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robello C.; Navarro P.; Castanys S.; Gamarro F. A pteridine reductase gene ptr1 contiguous to a P-glycoprotein confers resistance to antifolates in Trypanosoma cruzi. Mol. Biochem. Parasitol. 1997, 90, 525–535. [DOI] [PubMed] [Google Scholar]
- Hardy L. W.; Matthews W.; Nare B.; Beverley S. M. Biochemical and genetic tests for inhibitors of Leishmania pteridine pathways. Exp. Parasitol. 1997, 87, 158–170. [PubMed] [Google Scholar]
- Dawson A.; Gibellini F.; Sienkiewicz N.; Tulloch L. B.; Fyfe P. K.; McLuskey K.; Fairlamb A. H.; Hunter W. N. Mol. Microbiol. 2006, 61, 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley D. G.; Schuttelkopf A. W.; Leonard G. A.; Luba J.; Hardy L. W.; Beverley S. M.; Hunter W. N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 2001, 8, 521–525. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; Hardy L. W.; Beverley S. M.; Hunter W. N. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J. Mol. Biol. 2005, 352, 105–116. [DOI] [PubMed] [Google Scholar]
- Mpamhanga C. P.; Spinks D; Tulloch L. B.; Shanks E. J.; Robinson D. A.; Collie I. T.; Fairlamb A. H.; Wyatt P. G.; Frearson J. A.; Hunter W. N.; Gilbert I. H.; Brenk R. One scaffold, three binding modes: novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J. Med. Chem. 2009, 52, 4454–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B.; Langan P.; Coates L.; Mustyakimov M.; Schoenborn B.; Howell E. E.; Dealwis C. Neutron diffraction studies of Escherichia coli dihydrofolate reductase complexed with methotrexate. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 18493–18498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis G. W.; Murcko M. A. Properties of known drugs. 2. Side chains. J. Med. Chem. 1999, 42, 5095–5099. [DOI] [PubMed] [Google Scholar]
- Gibson C. L.; Huggan J. K.; Kennedy A.; Kiefer L.; Lee J. H.; Suckling C. J.; Clements C.; Harvey A. L.; Hunter W. N.; Tulloch L. B. Diversity oriented syntheses of fused pyrimidines designed as potential antifolates. Org. Biomol. Chem. 2009, 7, 1829–1842. [DOI] [PubMed] [Google Scholar]
- Cavazzuti A.; Paglietti G.; Hunter W. N.; Gamarro F.; Piras S.; Loriga M.; Allecca S.; Corona P.; McLuskey K.; Tulloch L.; Gibellini F.; Ferrari S.; Costi M. P. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe J. J.; McCormack J. J.; Gutteridge W. E. Dihydrofolate reductases within the genus Trypanosoma. Exp. Parasitol. 1969, 25, 311–318. [DOI] [PubMed] [Google Scholar]
- Sirawaraporn W.; Sertsrivanich R.; Booth R. G.; Hansch C.; Neal R. A.; Santi D. V. Selective inhibition of Leishmania dihydrofolate reductase and Leishmania growth by 5-benzyl-2,4-diaminopyrimidines. Mol. Biochem. Parasitol. 1988, 31, 79–86. [DOI] [PubMed] [Google Scholar]
- Davies G. D.; Noell C. W.; Robins R. K.; Koppel H. C.; Beaman A. G. Potential purine antagonists. XXII. The preparation and reactions of certain derivatives of 2-amino-6-purinethiol. J. Am. Chem. Soc. 1960, 118, 2633–2640. [Google Scholar]
- Taylor E. C.; Liu B. A New and efficient synthesis of pyrrolo[2,3-d]pyrimidine anticancer agents: alimta (LY231514, MTA), homo-alimta, TNP-351, and some aryl 5-substituted pyrrolo[2,3-d]pyrimidines. J. Org. Chem. 2003, 68, 9938–9947. [DOI] [PubMed] [Google Scholar]
- SigmaPlot; Systat Software Inc.: Hounslow, London, U.K. [Google Scholar]
- Morrison J. F. Kinetics of the reversible inhibition of enzyme-catalyzed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 1969, 185, 269–286. [DOI] [PubMed] [Google Scholar]
- Wirtz E.; Leal S.; Ochatt C.; Cross G. A. M. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 99, 89–101. [DOI] [PubMed] [Google Scholar]
- Mikus J.; Steverding D. A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar blue. Parasitol Int. 2000, 48, 265–269. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994, D50, 760−763. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. The integration of macromolecular diffraction data. Acta Crystallogr. 2006, D62, 48–57. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr. 2006, D62, 72–82. [DOI] [PubMed] [Google Scholar]
- Kabsch W.X-ray Detector Software, 2004. http://www.mpimf-heidelberg.mpg.de/∼kabsch/xds.
- Vagin A.; Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar]
- Murshudov G. N.; Vagin A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997, D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004, D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.The PYMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, 2002. [Google Scholar]
- Scott D. A.; Hickerson S. M.; Vickers T. J.; Beverley S. M. The role of the mitochondrial glycine cleavage complex in the metabolism and virulence of the protozoan parasite Leishmania major. J. Biol. Chem. 2008, 283, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.