

Abstract
Motivation: Microarray-based gene expression data have been generated widely to study different biological processes and systems. Gene co-expression networks are often used to extract information about groups of genes that are ‘functionally’ related or co-regulated. However, the structural properties of such co-expression networks have not been rigorously studied and fully compared with known biological networks. In this article, we aim at investigating the structural properties of co-expression networks inferred for the species Saccharomyces Cerevisiae and comparing them with the topological properties of the known, well-established transcriptional network, MIPS physical network and protein–protein interaction (PPI) network of yeast.
Results: These topological comparisons indicate that co-expression networks are not distinctly related with either the PPI or the MIPS physical interaction networks, showing important structural differences between them. When focusing on a more literal comparison, vertex by vertex and edge by edge, the conclusion is the same: the fact that two genes exhibit a high gene expression correlation degree does not seem to obviously correlate with the existence of a physical binding between the proteins produced by these genes or the existence of a MIPS physical interaction between the genes. The comparison of the yeast regulatory network with inferred yeast co-expression networks would suggest, however, that they could somehow be related.
Conclusions: We conclude that the gene expression-based co-expression networks reflect more on the gene regulatory networks but less on the PPI or MIPS physical interaction networks.
Contact: hongzhe@mail.med.upenn.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Gene co-expression networks are constructed from data of gene expression microarray experiments by using different correlation-based inference methods. The vertices of these networks represent genes, while their edges are related to the values of the pairwise correlation coefficient that is calculated from the expression data of the genes. Co-expression networks, in contrast with other networks whose edges represent well-defined biological interactions, are composed of edges that show co-expression patterns of genes over different experimental conditions (Stuart et al., 2003). Groups of genes derived from modular analysis on such co-expression networks often show enrichment of certain Gene Ontology categories (Horvath and Dong, 2008; Stuart et al., 2003; Yan et al., 2007; Zhang and Horvath, 2005), indicating that the edges of such networks indeed contain some biological meaning. This, in turn, suggest that co-expression networks have to be biologically meaningful by themselves. However, it is still unclear how co-expression networks are related to true biological networks.
The meaning of the edges is a relevant question when talking about network analysis. Graphs, by their own nature, are abstract representations of the pairwise interactions or relationships between the different parts or subunits of a complex system. Thus, there exists an important difference between analyzing co-expression data by using networks tools and analyzing networks constructed from co-expression data. In the first case, the focus of the analysis lies (or have been lain so far) on the statistical study of sets of genes which are interesting due to whatever statistical-biological reason (set enrichment analysis, for example), regardless of the pairwise interactions among the genes in the sets. In the second case, the focus of the analysis is on the structure of the pairwise interactions and the meaning of this structure. Both types of analysis are relevant and biologically interesting. We concentrate here on the second type of analysis, the structure of the pairwise interactions or correlations.
In this article, in order to attack the question of the meaning of co-expression edges, co-expression networks inferred from a yeast gene expression microarray dataset are compared with available, well-established network data of the same organism, the yeast. The yeast networks that are compared with are the following: the yeast protein-protein interaction (PPI) network (Breitkreutz et al., 2008; Jensen et al., 2009; Steffen et al., 2002), the yeast MIPS physical interaction network (Munich Information center for Protein Sequences) and the yeast regulatory network reflecting transcription factor (TF)-DNA binding (Harbison et al., 2004). The ultimate purpose of this comparison will be to determine whether the edges of a co-expression network may (or may not) represent (i) a physical interaction between those proteins resulting from the expression of the genes, (ii) some type of biological regulation or (iii) something similar to what edges of the MIPS network represents.
The article is organized as follows. Section 2 describes (i) the procedures that we employ to infer the co-expression networks, (ii) the method we use to compare the different topologies between the networks and (iii) the statistical method we apply to estimate the accuracy of our analysis. The results of the comparisons between the networks are presented in Sections 3 and 4. Finally, some conclusions are drawn in Section 5.
2 METHODS
2.1 Construction of co-expression networks
When inferring co-expression networks from high-throughput gene expression data, one usually takes as primary input the data from a set of n independent measurements of the mRNA gene expression levels and then, by using whatever correlation-based inference method, constructs the corresponding network. The mRNA measurements are carried out by means of microarray techniques, and each measurement, which is able to collect information of a very big number (p) of genes, corresponds to a particular group of cells of a certain individual. In this article, our main experiment is based on a recent genome-wide study on expression variation by crossing two yeast strains (Brem et al., (2002, 2005), where 112 segregants were individually genotyped at 2956 marker positions and 6228 gene expressions were measured for each segregant. Our analysis only uses the gene expression data. The reason for choosing this particular gene expression dataset is that the 112 yeast segregants studied in Brem et al. (2002, 2005) are randomly assigned genotypes (Mendelian randomization), which allows us to consider them as independent and identically distributed (i.i.d.) samples from the population of all segregants. Given that valid inferences on correlations require the assumption that the observations are i.i.d., the choice of the above dataset ensures the legitimacy of the standard correlation calculations and inferences. In contrast, pooling data from different experiments—i.e. measured under different biological conditions—may, first, violate the i.i.d. assumption, and secondly, may result in very different co-expression patterns that could either mask the true co-expressions or even introduce false ones. Time course experiments, on the other hand, might yield non-independent observations.
The more basic co-expression inference network model that one can find in the literature consists in calculating first the linear pairwise correlation coefficient r of all possible pairs of genes (using for this purpose the data of the n=112 microarray measurements), and then, establishing a link between those gene pairs that show a ‘large enough’ value of r. The natural assumption behind this construction process is that a large value of the correlation coefficient signifies some functional relationship among the pair of genes involved. Of course, an important aspect that needs to be precisely established is the meaning of ‘large enough’. When inferring co-expression networks, people working in the field usually address this question by fixing a cutoff (r2cf) for the squared values of r, so that, if r2 is larger than the cutoff, then a link between the pair of genes is established, and if r2 is smaller, the gene pair remains unlinked. This solution shifts certainly the problem to the question of what cutoff's value should be imposed.
The inference methods we employ to construct our co-expression networks are based on the above-mentioned co-expression inference network model, but specially adjusted to generate networks with specific properties so that these co-expression networks can ‘fairly’ be compared with the available yeast network data. Our approach to the subject is 2-fold. On the one hand, we suggest to modify the basic model in such a way that the resulting co-expression networks are composed of a desired number of vertices and edges. The rationale for this constraint is that a network needs to have the same number of vertices and edges as the network which it has to be compared with. We will refer to this procedure as network inference procedure I. On the other hand, we propose as a second procedure to use the basic inference model directly, without modifications, but compelling r2cf to take large values only. The idea behind this second method is now to be able to generate networks containing only highly trustworthy edges. We will refer to it as network inference procedure II.
The co-expression networks thus constructed are then compared with the following yeast networks: (i) The PPI network compiled by (Steffen et al., 2002), which contains 3775 proteins and 5983 protein interactions. (ii) The MIPS physical interaction network (MIPS) from the Munich Information center for Protein Sequences, which contains 4139 vertices and 7377 edges. And (iii), the transcriptional regulatory network (REG) based on the TF-DNA binding data from (Harbison et al., 2004), where 203 TFs were tested for their binding profiles in yeast. By using P<0.001 as threshold for positive binding (as the authors do in the original article), this REG network contains 167 genes and 429 edges. All these data correspond to the networks considered as simple graphs, i.e. once the few loops contained in all three datasets are eliminated. Since the data of these three networks have extensively been verified by experiments, the above networks may be thought of being highly reliable networks. We will call them throughout the paper true yeast networks.
As can be noted, the number of genes considered by the different datasets is considerably different. Indeed, many genes belonging to the co-expression network dataset are missing in the ‘true’ PPI, REG or MIPS networks, and a few nodes present in them are absent in the co-expression data. In order to carry out even comparisons, we only consider the larger subgraphs contained in the network datasets that can be generated by the vertices (genes) that belong to both datasets. Thus, the number of genes that can be found in both the PPI dataset and gene expression dataset is N=3711, and the number of edges belonging to the PPI network subgraph generated by these 3711 genes is M=5869. In the MIPS case, the number of vertices belonging to both the MIPS and expression datasets is N=4112, and the number of edges in the subgraph is N=7327. In the REG network case, the number of vertices belonging to both the reg and expression datasets is N=166, and the number of edges in the subnetwork, N=427. From here on, when we talk about the inferred co-expression networks or the true PPI, MIPS and REG networks, we will always refer to the above (sub)networks.
Network inference procedure I consists explicitly in calculating r2 for all pairs of genes, and taking then the m largest found values of r2, where m is the number of edges of the true PPI, MIPS or REG (sub)network, respectively. As a result, the graphs to be compared have exactly the same number of nodes and edges, which allows a correct comparison from a topological viewpoint. Note that this way to proceed indirectly fixes a cutoff too.
Network inference procedure II works by directly fixing r2cf to take high values only. Given the small probability that large values of r2 appear by chance, one expects that this way to proceed generates co-expression networks containing only trustworthy edges, i.e. edges that represent ‘reliable’ relationships among genes. Unfortunately, as a side effect, the networks thus inferred tend to contain a small number of edges, which results in a big number of isolated network vertices. These isolated nodes can safely be excluded of our analysis since they provide no information about the gene pairwise interactions. Thus, in order to perform the comparisons, we proceed as follows: from the co-expression network constructed, we remove the isolated vertices, leaving the rest of the network unchanged. From the corresponding true network, we extract that subnetwork generated by the genes that belong to both the true network and the set of non-isolated co-expression nodes. The co-expression network remaining after eliminating the isolated vertices and the mentioned extracted true subnetwork will be the graphs that we will compare. The above procedure answers, of course, the purpose of getting networks having exactly the same number of vertices. Note, however, that the inferred co-expression network and the corresponding true network may have a different number of edges.
2.2 Comparison of different network structures
Depending on what network aspect one focuses on, two networks can be compared in several ways. One usual way is to focus on their structural features, regardless of the name or label of the network vertices. In this case, the focus of the comparison falls on network topologies such as the average shortest path length, the mean clustering coefficient, the degree distribution, etc. Another way to compare two networks is to compare them node by node and edge by edge. In this case, the purpose is to know whether an edge or vertex belongs to both networks or not. Note that an edge is defined by the vertices to which it is attached, which entails that, if one edge belongs to both networks, then the vertices ‘defining’ the edge have necessarily to belong to both networks too. In this second case, two networks are said to be equal if they contain the same set of nodes and the same set of edges.
We apply here both comparison methods. The structural one, because topological similarities (or differences) between networks can provide useful information about the strengths (and weaknesses) of the association model used to infer the co-expression network. The second one, because high degrees of network similarity mean in this case that most nodes and edges can be found in both networks, which, in turn, supports the thesis that the nodes and edges of both networks may share a common meaning in biological terms.
Regarding the structural method, the following network topologies are investigated in this study [see (Albert and Barabási, 2002; Newman, 2003a) for a review].
The average shortest path length (l), defined as the mean distance between each two vertices of a network, being the distance between any two vertices the number of edges along the shortest path connecting them.
Network diameter (d), which is the distance between the two vertices which are furthest from each other. (Note that both previous definitions assume the network to be completely connected. If this is not the case, both l and d are, respectively, defined as the average path length and diameter of the network largest component.)
The degree distribution (P(k)), which gives the probability that a randomly selected node of a network has degree k, i.e. that it is connected to k other different vertices. Most real networks are scale-free, meaning the P(k) ∼ k−γ, where γ is a constant usually between 1 and 3.
The mean clustering coefficient (C) and the local or degree-dependent clustering coefficient (C(k)), which are both related to the meaning of clustering coefficient of a vertex, which, in turn, is defined as the ratio between the number of connections existing among its neighbors and the maximal number of edges that can exist among them (Watts and Strogatz, 1998). C and C(k) are then, respectively, defined as the average of the clustering coefficients over all network vertices (or, more correctly, over all vertices having a degree equal or larger than two) and over all network vertices of degree k. Note that biological networks have been found to be highly interconnected and therefore have a high mean clustering coefficient.
The nearest neighbor average degree function (
 ), which can be written as
), which can be written as 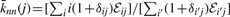 , and provides a convenient measure of the degree-degree correlations of a network. Here, ℰij is the degree-degree correlation function, which gives the probability that a randomly selected edge connects one vertex of degree i to another of degree j. Degree-degree correlations, apart from being an essential measure for characterizing the topology of networks, have became important as a result of the discovery that biological networks are dissortative (high-degree vertices tend to connect to low-degree vertices), while social networks are assortative (high-degree vertices attach preferably other highly connected vertices) (Newman, 2003b). Function
, and provides a convenient measure of the degree-degree correlations of a network. Here, ℰij is the degree-degree correlation function, which gives the probability that a randomly selected edge connects one vertex of degree i to another of degree j. Degree-degree correlations, apart from being an essential measure for characterizing the topology of networks, have became important as a result of the discovery that biological networks are dissortative (high-degree vertices tend to connect to low-degree vertices), while social networks are assortative (high-degree vertices attach preferably other highly connected vertices) (Newman, 2003b). Function  takes constant value
takes constant value  if no type of network degree-degree correlation exists (i.e. when ℰij=(2−δij)iP(i)jP(j)/〈i〉2), while it is a decreasing (increasing) function if dissortative (assortative) mixing is present (Vázquez et al., 2002).
if no type of network degree-degree correlation exists (i.e. when ℰij=(2−δij)iP(i)jP(j)/〈i〉2), while it is a decreasing (increasing) function if dissortative (assortative) mixing is present (Vázquez et al., 2002).In addition to all these quantities, we also inquire into the network maximum degree, which is the degree of that network vertex that has the maximum degree, and the number of nodes (or order) of the network largest component (lco).
With respect to the second method of comparison, we introduce only one quantity to measure the degree of similarity of two networks. In order to explain this quantity, we introduce first some notation. Consider that the networks to be compared, say, network a and network b, have exactly the same set of nodes. Let A be the set of edges of network a, and B, the set of edges of network b. Finally, let #(S) mean the number of elements of a set S. Then, the degree of similarity of two networks is said to be
| (1) |
where fa=(#(A∩B))/(#A) and fb=(#(A∩B))/(#b). Notice a few things about this definition. First, s takes the value 1 when both networks are equal, and vanishes when the number of edges belonging to both networks is zero. Second, s can be expressed as a function of fractions: fa and: fb, which indicates that s does not depend on the absolute values #(A) and #(B). Hence, it does not depend on the size of the networks, but on the proportion of edges that belong to both networks. Third, the definition of s is suitable for only ‘deterministic’ networks, i.e. network whose edges are present with probability either one or zero.
2.3 The bootstrap procedure
The co-expression network model takes the measurements of n independent microarray measurements, each of them corresponding to a certain individual, and constructs a co-expression network by assuming that high linear correlations reflect some type of functional gene relationships. The result is one only graph which intends to rephrase in network terms the biology of the system under study. However, because this network construction process is based on a small sample n of measurements, it can only provide estimates of the network properties. The question that arises then is how accurate these estimates are.
We address this question by using the bootstrap method (Efron and Tibshirani, 1993). In our case, the randomly sampled original data points are the n microarray runs, each of them containing p expression values corresponding to the p genes analyzed by the microarray. Every bootstrap sample is a random sample of size n drawn, with replacement, from the original n microarray runs. For each bootstrap run, we recalculate the network statistics listed in previous section and then obtain the standard error of the estimated statistics based on the original data.
It is worth to mention that statistics comes to the problem not because of the model used to infer the network—which, indeed, is a deterministic model—but due to the fact that the raw data, the microarrays, represent a small sample of the population of all individuals.
3 RESULTS
Tables 1, 2 and 3 show the results of comparing the inferred co-expression networks with, respectively, the ‘true’ PPI, MIPS and REG networks. The tables are divided in sections, each of them corresponding to one of the two network inference procedures described in Section 2. All three tables show the obtained values of the following quantities: number of nodes (nodes), number of links (edges), average shortest path length (l), diameter (d), mean clustering coefficient (C), network maximum degree (max. deg.), number of nodes of the largest component (lco), network similarity (s), cutoff (r2cf) and coefficient of resemblance (ℛ). The last quantity will be discussed in Section 3.3. Standard errors are displayed in brackets. They are estimated by using 10 000 independent bootstrap replications in all cases.
Table 1.
Comparison of the co-expression network and the PPI network based on different procedures
| Procedure I | Procedure II: r2cf=0.7 | Procedure II: r2cf=0.9 | ||||
|---|---|---|---|---|---|---|
| Quantity | PPI | Co-expression | PPI | Co-expression | PPI | Co-expression |
| Edges | 5869 | 5869 | 116 (46.7) | 2234 (1080) | 0 (2.46) | 17 (14.3) |
| Nodes | 3711 | 3711 | 497 (72.4) | 497 (72.4) | 30 (12.7) | 30 (12.7) |
| l | 5.940 | 3.427 (0.788) | 3.07 (1.09) | 3.745 (0.764) | 0 (0.425) | 1.333 (0.619) |
| d | 15 | 14 (3.06) | 7 (3.03) | 10 (2.94) | 0 (0.678) | 2 (1.75) |
| C | 0.1181 | 0.6479 (0.0268) | 0.4 (0.0455) | 0.6274 (0.0239) | 0 (0.304) | 0.778 (0.209) |
| Max. deg. | 99 | 122 (19.3) | 7 (2.92) | 47 (25.9) | 0 (0.931) | 3 (2.04) |
| lco | 3178 | 418 (137) | 11 (16.4) | 245 (88.5) | 1 (0.931) | 4 (5.23) |
| Similarity | 0.003448 (0.000295) | 0.00988 (0.00139) | 0.0000 (0.0283) | |||
| Cutoff | 0.6242 (0.0241) | 0.70 | 0.90 | |||
| ℛ | 0.002823 (0.000471) | 0.00659 (0.00430) | 0.0423 (0.0284) | |||
l: average shortest path length; d: network diameter; C: mean clustering coefficient; max.deg: network maximum degree; lco; number of nodes of the network largest component; s: network similarity; ℛ: coefficient of resemblance. Numbers in the parentheses are the standard errors based on 10 000 bootstrap samples.
Table 2.
Comparison of the co-expression network and the MIPS physical interaction network based on different procedures
| Procedure I | Procedure II: r2cf=0.7 | Procedure II: r2cf=0.9 | ||||
|---|---|---|---|---|---|---|
| Quantity | MIPS | Co-expression | MIPS | Co-expression | MIPS | Co-expression |
| Edges | 7327 | 7327 | 198 (61.8) | 3147 (1507) | 0 (2.80) | 26 (20.9) |
| Nodes | 4112 | 4112 | 589 (83.8) | 589 (83.8) | 39 (15.4) | 39 (15.4) |
| l | 4.843 | 3.491 (0.823) | 4.67 (1.04) | 3.716 (0.829) | 0 (0.373) | 1.533 (0.793) |
| d | 13 | 14 (3.18) | 13 (3.37) | 9 (3.26) | 0 (0.623) | 3 (2.13) |
| C | 0.0998 | 0.6418 (0.0248) | 0.2788 (0.0313) | 0.6350 (0.0226) | 0 (0.295) | 0.611 (0.133) |
| Max. deg. | 288 | 134 (17.4) | 26 (3.07) | 61 (29.6) | 0 (1.01) | 4 (1.87) |
| lco | 3848 | 473 (153) | 95 (55.0) | 293 (108) | 1 (1.04) | 6 (7.39) |
| Similarity | 0.003767 (0.000304) | 0.008745 (0.00131) | 0.0000 (0.0202) | |||
| Cutoff | 0.6338 (0.0242) | 0.70 | 0.90 | |||
| ℛ | 0.003154 (0.000469) | 0.00527 (0.00460) | 0.0294 (0.0297) | |||
l: average shortest path length; d: network diameter; C: mean clustering coefficient; max.deg: network maximum degree; lco; number of nodes of the network largest component; s: network similarity; ℛ: coefficient of resemblance. Numbers in the parentheses are the standard errors based on 10 000 bootstrap samples.
Table 3.
Comparison of the co-expression network and the regulatory network based on different procedures
| Procedure I | Procedure II: r2cf=0.5 | Approach II: r2cf=0.6 | Procedure II: r2cf=0.7 | |||||
|---|---|---|---|---|---|---|---|---|
| Quantity | REG | Co-expression | REG | Co-expression | REG | Co-expression | REG | Co-expression |
| Edges | 427 | 427 | 3 (4.05) | 12 (7.47) | 1 (0.779) | 1 (2.76) | 1 (0.118) | 1 (0.564) |
| Nodes | 166 | 166 | 12 (5.15) | 12 (5.15) | 2 (2.81) | 2 (2.81) | 2 (0.781) | 2 (0.781) |
| l | 3.352 | 3.037 (0.128) | 1.333 (0.590) | 1.3 (0.558) | 1 (0.165) | 1 (0.294) | 1 (0.0798) | 1 (0.101) |
| d | 8 | 8 (1.04) | 2 (1.68) | 2 (1.53) | 1 (0.482) | 1 (0.840) | 1 (0.0879) | 1 (0.298) |
| C | 0.1270 | 0.3919 (0.0424) | 0 (0.121) | 0.571 (0.190) | 0 (0.0198) | 0 (0.385) | 0 (0.000) | 0 (0.127) |
| Max. deg. | 17 | 27 (3.05) | 2 (1.14) | 4 (1.42) | 1 (0.329) | 1 (0.993) | 1 (0.0879) | 1 (0.327) |
| lco | 160 | 117 (7.63) | 3 (3.09) | 5 (4.29) | 2 (0.519) | 2 (1.38) | 2 (0.0879) | 2 (0.346) |
| Similarity | 0.02768 (0.00505) | 0.0714 (0.0304) | 1.000 (0.233) | 1.000(0.227) | ||||
| Cutoff | 0.2004 (0.0165) | 0.50 | 0.60 | 0.70 | ||||
| ℛ | 0.0160 (0.0358) | 0.013 (0.207) | 0.262 (0.441) | 0.879(0.283) | ||||
l: average shortest path length; d: network diameter; C: mean clustering coefficient; max.deg: network maximum degree; lco; number of nodes of the network largest component; s: network similarity; ℛ: coefficient of resemblance. Numbers in the parentheses are the standard errors based on 10 000 bootstrap samples.
3.1 Comparison based on procedure I
The comparison of the different results shown in the tables indicate that, with respect to the first inference procedure, procedure I, the inferred co-expression networks substantially differ from the PPI, MIPS and REG networks. As can be seen, they differ in, mainly, the mean clustering coefficient and the number of nodes belonging to the largest network component. The comparison of the average path lengths and diameters does not provide any fundamental information (specially in the PPI and MIPS cases), due to the considerable difference in size of the corresponding largest network components. Another evident discrepancy between the networks can be found in the value of their maximum degree. Finally, the degree of network similarity, s, demonstrate that ‘true’ and co-expression networks have only an insignificant percentage of edges in common.
The analysis of the non-scalar measures seems to lead to the same conclusion. An inspection of the corresponding degree distributions indicates that they are sensibly different from each other (in spite of all of them approximately decaying as power law functions). As an example, Figure 1 illustrates the difference in degree distribution between the true PPI network and the corresponding co-expression network. Although not shown, similar results are found for the MIPS and REG networks. Further, the degree-degree correlations appear also to be notably different, principally in the PPI and MIPS cases. For example, the nearest neighbors average degree functions of the true MIPS network and the corresponding co-expression network are plotted in Figure 2. It can easily be seen in the figure that the MIPS network shows a clear dissortative behavior, while the co-expression network exhibits an explicitly assortative mixing. Again, similar results can be found for the PPI and REG networks. Regarding the local clustering coefficients, the results also indicate that the networks are different. Plots of the degree-dependent clustering coefficients of the true PPI, MIP and REG networks remarkably differ from those corresponding to the co-expression networks. Interestingly, C(k) shows in no case an explicit power law decay of the form P(k) ∼ k−β. As an example, Figure 3 compares C(k) for the PPI and the corresponding co-expression network.
Fig. 1.

Degree distribution, P(k), of the true PPI network (cycles) and the corresponding co-expression network inferred by network inference procedure I (squares) as a function of k. The inset panel displays P(k) in double logarithmic scales, showing that P(k) approximately decays as a power law. These results indicate that, in spite of being both networks approximately scale free (at least, for all k : k>1), their degree distribution is different. For the co-expression network, the standard errors are plotted based on 104 bootstrap samples.
Fig. 2.

Nearest neighbors average degree function,  , of the true MIPS network (cycles) and the corresponding co-expression network inferred by network inference procedure I (squares), indicating that the MIPS network is dissortative while the co-expression network is assortative. For the association network, the standard errors are also plotted based on 104 bootstrap samples.
, of the true MIPS network (cycles) and the corresponding co-expression network inferred by network inference procedure I (squares), indicating that the MIPS network is dissortative while the co-expression network is assortative. For the association network, the standard errors are also plotted based on 104 bootstrap samples.
Fig. 3.

Degree-dependent clustering coefficient, C(k), of the true PPI network (cycles) and the corresponding co-expression network inferred by network inference procedure I (squares). The picture shows that both C(k) functions are different, being the local clustering coefficient of the co-expression network always substantially larger than that of the yeast PPI network. Note that C(k) does not decay as C(k) ∼ k−β (β a constant, usually close to 1). For the co-expression network, the standard errors are also plotted based on 104 bootstrap samples.
The results suggest that, at least with relation to inference procedure I, co-expression networks differ remarkably from both the PPI and MIPS networks, and considerably from REG networks. Note that these findings are specially relevant in view of the fact that, from both construction procedures, procedure I is the one that can provide more conclusive results in terms of topological network comparison. The reason, of course, is that the compared networks have in this case, by construction, the same number of vertices and links. This important condition should not be underestimated when comparing networks, since two networks having different number of vertices and/or edges can exhibit different values of relevant topological measures—such as the average path length, the mean clustering coefficient, etc—even if both networks derive from the same generating network model.
3.2 Comparison based on procedure II
When inference procedure II is used, the situation seems to change slightly. The results still appear to indicate that co-expression networks are substantially different from the PPI and MIPS networks. However, they suggest that co-expression networks may somehow be related to regulatory networks.
Let us first comment on the results corresponding to the PPI and MIPS networks (Tables 1 and 2). As we can see from both tables, when cutoff r2cf takes the value r2cf=0.7 or r2cf=0.9, the number of edges of the co-expression networks is always much more larger than the number corresponding to PPI or MIPS networks. Bearing in mind that the amount of edges of a network plays a deciding role on its structure, this unique difference would be enough to conclude that these networks may not come from the same generating network model. The values of lco and similarity s seem only to confirm this statement. (Note that a comparison of measures l, d and C is not suitable here because these measures are very sensible to differences in the number of network edges.)
Functions P(k), ℰij and C(k) does not provide any additional, relevant information either. The reason is that the domain of these functions (or, in other words, the maximum degree of the networks) is too small to make any indubitable, statistical conclusion. Nevertheless, the difference in the function domain between true and co-expression networks would already show that both PPI and MIPS networks are different from the corresponding co-expression networks.
Table 1 shows an illustrative example of how different co-expression and PPI networks can be when r2cf=0.9. In this case, the co-expression network inferred by procedure II contains 30 nodes and 17 edges. The structure of this network is the following: 26 of the 30 nodes are making up 13 isolated components consisting of two vertices and only one link joining them. The other four vertices are joined together in a unique component whose form is a ‘triangle with a tail’ (two vertices of degree 2, one of degree 3 and one of degree 1). In fact, C is so large in this network because the vertices having a degree >2 are those belonging to the ‘triangle’ of the last described component. Note also that, by construction, no isolated vertices can appear in this network. Let us further remark the obvious fact that this network is really simple, so simple that it seems to be uninteresting! Remember, however, that networks constructed by using high cutoff values in procedure II are interesting to us not because of its structural complexity (or simplicity) but because of the fact that all edges of these networks are presumably highly reliable. Next, we identify the 30 nodes of the network, look for these same nodes within the PPI network and analyze the subnetwork generated by them. What it can be observed is that no edges among these 30 nodes are present in the PPI subnetwork. In other words, all vertices of the PPI subnetwork thus generated are isolated. Table 2 show similar results regarding the MIPS and the corresponding co-expression network when r2cf=0.9. When one takes into account the standard errors associated with two different topologies, the picture does not substantially changes.
The comparison with the regulatory suggests, however, certain relationship between reg and co-expression networks. Table 3 shows the results of the corresponding reg and co-expression networks when the selected cutoff is, respectively, r2cf=0.5, r2cf=0.6 and r2cf=0.7. (No gene pairs of the gene expression dataset exhibits a squared pairwise correlation coefficient larger than r2=0.77.) In the two last cases, both the co-expression network and the corresponding REG subnetwork coincide. When r2cf=0.5, however, the networks have only one edge in common. In all cases, the networks are too small to gain any topological information of them. The results, specially when r2cf≥0.6, would appear to provide relevant information, but the small size of the networks involved does not make reliable this conclusion (when r2cf≥0.6, the networks only contain two vertices and one link).
Network similarity s seems to be a more promising measure when comparing networks being small to show any complex structure. The results of network similarity, s, tell us that PPI and MIPS networks are not related with their corresponding co-expressed counterparts, while the REG network coincide with the inferred co-expression network when r2cf≥0.6. Unfortunately, the small size of the networks in the last case does not seem to make that result reliable enough.
3.3 Coefficients of resemblance
In order to better grasp the variance of the different networks, we introduce the coefficient of resemblance (ℛ). The idea behind this coefficient is to provide a network measure similar to s, but more suitable for capturing the statistical aspect of the networks. This coefficient should improve similarity s in the following aspects: (i) in being able to somehow capture the variance shown by standard errors and (ii) correcting the value of s to be zero when the networks are positively non-related, i.e. when the number of common edges that they share is due to simple chance (in this respect, note that the estimated values of s might appreciably be distorted if the networks are small). The coefficient we propose involves substituting estimators 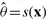 , which are obtained from the original data points x = {x1, x2,···, xn} (i.e. obtained from the one only network generated from the n = 112 individuals data), for the mean of the bootstrap estimators
, which are obtained from the original data points x = {x1, x2,···, xn} (i.e. obtained from the one only network generated from the n = 112 individuals data), for the mean of the bootstrap estimators  , which is obtained from the average over all bootstrap networks generated. Thus, analogously as the definition of s, we estimate the bootstrap degree of similarity of two probabilistic networks as
, which is obtained from the average over all bootstrap networks generated. Thus, analogously as the definition of s, we estimate the bootstrap degree of similarity of two probabilistic networks as
| (2) |
where A*i and B*i are the sets of edges of the bootstrap networks a*i and b*i. Bearing also in mind that the final coefficient must vanish when no causal connection between the networks exists, we define the coefficient of resemblance, ℛ, as
| (3) |
where (i) s′ is the bootstrap degree of similarity between the PPI, MIPS or REG networks and the corresponding co-expression network model and (ii) s′rand is the bootstrap degree of similarity between the PPI, MIPS or REG networks and the ‘randomized’ corresponding co-expression network. Randomized co-expression networks can be obtained by interchanging in the gene expression dataset the expression values of two genes whatever, regardless whether they belong to the same individual or not. This process, when repeated sufficiently many times, destroys any pairwise correlations in the dataset, and consequently, the co-expression networks that result from the process cannot be biologically related to the PPI, MIPS or REG networks.
We randomize the gene expression dataset by repeating the gene expression value interchange process 109 times. Then, by applying procedures I and II to this randomized dataset, we construct the corresponding randomized co-expression networks. These networks, of course, have no significance, meaning that their edges do not represent any physical or biological relationship between genes, but they will permit us to quantify how large s′ is when no relationship between the networks exists. The randomization process is carried out 10 000 times, and each time procedures I and II are applied. As a result, 10 000 randomized realizations of the networks are obtained. These randomized networks are compared with the corresponding PPI, MIPS and REG networks to estimate s′rand.
The coefficients of resemblance shown in Tables 1, 2 and 3 indicate that the inferred yeast co-expression networks are not related with the true PPI or MIPS networks. However, it does seem to suggest that some type of connection between regulation and co-expression networks may does exist. According to the results, this connection would be more evident between the genes showing a very high linear correlation coefficient. As the values of the correlation coefficient decrease, more and more edges appear that would not represent a direct regulation among the genes involved.
4 THE PPI NETWORK CASE REVISITED
Given the current importance of PPI networks in the field, in this section we extend the previous analysis to include two additional, currently updated and well-established PPI yeast networks: the STRING PPI network (Jensen et al., 2009) and the BioGRID network (Breitkreutz et al., 2008).
We repeat the analysis with these two additional networks to confirm whether the previous PPI results presented in the last section remain valid when more updated PPI networks are considered. The STRING database provides a quality-controlled collection of protein–protein associations for a large number of organisms (the yeast among them), where a protein–protein association can mean either a direct physical binding or an indirect interaction such as participation in the same metabolic pathway or cellular process. The associations are derived from high-throughput experimental data, from the mining of databases and literature and from predictions based on genomic context analysis. Thus, STRING takes a more generalized perspective on protein and their associations than other databases whose main purpose is to collect and curate direct experimental evidence about protein–protein physical interactions. Because the STRING network explicitly includes indirected protein–protein associations from high-throughput co-expression data, it is expected that a comparison with the corresponding inferred co-expression networks yields a much higher degree of likeness between the networks than what we obtained using the PPI network of (Steffen et al., 2002).
To extract from the STRING database the analyzable PPI network of yeast, we take the STRING file protein.links.v8.1.txt.gz (Jensen et al., 2009) (which is the current release of the protein–protein network database), and select from it the interactions for all proteins starting with the ID assigned to S.Cerevisiae, ID=4932. From the set of all interactions thus obtained, we pick next those interactions having a combined score ≥ 900 (von Mering et al., 2005). This process results in a preliminary network of 3622 proteins and 17 684 interactions, which, after eliminating all proteins/nodes which are not present in the co-expression data (Brem et al., 2005, 2002) (together with the edges attached to them), gives us a final PPI network of 3590 nodes and 17 514 edges. Using this new PPI network, we repeat then the analysis described in the preceding sections. The results of this analysis can be found in Supplementary Table S1 and Supplementary Figure 4(1c), 4(2c) and 4(3c). It is clear to observe that the new results indicate that the compared networks are certainly not equal, but much more similar than when the Steffen's PPI network was used for the comparison. The results are expected since the STRING PPI network also includes the pairs derived from co-expression analysis. The results further confirm the validity of using the co-expression data (Brem et al., 2005, 2002) for carrying out our analysis.
Fig. 4.

Panels (1a–c): degree-dependent clustering coefficient, C(k), of the true PPI networks (cycles) and the corresponding co-expression networks inferred by procedure I (squares). Panels (2a–c): degree distribution, P(k), of the true PPI networks (cycles) and the corresponding co-expression networks inferred by procedure I (squares). Panels (3a–c): nearest neighbors average degree function,  , of the true protein-protein interaction networks (cycles) and the corresponding co-expression networks inferred by procedure I (squares). Note that panels (a) correspond to the direct physical BioGRID PPI network, panels (b) correspond to the entire BioGRID PPI network, and panels (c) correspond to the STRING PPI network (see text for details). For the co-expression networks, the standard errors are plotted based on 2000 bootstrap samples. The plots show (i) that the direct physical BioGRID PPI network is topologically different from its associated co-expression network (they show, for example, different types of degree-degree correlation and a big difference in C(k)), (ii) the topological differences between the entire BioGRID network and its associated co-expression networks are sensibly smaller and (iii) those differences, although still appreciable, begin to disappear when the compared networks are the STRING PPI and its associated co-expression network.
, of the true protein-protein interaction networks (cycles) and the corresponding co-expression networks inferred by procedure I (squares). Note that panels (a) correspond to the direct physical BioGRID PPI network, panels (b) correspond to the entire BioGRID PPI network, and panels (c) correspond to the STRING PPI network (see text for details). For the co-expression networks, the standard errors are plotted based on 2000 bootstrap samples. The plots show (i) that the direct physical BioGRID PPI network is topologically different from its associated co-expression network (they show, for example, different types of degree-degree correlation and a big difference in C(k)), (ii) the topological differences between the entire BioGRID network and its associated co-expression networks are sensibly smaller and (iii) those differences, although still appreciable, begin to disappear when the compared networks are the STRING PPI and its associated co-expression network.
The second network we consider is the BioGRID PPI network (Breitkreutz et al., 2008). The BioGRID database provides curate evidence of physical (direct and indirect) and genetic PPIs, and has the peculiarity of being organized in such a way that extracting a subnetwork composed only of direct physical protein–protein interactions can quite easily be achieved. BioGRID data, however, does not tell the user which physical interactions are known to be direct and which to be indirect, but only the method used to demonstrate the interaction. It is left to the user to decide based on all the evidence codes annotated for a given interaction how likely that interaction is to occur and how likely it is to be direct. That said, the Affinity Capture methods and Co-fractionation, Co-purification and Co-localization methods (Breitkreutz et al., 2008) are generally accepted to be much more likely to show co-complex (indirect) interactions than the other physical methods. Thus, in order to select those interactions that can more likely be direct physical protein–protein bindings, we select from all PPIs listed on the last release of BioGRIG (BIOGRID-ORGANISM-2.0.55-tab.zip, S.Cerevisiae) those yeast interactions recordered only under the following experimental systems: Biochemical activity, Co-crystal structure, Far Western, FRET, Protein-peptide, Protein-RNA, Reconstituted Complex and Two-hybrid (Breitkreutz et al., 2008). The result is a PPI network of 4442 proteins and 23 553 interactions. After removing from this network, all proteins which are absent in the co-expression data of Brem et al. (2005, 2002) (together with the edges attached to them), the resulting network contains 4315 nodes and 17 446 edges. Supplementary Table S2 and Supplementary Figure 4(1a), 4(2a) and 4(3a) shows the comparison results between this final network and the corresponding inferred co-expression networks. These results clearly indicate that just like the PPI network of Steffen et al. (2002), the co-expression network is very different from the PPI network in structures and in topology.
As a last issue, we investigate what the comparison results would be if the complete yeast BioGRID PPI network, i.e. the network which includes both physical and genetic PPIs listed on file BIOGRID-ORGANISM-2.0.55-tab.zip, would be the chosen PPI network. In this case, the PPI network would contain 5601 proteins and 94 246 interactions. After removing all proteins which are not produced by the genes present in our co-expression data (together with the edges attached to them), the resulting network is made up of 5387 nodes and 93 239 edges. The comparison results between this complete BioGRID PPI network and the corresponding co-expression networks are shown in Supplementary Table S3 and Supplementary Figure 4(1b), 4(2b) and 4(3b). Interestingly, the results indicate now a better match between the networks than in the purely direct physical PPI case [Supplementary Table S2 and Supplementary Figure 4(1a), 4(2a) and 4(3a)]. In spite of matching better, the compared networks keep showing, however, some essential differences between them, differences that become even more important in view of the likeness between the STRING PPI and corresponding inferred co-expression networks.
Taken together, the conclusion that could be extracted from the whole presented PPI analysis is that co-expression networks does not seem to reflect on PPI networks whose edges represent direct physical protein-protein bindings, but they seem to better and better reflect on the PPI networks as indirect physical, genetic and predicted interactions are progressively included in the definition of the PPI network.
5 CONCLUSIONS AND DISCUSSION
We compare the co-expression networks inferred from yeast gene expression data with three well-established yeast networks whose biological meaning is well-known and manifest in terms of the biological pairwise interactions of their elements. The networks are the yeast PPI network, the yeast MIPS physical interaction network and the undirected yeast regulatory network.
The comparisons indicate that co-expression networks are not distinctly related, in any sense, with either PPI or the MIPS networks. The very basic structure of the networks would be the usual structure that can be found in most biological networks, i.e. scale-free character, small-world behavior and large degrees of clustering. However, their very specific structure explicitly shows important topological differences between them.
When focusing on a more literal comparison, vertex by vertex and edge by edge, the conclusion is the same: the fact that two genes exhibit a high gene expression correlation degree does not seem to obviously correlate with the existence or not of a protein–protein or MIPS interaction between these genes. In fact, we only observed that a few large protein complexes such as ribosome and proteasome appeared in both PPI and the co-expression networks. Our observations largely agree with reports in literature on relationships between gene expression and PPIs. For example, Ge et al. (2001) showed that interacting protein pairs are more likely to be in the same expression cluster than random pairs for yeast. However, when the self-interactions or homodimers were removed from their analysis, Mrowka et al. (2003) observed that the number of intracluster protein pairs did not differ significantly from the random expectation. Similarly, Jansen et al. (2002) observed strong correlations in expression for protein pairs in permanent protein complexes, but very weak overall relationship when all the interacting protein pairs are considered. Similar weak correspondence between gene expression and PPIs was also reported in Bhardwaj and Lu (2005) for yeast.
The comparison of the yeast regulatory network with inferred co-expression networks would suggest, however, that they could somehow be related. Thus, an edge by edge network comparison seems certainly to indicate that high values of gene expression correlation coefficients correlate to some extent with the existence of gene regulations among the corresponding genes. This correlation would, however, rapidly fall as the values of the pairwise correlation coefficient decrease.
From a structural point of view, topologies such as the mean clustering coefficient (C) and the largest component vertex number lco provide suggesting information about the relationship between REG and co-expression networks. The findings are the following. When both networks are similar in size, C is much larger, and lco much smaller, in our yeast co-expression network than in the yeast regulatory network. These topological features, together with the presumable connection between both networks when only highly ‘reliable’ co-expression edges are present, suggest that a number of co-expression edges could represent no direct gene regulations. The reason would be that genes that are not directly connected in the regulatory network could indirectly be connected through a small regulatory pathway. This indirect gene regulation could be rephrased in the co-expression network in the existence of a co-expression edge between the genes.
There are, however, caveats in interpreting our observations. First, note that, when constructing a co-expression network, only the degree of linear correlatedness of the gene expression values is considered. No reason exists, however, for thinking that non-linear correlations are not significant, which means that they should possibly be considered in the analysis. Secondly, two genes can get correlated simply by chance. Indeed, the distribution of the values of the correlation coefficient r (after Fisher's transformation) produced by chance have been proved to be Gaussian (Anderson, 2003). As a result, some co-expression edges might be established by simple chance, meaning that they do not represent any biological correlation. The latter is more and more probable as smaller the correlation value between the pair of genes is. Thirdly, the fact that microarray gene expression values result from averaging the gene expression values over a large number of cells could also distort the whole co-expression analysis. Fourthly, the networks that we compare to are known to be incomplete and may include false edges or interactions, which may affect our results. It should, however, be noted that this is a limitation of all methods that utilize or analyze existing networks. Lastly, we only considered co-expression networks based on pairwise correlations. Such co-expression networks involve few statistical assumptions and are therefore widely used in analysis of gene expression data. It would, however, be interesting to use gene expression networks constructed by other methods, such as mutual information-based or Gaussian graphical model-based methods. Mutual information constructions, however, often depend, especially when dealing with very high dimensional gene expression data, on the way of discretizing the continuous gene data or on the particular parametric assumption one makes on the distribution functions used (Qiu et al., 2009). Similarly, sparse Gaussian graphical models developed in recent years to analyze gene expression data in high dimensional settings (Li and Gui, 2006; Mainshausen and Buhlmann, 2006; Schafer and Strimmer, 2005a, b) still heavily depend on the procedures used and the tuning parameters chosen. In contrast, co-expression networks are based on unambiguous procedures which only need the estimation of pairwise correlations.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Lars J. Jensen, from the STRING Database, and Andrew Winter, from the BioGRID Database, for their helpful comments and indications on their respective databases.
Funding: NIH grants (R01ES009911 and R01CA127334).
Conflict of Interest: none declared.
REFERENCES
- Albert R, Barabási AL. Statistical mechanics of complex networks. Rev. Mod. Phys. 2002;74:47. [Google Scholar]
- Anderson TW. An Introduction to Multivariate Statistical Analysis. 3rd. Hoboken, NJ: John Wiley and Sons; 2003. [Google Scholar]
- Bhardwaj N, Lu H. Correlation between gene expression profiles and protein-protein interactions within and across genomes. Bioinformatics. 2005;21:2730–2738. doi: 10.1093/bioinformatics/bti398. [DOI] [PubMed] [Google Scholar]
- Breitkreutz B-J, et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res. 2008;36:D637–D640. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, et al. Genetic interactions between polymorphisms that affect gene expression in yeast. Nature. 2005;436:701–703. doi: 10.1038/nature03865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, et al. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani RJ. Monographs on Statistics and Applied Probability. Vol. 57. New York: Chapman and Hall; 1993. An Introduction to the Bootstrap. [Google Scholar]
- Ge H, et al. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat. Genet. 2001;49:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Harbison CT, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008;4:e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, et al. Relating whole-genome expression data with protein-protein interactions. Genome Res. 2002;12:37–46. doi: 10.1101/gr.205602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ, et al. STRING 8- a global view on protein in their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Gui J. Gradient directed regularization for sparse Gaussian concentration graphs, with applications to inference of genetic networks. Biostatistics. 2006;7:302–317. doi: 10.1093/biostatistics/kxj008. [DOI] [PubMed] [Google Scholar]
- Mainshausen N, Buhlmann P. High-dimensional graphs and variable selection with the lasso. Annals of Statistics. 2006;34:1436–1462. [Google Scholar]
- Mrowka R, et al. Does mapping reveal correlation between gene expression and protein-protein interaction? Nat. Genet. 2003;33:15–16. doi: 10.1038/ng0103-15. [DOI] [PubMed] [Google Scholar]
- Newman MEJ. The structure and function of complex networks. SIAM Review. 2003a;45:167–256. [Google Scholar]
- Newman MEJ. Mixing patterns in networks. Phys. Rev. E. 2003b;67:026126. doi: 10.1103/PhysRevE.67.026126. [DOI] [PubMed] [Google Scholar]
- Qiu P, et al. Fast calculation of pairwise mutual information for gene regulatory network reconstruction. Comput. Methods Programs Biomed. 2009;94:177–180. doi: 10.1016/j.cmpb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Schafer J, Strimmer K. An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics. 2005a;21:754–764. doi: 10.1093/bioinformatics/bti062. [DOI] [PubMed] [Google Scholar]
- Schafer J, Strimmer K. A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat. Appl. Genet. Mol. Biol. 2005b;4:32. doi: 10.2202/1544-6115.1175. [DOI] [PubMed] [Google Scholar]
- Steffen M, et al. Automated modelling of signal transduction networks. BMC Bioinformatics. 2002;3:34. doi: 10.1186/1471-2105-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, et al. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Vázquez A, et al. Large-scale topological and dynamical properties of the Internet. Phys. Rev. E. 2002;65:066130. doi: 10.1103/PhysRevE.65.066130. [DOI] [PubMed] [Google Scholar]
- von Mering C, Jensen LJ, et al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33:D433–D437. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts DJ, Strogatz SH. Collective dynamics of ‘small-world’ networks. Nature. 1998;393:409–410. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- Yan X, et al. A graph-based approach to systematically reconstruct human transcriptional regulatory modules. Bioinformatics (ISMB 2007) 2007;23:i577–i586. doi: 10.1093/bioinformatics/btm227. [DOI] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article 17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.