

Abstract
The high-affinity binding of tissue inhibitors of metalloproteinases (TIMPs) to matrix metalloproteinases (MMPs) is essential for regulation of the turnover of the extracellular matrix during development, wound healing, and progression of inflammatory diseases such as cancer, atherosclerosis, and arthritis. Bacterially expressed N-terminal inhibitory domains of TIMPs (N-TIMPs) have been used extensively for biochemical and biophysical study of interactions with MMPs. Titration of N-TIMP-1 expressed in E. coli indicates, however, that only about 42% of the protein is active as an MMP inhibitor. The separation of inactive from fully active N-TIMP-1 has been achieved both by MMP affinity and by high-resolution cation exchange chromatography at an appropriate pH, based on a slight difference of charge. Purification by cation exchange chromatography with a Mono S column enriches the active portion of N-TIMP-1 to > 95%, with Ki of 1.5 nM for MMP-12. Mass spectra reveal that the inactive form differs from active N-TIMP-1 in being N-terminally acetylated, underscoring the importance of the free α-NH2 of Cys1 for MMP inhibition. Nα-acetylation of the CTCVPP sequence broadens the N-terminal sequence motifs reported to be susceptible to α-amino acetylation by E. coli N-acetyl transferases.
Keywords: N-acetylation, MS/MS sequencing, MMP inhibitor
INTRODUCTION
The matrix metalloproteinases (MMPs) catalyze the breakdown of polypeptide components of the extracellular matrix and have important roles in tissue remodeling, wound healing, embryo implantation, cell migration, and shedding of cell surface proteins 1,2. These activities are tightly regulated in vivo by a family of endogenous protein inhibitors, the tissue inhibitors of metalloproteinases (TIMPs). Four mammalian TIMPs (TIMP-1 to TIMP-4) have been identified and can form high affinity 1:1 complexes with virtually all MMPs. Besides MMPs, some members of the distantly related disintegrin metalloproteinase (ADAM) and disintegrin metalloproteinase with thrombospondin type 1 motif (ADAMTS) families are also inhibited by certain TIMPs. A loss of balance between the TIMPs and their target proteases is linked to diseases such as cancer and arthritis 2,3.
TIMPs are slow, tight-binding inhibitors of the MMPs with Ki values typically in the sub- to low nanomolar range 3. The four mammalian TIMPs are two-domain proteins. The larger N-terminal domain of about 125-residues can be expressed separately and carries the MMP-inhibitory activity 2,3. TIMPs from some invertebrates lack the C-terminal domain 3. The inhibitory domain of TIMP has an OB fold, featuring a 5-stranded β-barrel shaped like a cone with two short helices packed on the exterior 4–6. In atomic-resolution structures of the MMP-3/TIMP-1 4,7, MMP-1/N-TIMP-1 8, MT1-MMP/TIMP-2 9, and MMP-13/TIMP-2 10 complexes, most of the contacts with the MMP are contributed by the N-terminal domain of the TIMP, specifically the N-terminus and loops joining beta-strands A with B, C with D, and E with F 4,7–10. N-terminal residues 1 to 5 insert into the active site of the MMPs, positioning the α-amino group and carbonyl oxygen of Cys1 to coordinate the catalytic zinc of the MMPs 4,8–10. Disruption of this part of the interaction site, either by addition of an alanine to the N-terminus 11,12 or carbamylation of the α-amino group 13, radically reduces the inhibitory activities of TIMPs for MMPs. However, the Ala extension mutation in N-TIMP-3 has little effect on ADAM17 inhibition 12. Myeloperoxidase generates HOCl that oxidizes Cys1 of TIMP-1 to inactivate it, both in vitro and in the bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome 14.
The isolated N-terminal domains of TIMPs (N-TIMPs), expressed in E. coli and folded in vitro, have been extensively mutated with the aim of introducing selectivity among MMPs and TACE to enhance the therapeutic potential of the engineered N-TIMPs 12,15–23. However, repeated calorimetric titrations of N-TIMP-1 (from multiple preparations) by the catalytic domain of MMP-3 (MMP-3(ΔC)) showed that 56 to 62% of N-TIMP-1 generated by this method is inactive 24. Nonetheless, N-TIMP-1 preparations consistently have sufficient actively MMP-inhibitory protein to characterize N-TIMP-1/MMP interactions 15,25–27, including the structures of N-TIMP-1/MMP complexes 7,8. Williamson and coworkers found a modified form of N-TIMP-2 with a 42.3 Da addition that was enriched to 35% of the N-TIMP-2 at the leading edge of the peak eluting from a Resource-S column. They hypothesized the modification to be N-acetylation, but did not comment on the activity of the acetylated form 28. 1.31 equivalents of N-TIMP-2 folded from E. coli inclusion bodies were required to inhibit one equivalent of MMP-13 28. This suggests that perhaps 24% of that particular purified N-TIMP-2 fraction may have been inactive. The inactive fraction of TIMP-4 folded from E. coli inclusion bodies contains forms with either a 42 Da addition consistent with acetylation or a methionine added to the N-terminal tryptic fragments 29. Recognition and removal of the inactive fraction of N-TIMPs is desirable in preparing TIMP variants targeted to inhibit individual MMPs or groups of MMPs for detailed biophysical experiments and for exogenous additions to treat disease states linked to unregulated MMP activities 16. We have achieved separation of bacterially expressed N-TIMP-1, folded in vitro, into active and inactive fractions by either MMP affinity or by high resolution cation exchange chromatography. We demonstrate by mass spectrometry that the inactive fraction of the inhibitor is acetylated at its N-terminus. This underscores the importance of the free α-amino group of Cys1 in TIMP interactions with MMPs. The modification also broadens the sequence patterns of Nα-acetylation identified in recombinant proteins expressed in E. coli.
MATERIALS AND METHODS
Expression, Purification and Folding of N-TIMP-1
E. coli BL21(DE3) was transformed with the expression plasmid pET-3a::Ntimp-1 25. Three hours after inducing expression with IPTG, the 3-L culture was harvested by centrifugation and frozen at −70 °C overnight. The frozen cell pellets were thawed, resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 1 mM EDTA and 100 mM NaCl), centrifuged, and resuspended again in lysis buffer. The suspension was subsequently passed through a French press three times at 1000 psi to break the bacterial cell wall. The inclusion bodies were harvested by centrifugation and washed three times with lysis buffer containing 0.5% Triton X-100. The protein was extracted from the inclusion bodies with 30 mL of 20 mM Tris-HCl, pH 8.5 containing 6 M guanidine-HCl and 10 mM DTT. The insoluble cell remnants were removed by centrifugation. The unfolded N-TIMP-1 was purified by gel filtration with S-300 and folded in vitro as described 25. After folding, precipitated protein was removed from the protein solution by filtration through glass wool. The soluble protein was concentrated and purified by CM-52 cation exchange chromatography 25. Fractions containing the folded N-TIMP-1 were identified by SDS-PAGE and pooled. The sources of other materials have been reported 16,30.
MMP-Affinity Purification of N-TIMP-1
The active component of N-TIMP-1 was isolated based on its affinity for MMP-3. N-TIMP-1 and wild-type MMP-3(ΔC) were mixed, and the complex separated from the unbound proteins by gel filtration with Superdex 75. The complex was dissociated by incubation at pH 4.5 with 1 mM EDTA and active N-TIMP-1 was isolated by cation exchange chromatography with CM-52. Inactive N-TIMP-1 was isolated by collecting the slowly migrating fractions from the Superdex 75 and separating the inactive N-TIMP-1 from MMP-3(ΔC) using CM-52. The active and inactive fractions of N-TIMP-1 were infused by electrospray on a Finnigan LCQ Classic Ion-Trap mass spectrometer.
Chromatographic Separation of Active from Inactive forms of N-TIMP-1
After purification by cation exchange chromatography with CM-52, N-TIMP-1 was dialyzed overnight against 15 volumes of 20 mM Bis-Tris-HCl, pH 5.5. The dialyzed protein solution was subsequently fractionated on a cation exchange Mono S HR 5/5 column (Amersham Biosciences) previously equilibrated with the same buffer using a Bio-Rad Biologic DuoFlow medium-pressure chromatography system. The protein was eluted with a linear salt gradient of 0–0.5 M NaCl over 60 min at a flow rate of 1 mL/min. Two slightly overlapping, similarly sized peaks were obtained. To compensate for the Mono S resolution being diminished by normal usage, the gradient was decreased to 0 to 0.25 M NaCl over 240 min. The activities of fractions were measured by fluorescence assay of the proteolytic activity of MMP-3(ΔC) against NFF-3 substrate, or alternatively of MMP-12(ΔC) against FS-6 substrate, as described below. Fractions from the second peak, containing the MMP-3 inhibitory activity, were pooled and titrated against MMP-3(ΔC).
Fluorescence Assays for TIMP Activity
Fluorescence assays for TIMP activity were carried out with either a Perkin Elmer LS50B luminescence spectrometer or an SLM Aminco 8100 spectrofluorometer with PC1 ™ Photon counting upgrade (ISS Inc.,Champaign, IL, USA). Proteins were dissolved in TNC buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM CaCl2, and 0.02% Brij-35) which was used in all assays. For determination of N-TIMP-1 activity, 1 nM MMP-3(ΔC) was pre-incubated with or without different concentrations of inhibitor for 4 hours at 37 °C and assayed at 37 °C using NFF-3 substrate (Mca-Arg-Pro-Lys-Pro-Val-Glu-Nva-Trp-Arg-Lys(Dnp)-NH2) 31 at a final concentration of 1.5 μM. C-terminally truncated MMP-3 [MMP-3(ΔC)] and pET-3a-Mmp-1cd encoding MMP-1(ΔC) were kindly provided by Dr. H. Nagase, Kennedy Institute of Rheumatology, Imperial College, U.K. Reaction velocities were measured as the slope of the linear portion of the fluorescence curve. The percentage of residual MMP activity was calculated by dividing the velocities measured with inhibitor by the velocities measured without inhibitor (v/v0).
For titration of N-TIMP-1, various concentrations of the inhibitor were incubated with MMP-3(ΔC) (300 nM) for 4 hours at 37 °C, diluted 300-fold with TNC buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM CaCl2, and 0.02% Brij 35) and immediately assayed with 1.5 μM NFF-3 substrate as described above. Residual MMP activity (%) was calculated as described above and plotted against molar ratio of TIMP/MMP (0 to 4 in this case). The stoichiometry was determined by linear regression analysis of the first five data points (TIMP/MMP between 0 and 1).
Active site titration of MMP-12(ΔC) with N-TIMP-1 proceeded similarly, but with the following minor differences. Human MMP-12(ΔC) was prepared as described 32,33. The exact concentration of its active sites was determined by titration 34 using the tight-binding inhibitor known as galardin or GM6001 35 (EMD Biosciences). Mono S-purified N-TIMP-1 was pre-incubated with 55.6 nM MMP-12(ΔC) at least 1.5 hours at 32 °C before kinetic assay. MMP-12(ΔC) activity was monitored at 32 °C with FS-6, the soluble form of Knight’s FRET-labeled peptide at 4 μM 36. Initial velocities V as a function of N-TIMP-1 concentration I were fitted by non-linear regression using Origin 7.5 (Microcal, Northampton, MA) to the Morrison equation for tight inhibition 34,37 with the slight modification of replacing inhibitor concentration I by the product N*I, where N is the proportion of inhibitor that is active:
| eq 1 |
Since the active enzyme concentration Eo was already known by active site titration, N could then be fitted, along with Ki, to determine how much of the N-TIMP-1 was active after purification.
Mass Spectrometry
Intact proteins and protein digests were analyzed by nanospray quadrupole time of flight mass spectrometry (QqTOF MS) with an Applied Biosystems/MDS Sciex (Foster City, CA, USA) API QSTAR Pulsar instrument fitted with a Proxeon (Odense, Denmark) nanospray source. A stable spray was achieved at 800 V in the presence of nitrogen curtain gas for samples dissolved or diluted into 60/39/1 (v/v/v) acetonitrile/water/88% formic acid. Positive ion spectra were acquired in the profile MCA mode at a pulser frequency of 6.99 kHz. Collision-induced dissociation spectra (MS/MS) were acquired for peptides selected with a 3-amu wide precursor ion window. Nitrogen collision gas pressures and collision energy settings were adjusted to provide fragment ions across the mass range of 50–2000 Da. Instrument calibration was based on masses of MS/MS fragment ions from the standard peptide [Glu1]-Fibrinopeptide B. Intact protein spectra were deconvoluted with the ABI Bioanalyst Bayesian Reconstruct program using a 0.1 Da mass step size and 20 iterations.
Samples of N-TIMP-1 for MS analysis were desalted with C4 ZipTips (Millipore Corp., Bedford, MA, USA) that had been prepared by wetting with acetonitrile, washing with elution solvent, and equilibrating in loading solvent. Proteins were dissolved in 99/1 (v/v) water/88% formic acid and loaded onto the ZipTips. After sample loading, the ZipTips were washed with the loading solvent to remove contaminating salts. The adsorbed proteins were then eluted with 60/39/1 (v/v/v) acetonitrile/water/88% formic acid for analysis.
For MS/MS sequencing, N-TIMP-1 was reduced with DTT, alkylated with iodoacetamide to carboxamidomethylate cysteine residues, and then digested with sequencing grade trypsin (Promega, modified porcine, TPCK-treated). Trypsin digests of the proteins were also desalted using C18 ZipTips.
RESULTS
Separation of Active N-TIMP-1 from Inactive Form
Consistently, 38 to 44% of the N-TIMP-1 folded from inclusion bodies from multiple cultures of the E. coli expression host was found by titration calorimetry to be competent to bind to MMP-3(ΔC) 24. Electrospray mass spectra of the N-TIMP-1 indicated the largest peak to be at the molecular weight predicted for unmodified N-TIMP-1, heterogeneity featuring a second largest peak with a 42 Da addition, and other peaks of increased molecular weight. Integration of the peaks indicated the largest and unmodified N-TIMP-1 peak to be 42.5% of the sum of the integrated peaks, correlating with the 38 to 44% range of active N-TIMP-1 observed by titration calorimetry. In order to distinguish forms of N-TIMP-1 by activity as MMP inhibitors, we separated N-TIMP-1 that bound MMP-3(ΔC) from unbound N-TIMP-1. Complexes of N-TIMP-1 with MMP-3(ΔC) were separated from free N-TIMP-1 mostly incompetent to bind an MMP using Superdex 75 chromatography. Active N-TIMP-1 separated from MMP-3(ΔC) exhibited a predominant peak in the electrospray mass spectrum near the molecular weight predicted from the sequence (Fig. 1A). In the electrospray mass spectrum of the fraction of N-TIMP-1 mostly unable to bind MMP-3, peaks of higher molecular weight are observed, the largest of which was 42 Da larger in mass than the MMP-3-inhibitory fraction (Fig. 1B). This strongly suggests that the +42 Da modification is inactivating.
Figure 1.
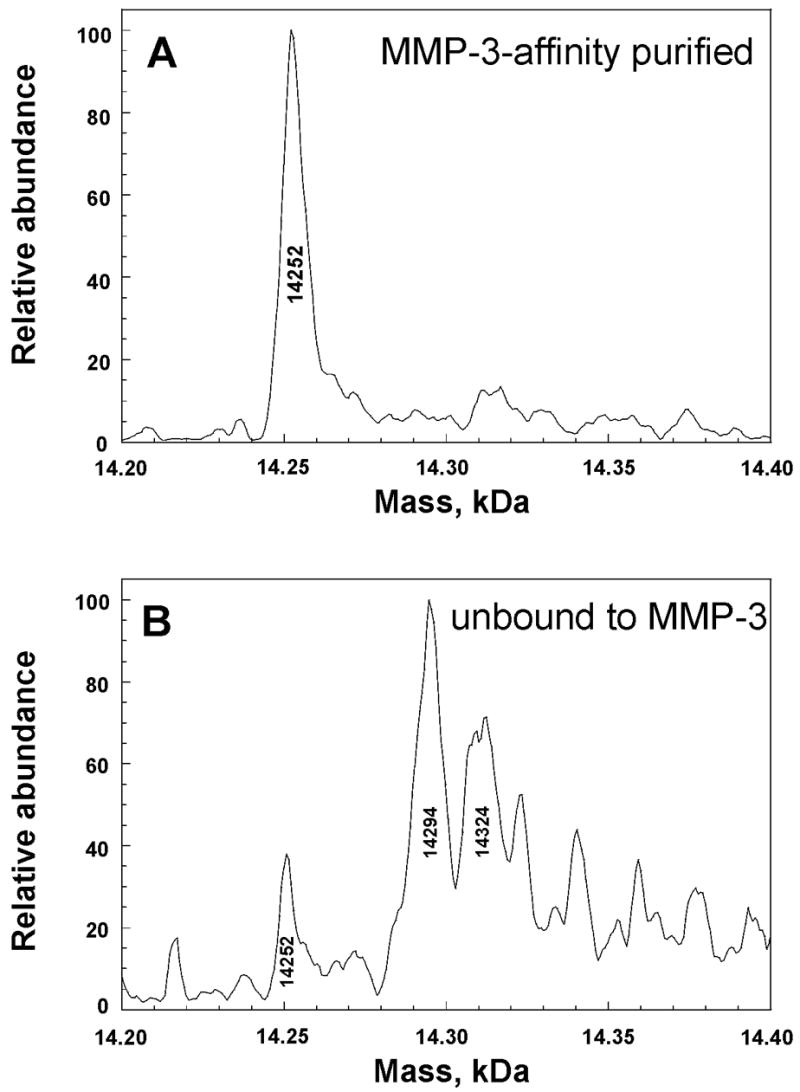
Electrospray mass spectra of MMP-3-affinity purified N-TIMP-1 (A) and N-TIMP-1 not bound to an MMP (B), separated by Superdex 75 size exclusion chromatography. Each fraction was infused by electrospray on a Finnigan LCQ Classic Ion-Trap mass spectrometer. Higher apparent masses than in Fig. 5 are attributed to lower mass accuracy of the Finnigan LCQ.
In an effort to separate the forms of N-TIMP-1 more easily than by affinity purification, the material from CM-52 purification was fractionated in buffers of different pH using a new medium-pressure, high resolution Mono S cation exchange column. Elution with a linear salt gradient at pH 5.5 separated the protein into two peaks (Fig. 2). The first contained little inhibitory activity for MMP-3(ΔC), whereas the second peak was highly active. Both peaks were collected, and titration with MMP-3(ΔC) showed that 85% of the N-TIMP-1 in the second peak is active by linear extrapolation, a substantial improvement over the 42% activity before purification (Fig. 3). Both peaks showed a single band on SDS gel electrophoresis with indistinguishable mobility consistent with the predicted size of N-TIMP-1.
Figure 2.

Chromatogram of the separation of N-TIMP-1 by high-resolution cation exchange chromatography. N-TIMP-1 (10 mg) purified by cation exchange chromatography with CM-52 was loaded onto a new Mono S HR 5/5 cation exchange column pre-equilibrated with buffer A (20 mM Bis Tris-HCl, pH 5.5). The column was then washed with 10 mL buffer A, and bound protein was eluted with a linear gradient of 0–100% buffer B (20 mM Bis Tris-HCl, pH 5.5, 0.5 M NaCl) over 60 minutes at a flow rate of 1mL/min. The two components are labeled peaks 1 and 2, respectively.
Figure 3.
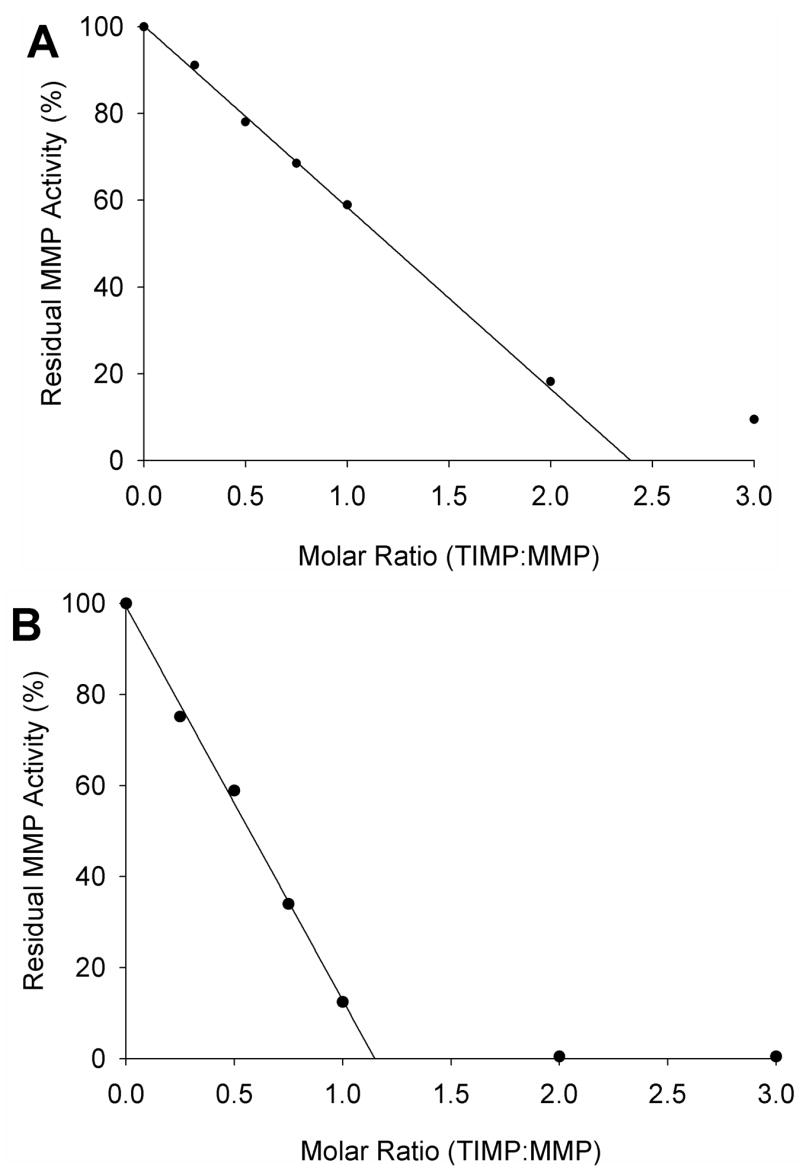
Titration of MMP-3(ΔC) with N-TIMP-1 before (A) and after (B) purification (peak 2 from Fig. 1). MMP-3(ΔC) (300 nM) was pre-incubated with 0 to 900 nM N-TIMP-1 for 4 hr at 37°C, diluted 300-fold with TNC buffer and immediately assayed with 1.5 μM NFF-3 substrate as described in Materials and Methods. Residual enzyme activity was plotted against the molar ratio of inhibitor/enzyme, and analyzed by linear regression. The fraction of active inhibitor was calculated as the reciprocal of the X-intercept.
Purification of N-TIMP-1 and active site titration was repeated in order to measure both its Ki for an MMP and its purity attainable, both key measures of the quality of the purified N-TIMP-1. A Mono S HR 5/5 column was used that had experienced repeated, normal use. To compensate for the diminished resolution routinely associated with more than a few uses of a Mono S column, the gradient was made shallower. About 40% of the trailing half of the second peak was collected (Fig. 4A) and titrated into MMP-12(ΔC) known to be 55.6 nM by active site titration (Fig. 4B). Simple linear extrapolation of the linear phase of the titration suggested 58.5 nM or 1.05 molar equivalents of N-TIMP-1 to be necessary to titrate the active MMP-12(ΔC) present (Fig. 4A). The MMP-12 velocities as a function of purified [N-TIMP-1] were nonlinearly fitted to eq. 1, the Morrison tight inhibition equation 34,37 modified in order to fit the proportion N of the inhibitor molecules that are active. This suggests 96 ± 3% of the purified N-TIMP-1 to be fully active in inhibiting MMP-12(ΔC), with Ki of approximately 1.5 nM (Fig. 4B). Within the 5% uncertainty of the BioRad protein assay of N-TIMP-1 concentration, this is equivalent to this portion of the second N-TIMP-1 peak being essentially 100% active.
Figure 4.

Purification of N-TIMP-1 by cation exchange chromatography (A) and demonstration of its high purity and activity by titration of MMP-12(ΔC) (B). Panel (A): The separation was performed similarly as in Fig. 2, except that the Mono S HR 5/5 cation exchange had experienced normal, repeated usage and that the gradient was lengthened to 240 min and the maximum [NaCl] was decreased to 0.25 M. The portion of peak 2 marked gray was collected for active site titration. Panel (B): The gray portion of peak 2 was titrated into MMP-12(ΔC) having 55.6 nM intact active sites by titration with GM6001. MMP-12(ΔC)’s initial velocities as a function of total, purified [N-TIMP-1] were fitted to the slightly modified Morrison tight-binding inhibition equation shown (eq. 1), in order to determine N, the proportion of the inhibitor N-TIMP-1 that is active, along with Ki. The uncertainties listed are the fitting errors. Experimental uncertainties may be 5% or more.
The lack of MMP-inhibitory activity of the protein in the first peak from Mono S chromatography could conceivably arise from either misfolding or inactivating covalent modification. However, the homogeneity of the NMR spectra of N-TIMP-1 samples that contain both the active and inactive components 6,38–40 implies that a misfolded form of N-TIMP-1 is not present. Rather, the fact that the two components can be resolved by chromatography at pH 5.5 is more consistent with a small difference in charge between the active and inactive fractions. These observations suggested the hypothesis that the inactivation of N-TIMPs could result from a chemical modification that increases molecular mass by 42 Da, and also reduces the overall positive charge of the N-TIMP-1 molecule.
QqTOF MS Analysis of Inactivating Modification of +42 Da
To test this hypothesis and determine the type and location of the adduct that abolishes the inhibitory activity of N-TIMP-1, both the inactive and active peaks of N-TIMP-1 were desalted and examined by MS analyses. The reconstructed mass spectrum obtained from the nanospray QqTOF MS multiple-charge ion spectrum shows that the major component of the 85% active fraction has a molecular weight of 14245.9 Da. A secondary peak at 14287.6 Da, and lesser peaks at 14263.2 and 14344.4 Da (Fig. 5A) were also present. The uncertainty in mass was determined to be ± 1 Da using the mass standard of cytochrome C (equine heart, 12360.1 Da). The mass of the principal peak of 14245.9 Da agrees, within this uncertainty, with the mass of 14246.27 Da calculated from the sequence of N-TIMP-1 assuming 3 disulfide bridges. The minor species at 14263.2 Da (Fig. 5A) and at 14304.2 Da (Fig. 5B) appear to be oxidized forms (+16.4 to +17.3 Da) of the two most abundant species at 14245.9 and at 14287.8 Da, respectively. The inactive fraction is composed mainly of the 14287.8 Da species (Fig. 5B), with the same mass within error as the secondary component present in the active fraction.
Figure 5.
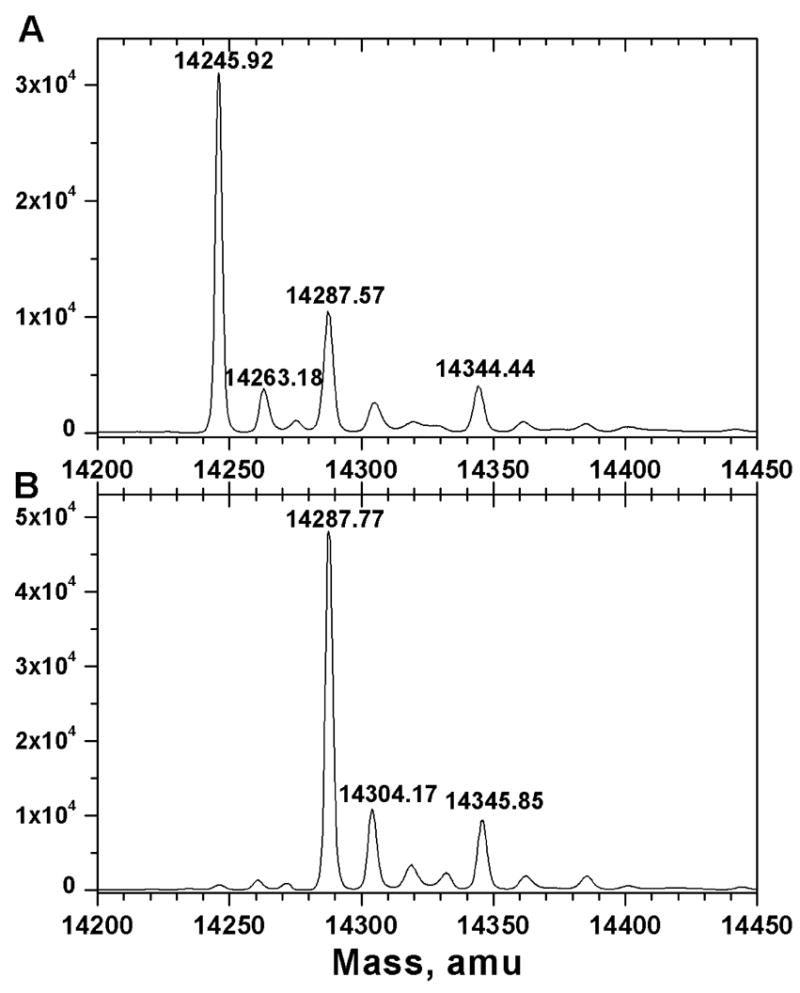
Reconstructed nanospray QqTOF MS spectra of (A) active fraction of N-TIMP-1 and (B) inactive N-TIMP-1. The samples were separated as in Figure 2 and desalted by dialysis against water and use of C4 ZipTips.
Initially, the nature of the approximately 42 Da difference between the predominant components of the active and inactive fractions was uncertain. Acetylation (+42.04 Da), trimethylation (+42.08 Da), or carbamylation (+43.02 Da) were possible candidates for the post-translational modification of the inactive fraction. Since trimethylation would not introduce negative charge to account for the faster elution of inactive peak 1 from the high-resolution cation exchange column (Fig. 2), it is unlikely to be the modification. Carbamylation is no risk when folding from guanidinium HCl solution by the method developed for N-TIMP-2 28. To check for any carbamylation of N-TIMP-1, we compared folding it from urea solution versus folding it from guanidinium HCl solution and found similar proportions of inactive N-TIMP-1, regardless of which denaturant and folding method was used. This implies that carbamylation of the α-amino group 13 by ammonium cyanate formed from urea is not the cause of partial inactivation. Therefore, it appears that the inactivating modification of N-TIMP-1 occurs during expression in E. coli. Since engineered substitutions of the α-amino group of N-TIMP-1 21, TIMP-2 11,13, and N-TIMP-3 12 are inactivating and oxidative modification of Cys1 of TIMP-1 is also inactivating 14, the modification was expected to involve the α-amino group. The identification of Nα-acetylation in a handful of recombinant proteins expressed in E. coli (Table 1) further suggested that the inactivating +42 Da covalent modification could be Nα-acetylation of N-TIMP-1.
Table I.
Proteins Nα-acetylated in E. coli
| N-acetylated protein | SequenceA | |
|---|---|---|
| Recombinant | Human N-TIMP-1 | CTCVPP |
| Human interferon A 50 | CDLPQT | |
| Human interferon γ51 | CYCQDP | |
| Human TIMP-4 29 | CSCAPA | |
| Human N-TIMP-2 B, 28 | CSCSPV | |
| Rat stathmin-like domains RB3, RB3′ 52 | ADMEVI | |
| Yersinia F1-V 49 | ADLTAS | |
| Human prothymosin α 53 | SDAAVD | |
| Eglin c 54 | TEFGLE | |
| Native E. coli | Ribosomal S5 47 | AHIEKQ |
| Ribosomal S18 47 | ARYFRR | |
| Ribosomal L12 46 | SITKDQ | |
| Elongation factor Tu 48 | SKEKFE | |
| SecB B, 55 (Randall and Smith, personal communication) | SEQNNT |
Confirmation of Nα-Acetylation as the Inactivating Modification by MS/MS Sequencing
To identify the site of the inactivating post-translational modification and to differentiate acetylation from the alternative modification of carbamylation (+43 Da), tryptic digests of the reduced and carboxamidomethylated active and inactive fractions of N-TIMP-1 were analyzed by nanospray QqTOF MS. A double-charge ion with the expected m/z (1186.5) for the unmodified N-terminal tryptic fragment, CTCVPPHPQTAFCNSDLVIR (monoisotopic [M+H]+ 2372.0950 Da), is enriched in the active N-TIMP-1 sample. However, a double-charge ion with the expected m/z (1207.6) of the acetylated N-terminal tryptic fragment, containing residues 1 to 20, is observed at substantially higher signal intensity in the inactive N-TIMP-1 sample. Furthermore, the relative abundance of these two peptides (and other charge states thereof) constitutes the most significant difference in the MS spectra for the digests of the two samples. No carbamylated tryptic fragments are observed in either sample. No peptides in either of the digests experienced blockage of trypsin cleavage by internal trimethylated lysine or arginine residues.
Collision-induced dissociation mass spectra (MS/MS) obtained for the ions at 1186.5 Da and 1207.6 Da confirm that they are from the respective unmodified and acetylated N-terminal tryptic peptides, respectively. The fragment ions (< 0.03 Da mass uncertainty) are assigned to the N-terminal ions b2 through b9 and b11 through b14 (nomenclature of ref 41) of the unmodified N-terminal tryptic peptide (parent ion 1186.5 Da) of the active protein sample. (For simplicity, only the b-ions of the low mass region are illustrated in Fig. 6.) These fragment ions are not observed in the MS/MS of the 1207.6 Da double-charge peptide ion found in the digest of the inactive protein sample. Instead, fragment ions corresponding to acetylated b-ions, that is, b-ions + 42.01 Da, are observed from b2 through b7, b9 through b12, b14, and b15. Furthermore, all of the expected y′-ions, including the penultimate y′19, are present without modification in the spectrum (not shown). Therefore the 42.0 Da addition, diagnostic of acetylation, is localized on Cys1.
Figure 6.
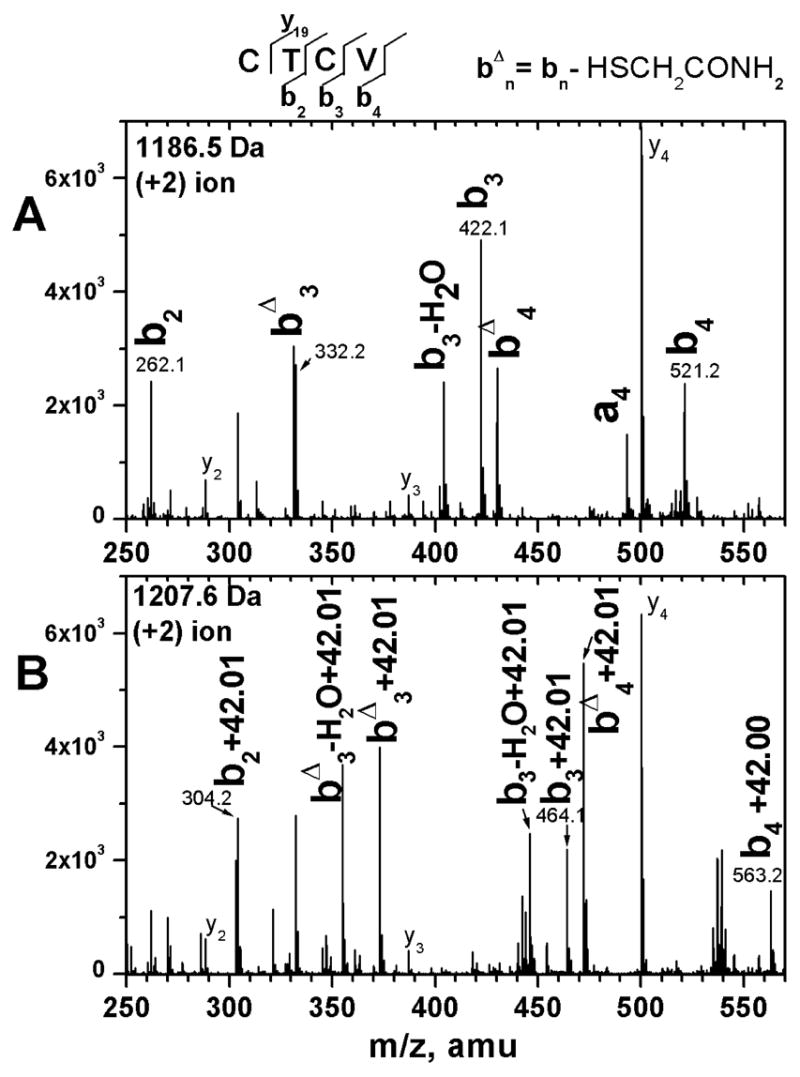
Assigned collision-induced dissociation MS/MS spectra of the N-terminal tryptic fragments of (A) active N-TIMP-1 and (B) inactive N-TIMP-1. The N-terminal tryptic peptide fragment of N-TIMP-1 has the sequence: CTCVPPHPQTAFCNSDLVIR. The low mass region of the MS/MS spectrum for the double-charged ion in the digest of the active N-TIMP-1 at m/z 1186.55 Da (unmodified peptide) is shown in panel A. The systematically different MS/MS spectrum for the double-charge ion in the digest of the inactive N-TIMP-1 at m/z 1207.6 Da (predicted to be the acetylated N-terminal tryptic peptide) is shown in panel B. Assigned b-ions, containing the intact or modified α-amino group, are labeled in bold. The peak at 332.2 Da is assigned to the internal fragment with sequence PPH or PHP. The peak assigned to the a4 ion could alternatively be the internal fragment with sequence AFCN. The superscript Δ refers to loss of mercaptoacetamide.
Other Modifications
In the tryptic digests of both active and inactivated N-TIMP-1, both unoxidized and oxidized forms of the fragment containing residues 60–75 (FVYTPAMESVCGYFHR) are observed. (This is the only methionine-containing fragment observed in the MS spectra of the digests.) Oxidation of Met66 accounts for the minor, oxidized components in the mass spectra of intact N-TIMP-1 (Fig. 3). The carboxamidomethylated cysteine-containing tryptic peptides sequenced exhibited 91 Da loss of mercaptoacetamide (Fig. 6). Loss of mercaptoacetamide from carboxamidomethylated cysteine residues has been observed previously 42.
DISCUSSION
Our results show that the inactivation of N-TIMP-1 as an inhibitor of MMPs correlates with α-N-acetylation of Cys1. Before this modification can occur, the initiating N-formyl methionine (fMet) has to be deformylated and the Met removed by methionine aminopeptidase 43. The preference of E. coli Met aminopeptidase for small residues, including cysteine, in the penultimate position facilitates the removal 44. Subsequent Nα-acetylation inactivates the MMP inhibitory activity of N-TIMP-1. It appears that many modifications of the Cys1 α-amino group can interfere in MMP inhibition, such as Nα-carbamylation ofTIMP-2 11, addition of alanine to the N-terminus of TIMP-2 or N-TIMP-3 11,12, or oxidation of Cys1 of TIMP-1 14. The apparent partial Nα-acetylation of N-TIMP-2 28 is likely to inactivate it as well. Nα-acetylation or N-terminal methionine is found among the inactive forms of TIMP-4 from E. coli 29. E. coli-expressed N-TIMP-3 elutes as a single peak from cation exchange chromatography in which the later fractions of the peak are fully active while the earlier fractions are only partially active 38. Since this elution pattern resembles N-TIMP-2 elution from cation exchange column in which the leading edge of the peak is enriched in the + 42 Da N-acetylated form 28, N-TIMP-3 can be hypothesized to be Nα-acetylated as well. Thus, Nα-acetylation appears to disrupt MMP-inhibitory activities of all four TIMP sequences expressed in E. coli. The ability of Mono S cation exchange chromatography to resolve acetylated from non-acetylated N-TIMP-1 may prove useful for enriching the specific activity of other TIMPs expressed in E. coli.
N-acetylation in E. coli
Acetylation of the N-terminal α-amino group is widespread in mammalian and yeast proteomes, but rare in E. coli where only three of 810 endogenous proteins examined were acetylated 45. The three reported to be acetylated are ribosomal S18, S5, and L12 proteins, with N-terminal sequences beginning AH, AR, and SI, respectively 46,47 (Table 1). A fourth Nα-acetylated E. coli protein is elongation factor EF-Tu with N-terminal sequence of SK 48. This N-terminal A/S motif is similar to that of eukaryotic substrates of the NatA N-acetyl transferase 45,49. The side chain of the second residue is bulky in most of these N-acetylated E. coli proteins (Table 1). Among recombinant proteins expressed in E. coli, there have been reports of partial N-acetylation of Cys and Thr in addition to Ala and Ser (Table 1). The second residue of each acetylated recombinant protein tends to be smaller than those in native E. coli proteins (Table 1). This second residue of N-acetylated recombinant proteins in E. coli usually has a side chain carboxyl group, suggestive of a specificity similar to that of yeast NatA’ N-acetyl transferase 45,49. Nα-acetylation of N-TIMP-1 diversifies the sequence trend with a β-branched Thr occupying position 2. Moreover, N-TIMP-1, N-TIMP-2, interferon A, and interferon differ from the other Nα-acetylated proteins in having Cys at the N-terminus (Table 1).
The removal of fMet from N-TIMP-2 was proposed to occur rapidly (or cotranslationally) before the N-TIMP aggregates into insoluble inclusion bodies to prevent access by methionine aminopeptidase 28. The ensuing Nα-acetylation of TIMPs by the N-acetyl transferase likewise seems more likely to occur in the initial window of solubility of newly expressed N-TIMP molecules before they aggregate to form inclusion bodies.
CONCLUSIONS
The active and inactive fractions of bacterially expressed N-TIMP-1 are separable by high resolution cation exchange chromatography with a Mono S column, or with more effort by affinity for an activated MMP. The purification of the active form is an advantage for in vitro assay of MMP inhibition and potential administration in vivo. Mass spectral evidence for Nα-acetylation of N-TIMP-1 correlates with its inactivation as an MMP inhibitor. This inactivation suggests that the apparent partial Nα-acetylation of bacterially expressed N-TIMP-2 28 should also be inactivating. However, based on the effects of other N-terminal modifications, N-acetylation is not expected to inactivate N-TIMP-3 as an inhibitor of ADAM-17 (TACE) 12. The partial acetylations of the α-amino groups of the CTCVPP, CSCSPV, CTCSPS, and CSCAPA N-terminal sequences of N-TIMP-1, -2, -3, and -4, respectively, broaden the array of N-terminal sequence motifs that may make recombinant proteins vulnerable to the N-acetylation that is otherwise unusual in E. coli.
Acknowledgments
The authors gratefully acknowledge The Charles Gehrke Proteomics Center at the University of Missouri for mass spectrometry measurements, the Washington University Mass Spectrometry Resource (supported by NIH grant P41RR0954) for spectra on the Finnigan LCQ, Valentyna Semenchenko for early preparations, and Prof. Scott Peck for critically reading the manuscript.
Grant sponsor: NIH grants R01GM57289 (to SRV) and R01 AR40994 (to KB)
The abbreviations used are
- MMP
matrix metalloproteinase
- MMP-3(ΔC)
stromelysin 1 catalytic domain
- TIMP
tissue inhibitor of metalloproteinase
- N-TIMP
N-terminal inhibitory domain of TIMP
- ADAM
a disintegrin and metalloproteinase
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin domains
- TACE
tumor necrosis factor a converting enzyme
- Ki
apparent inhibition constant
References
- 1.McCawley LJ, Matrisian LM. Curr Opin Cell Biol. 2001;13:534–540. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- 2.Lee MH, Murphy G. J Cell Sci. 2004;117:4015–4016. doi: 10.1242/jcs.01223. [DOI] [PubMed] [Google Scholar]
- 3.Brew K, Dinakarpandian D, Nagase H. Biochim Biophys Acta. 2000;1477:267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- 4.Gomis-Ruth FX, Maskos K, Betz M, Bergner A, Huber R, Suzuki K, Yoshida N, Nagase H, Brew K, Bourenkov GP, Bartunik H, Bode W. Nature. 1997;389:77–81. doi: 10.1038/37995. [DOI] [PubMed] [Google Scholar]
- 5.Muskett FW, Frenkiel TA, Feeney J, Freedman RB, Carr MD, Williamson RA. J Biol Chem. 1998;273:21736–21743. doi: 10.1074/jbc.273.34.21736. [DOI] [PubMed] [Google Scholar]
- 6.Wu B, Arumugam S, Gao G, Lee GI, Semenchenko V, Huang W, Brew K, Van Doren SR. J Mol Biol. 2000;295:257–268. doi: 10.1006/jmbi.1999.3362. [DOI] [PubMed] [Google Scholar]
- 7.Arumugam S, Van Doren SR. Biochemistry. 2003;42:7950–7958. doi: 10.1021/bi034545s. [DOI] [PubMed] [Google Scholar]
- 8.Iyer S, Wei S, Brew K, Acharya KR. J Biol Chem. 2007;282:364–371. doi: 10.1074/jbc.M607625200. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Catalan C, Bode W, Huber R, Turk D, Calvete JJ, Lichte A, Tschesche H, Maskos K. EMBO J. 1998;17:5238–5248. doi: 10.1093/emboj/17.17.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maskos K, Lang R, Tschesche H, Bode W. J Mol Biol. 2007;366:1222–1231. doi: 10.1016/j.jmb.2006.11.072. [DOI] [PubMed] [Google Scholar]
- 11.Wingfield PT, Sax JK, Stahl SJ, Kaufman J, Palmer I, Chung V, Corcoran ML, Kleiner DE, Stetler-Stevenson WG. J Biol Chem. 1999;274:21362–21368. doi: 10.1074/jbc.274.30.21362. [DOI] [PubMed] [Google Scholar]
- 12.Wei S, Kashiwagi M, Kota S, Xie Z, Nagase H, Brew K. J Biol Chem. 2005;280:32877–32882. doi: 10.1074/jbc.C500220200. [DOI] [PubMed] [Google Scholar]
- 13.Higashi S, Miyazaki K. J Biol Chem. 1999;274:10497–10504. doi: 10.1074/jbc.274.15.10497. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Rosen H, Madtes DK, Shao B, Martin TR, Heinecke JW, Fu X. J Biol Chem. 2007;282:31826–31834. doi: 10.1074/jbc.M704894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang W, Meng Q, Suzuki K, Nagase H, Brew K. J Biol Chem. 1997;272:22086–22091. doi: 10.1074/jbc.272.35.22086. [DOI] [PubMed] [Google Scholar]
- 16.Wei S, Chen Y, Chung L, Nagase H, Brew K. J Biol Chem. 2003;278:9831–9834. doi: 10.1074/jbc.M211793200. [DOI] [PubMed] [Google Scholar]
- 17.Butler GS, Hutton M, Wattam BA, Williamson RA, Knauper V, Willenbrock F, Murphy G. J Biol Chem. 1999;274:20391–20396. doi: 10.1074/jbc.274.29.20391. [DOI] [PubMed] [Google Scholar]
- 18.Lee MH, Rapti M, Murphy G. J Biol Chem. 2003;278:40224–40230. doi: 10.1074/jbc.M305678200. [DOI] [PubMed] [Google Scholar]
- 19.Lee MH, Rapti M, Knauper V, Murphy G. J Biol Chem. 2004;279:17562–17569. doi: 10.1074/jbc.M312589200. [DOI] [PubMed] [Google Scholar]
- 20.Lee MH, Rapti M, Murphy G. J Biol Chem. 2005;280:15967–15975. doi: 10.1074/jbc.M500897200. [DOI] [PubMed] [Google Scholar]
- 21.Hamze AB, Wei S, Bahudhanapati H, Kota S, Acharya KR, Brew K. Protein Sci. 2007;16:1905–1913. doi: 10.1110/ps.072978507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee MH, Rapti M, Murphy G. J Biol Chem. 2004;279:45121–45129. doi: 10.1074/jbc.M406611200. [DOI] [PubMed] [Google Scholar]
- 23.Rapti M, Knauper V, Murphy G, Williamson RA. J Biol Chem. 2006;281:23386–23394. doi: 10.1074/jbc.M604423200. [DOI] [PubMed] [Google Scholar]
- 24.Arumugam S, Gao G, Patton BL, Semenchenko V, Brew K, Van Doren SR. Journal of Molecular Biology. 2003;327:719–734. doi: 10.1016/s0022-2836(03)00180-3. [DOI] [PubMed] [Google Scholar]
- 25.Huang W, Suzuki K, Nagase H, Arumugam S, Van Doren SR, Brew K. FEBS Lett. 1996;384:155–161. doi: 10.1016/0014-5793(96)00304-3. [DOI] [PubMed] [Google Scholar]
- 26.Arumugam S, Hemme CL, Yoshida N, Suzuki K, Nagase H, Berjanskii M, Wu B, Van Doren SR. Biochemistry. 1998;37:9650–9657. doi: 10.1021/bi980128h. [DOI] [PubMed] [Google Scholar]
- 27.Arumugam S, Gao G, Patton BL, Semenchenko V, Brew K, Van Doren SR. J Mol Biol. 2003;327:719–734. doi: 10.1016/s0022-2836(03)00180-3. [DOI] [PubMed] [Google Scholar]
- 28.Williamson RA, Natalia D, Gee CK, Murphy G, Carr MD, Freedman RB. Eur J Biochem. 1996;241:476–483. doi: 10.1111/j.1432-1033.1996.00476.x. [DOI] [PubMed] [Google Scholar]
- 29.Troeberg L, Tanaka M, Wait R, Shi YE, Brew K, Nagase H. Biochemistry. 2002;41:15025–15035. doi: 10.1021/bi026454l. [DOI] [PubMed] [Google Scholar]
- 30.Wei S, Xie Z, Filenova E, Brew K. Biochemistry. 2003;42:12200–12207. doi: 10.1021/bi035358x. [DOI] [PubMed] [Google Scholar]
- 31.Nagase H, Fields CG, Fields GB. J Biol Chem. 1994;269:20952–20957. [PubMed] [Google Scholar]
- 32.Fu JY, Lyga A, Shi H, Blue ML, Dixon B, Chen D. Protein Expr Purif. 2001;21:268–274. doi: 10.1006/prep.2000.1376. [DOI] [PubMed] [Google Scholar]
- 33.Palmier MO, Van Doren SR. Anal Biochem. 2007;371:43–51. doi: 10.1016/j.ab.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knight CG. Methods Enzymol. 1995;248:85–101. doi: 10.1016/0076-6879(95)48008-0. [DOI] [PubMed] [Google Scholar]
- 35.Galardy RE, Grobelny D, Foellmer HG, Fernandez LA. Cancer Res. 1994;54:4715–4718. [PubMed] [Google Scholar]
- 36.Neumann U, Kubota H, Frei K, Ganu V, Leppert D. Anal Biochem. 2004;328:166–173. doi: 10.1016/j.ab.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 37.Williams JW, Morrison JF. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- 38.Kashiwagi M, Tortorella M, Nagase H, Brew K. J Biol Chem. 2001;276:12501–12504. doi: 10.1074/jbc.C000848200. [DOI] [PubMed] [Google Scholar]
- 39.Wu B, Arumugam S, Huang W, Brew K, Van Doren SR. Journal of Biomolecular NMR. 1999;14:289–290. doi: 10.1023/a:1008310807946. [DOI] [PubMed] [Google Scholar]
- 40.Gao G, Semenchenko V, Arumugam S, Van Doren SR. J Mol Biol. 2000;301:537–552. doi: 10.1006/jmbi.2000.3976. [DOI] [PubMed] [Google Scholar]
- 41.Biemann K. Biomed Environ Mass Spectrom. 1988;16:99–111. doi: 10.1002/bms.1200160119. [DOI] [PubMed] [Google Scholar]
- 42.Wirth U, Muller D, Schindler P, Lange J, van Oostrum J. Proteomics. 2002;2:1445–1451. doi: 10.1002/1615-9861(200210)2:10<1445::AID-PROT1445>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 43.Arfin SM, Kendall RL, Hall L, Weaver LH, Stewart AE, Matthews BW, Bradshaw RA. Proc Natl Acad Sci U S A. 1995;92:7714–7718. doi: 10.1073/pnas.92.17.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirel PH, Schmitter MJ, Dessen P, Fayat G, Blanquet S. Proc Natl Acad Sci U S A. 1989;86:8247–8251. doi: 10.1073/pnas.86.21.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polevoda B, Sherman F. J Mol Biol. 2003;325:595–622. doi: 10.1016/s0022-2836(02)01269-x. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka S, Matsushita Y, Yoshikawa A, Isono K. Mol Gen Genet. 1989;217:289–293. doi: 10.1007/BF02464895. [DOI] [PubMed] [Google Scholar]
- 47.Yoshikawa A, Isono S, Sheback A, Isono K. Mol Gen Genet. 1987;209:481–488. doi: 10.1007/BF00331153. [DOI] [PubMed] [Google Scholar]
- 48.Arai K, Clark BF, Duffy L, Jones MD, Kaziro Y, Laursen RA, L’Italien J, Miller DL, Nagarkatti S, Nakamura S, Nielsen KM, Petersen TE, Takahashi K, Wade M. Proc Natl Acad Sci U S A. 1980;77:1326–1330. doi: 10.1073/pnas.77.3.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bariola PA, Russell BA, Monahan SJ, Stroop SD. J Biotechnol. 2007;130:11–23. doi: 10.1016/j.jbiotec.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 50.Takao T, Kobayashi M, Nishimura O, Shimonishi Y. J Biol Chem. 1987;262:3541–3547. [PubMed] [Google Scholar]
- 51.Honda S, Asano T, Kajio T, Nishimura O. Arch Biochem Biophys. 1989;269:612–622. doi: 10.1016/0003-9861(89)90147-1. [DOI] [PubMed] [Google Scholar]
- 52.Charbaut E, Redeker V, Rossier J, Sobel A. FEBS Lett. 2002;529:341–345. doi: 10.1016/s0014-5793(02)03421-x. [DOI] [PubMed] [Google Scholar]
- 53.Wu J, Chang S, Gong X, Liu D, Ma Q. Biochim Biophys Acta. 2006;1760:1241–1247. doi: 10.1016/j.bbagen.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 54.Grutter MG, Marki W, Walliser HP. J Biol Chem. 1985;260:11436–11437. [PubMed] [Google Scholar]
- 55.Smith VF, Schwartz BL, Randall LL, Smith RD. Protein Sci. 1996;5:488–494. doi: 10.1002/pro.5560050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Polevoda B, Sherman F. J Biol Chem. 2000;275:36479–36482. doi: 10.1074/jbc.R000023200. [DOI] [PubMed] [Google Scholar]