

Abstract
Escherichia coli strain O157 produces an O-antigen with the repeating tetrasaccharide unit α-d-PerNAc-α-l-Fuc-β-d-Glc-α-d-GalNAc, preassembled on undecaprenyl pyrophosphate (Und-P-P). These studies were conducted to determine whether the biosynthesis of the lipid-linked repeating tetrasaccharide was initiated by the formation of GalNAc-P-P-Und by WecA. When membrane fractions from E. coli strains K12, O157, and PR4019, a WecA-overexpressing strain, were incubated with UDP-[3H]GalNAc, neither the enzymatic synthesis of [3H]GlcNAc-P-P-Und nor [3H]GalNAc-P-P-Und was detected. However, when membrane fractions from strain O157 were incubated with UDP-[3H]GlcNAc, two enzymatically labeled products were observed with the chemical and chromatographic properties of [3H]GlcNAc-P-P-Und and [3H]GalNAc-P-P-Und, suggesting that strain O157 contained an epimerase capable of interconverting GlcNAc-P-P-Und and GalNAc-P-P-Und. The presence of a novel epimerase was demonstrated by showing that exogenous [3H]GlcNAc-P-P-Und was converted to [3H]GalNAc-P-P-Und when incubated with membranes from strain O157. When strain O157 was metabolically labeled with [3H]GlcNAc, both [3H]GlcNAc-P-P-Und and [3H]GalNAc-P-P-Und were detected. Transformation of E. coli strain 21546 with the Z3206 gene enabled these cells to synthesize GalNAc-P-P-Und in vivo and in vitro. The reversibility of the epimerase reaction was demonstrated by showing that [3H]GlcNAc-P-P-Und was reformed when membranes from strain O157 were incubated with exogenous [3H]GalNAc-P-P-Und. The inability of Z3206 to complement the loss of the gne gene in the expression of the Campylobacter jejuni N-glycosylation system in E. coli indicated that it does not function as a UDP-GlcNAc/UDP-GalNAc epimerase. Based on these results, GalNAc-P-P-Und is synthesized reversibly by a novel GlcNAc-P-P-Und epimerase after the formation of GlcNAc-P-P-Und by WecA in E. coli O157.
Keywords: Cell/Wall/Bacteria, Enzymes/Lipid, Glycolipids, Lipid/Isoprenoid, Lipid/Synthesis, Organisms/Bacteria, Epimerase, Lipid Intermediate
Introduction
The Escherichia coli cell envelope contains an inner plasma membrane, a stress-bearing peptidoglycan layer, and an asymmetric outer membrane consisting of a phospholipid inner monolayer and an outer monolayer composed of bacterial lipopolysaccharide (LPS).2 LPS contains three components, the lipid A anchor, the 3-deoxy-d-manno-oct-2-ulosonic acid-containing core, and the O-antigen region (see Refs. 1–3 and references therein for reviews on the assembly of O-antigens of bacterial LPS).
The O-antigen components of bacterial LPS are large, extremely diverse polysaccharides that can be either homopolymeric, composed of a single repeating monosaccharide, or heteropolymeric, containing 10–30 repeats of 3–6 sugar units (4). O-antigens are, thus, the dominant feature of the bacterial cell surface and constitute important determinants of virulence and pathogenicity (5–8). Currently, E. coli strains with more than 180 individual O-serotypes, attributed to unique O-antigen structures, have been identified (8).
O-antigen repeat units are pre-assembled on the cytosolic face of the inner membrane attached to undecaprenyl pyrophosphate (Und-P-P). The lipid-linked repeat units diffuse transversely (flip-flop) to the periplasmic surface of the inner membrane and are polymerized before transport to the outer membrane and ligation to LPS. Most heteropolymeric O-antigen repeat units have either N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) at the reducing terminus, and it has been assumed that the biosynthesis of the lipid intermediates is initiated by the transfer of GlcNAc-P or GalNAc-P from their respective sugar nucleotide derivatives to Und-P catalyzed by WecA (3, 9–12). Although the properties and specificity of the GlcNAc-phosphotransferase activity of WecA have been characterized (13), the conclusion that WecA catalyzes the synthesis of GalNAc-P-P-Und is based on genetic studies (12). However, there is no direct enzymological evidence demonstrating that WecA utilizes UDP-GalNAc as a GalNAc-P donor.
E. coli O157 is an enterohemorrhagic strain responsible for approximately two-thirds of all recent cases of hemolytic-uremic syndrome and poses serious human health concerns (5, 12). E. coli O157 synthesizes an O-antigen containing the repeating tetrasaccharide with the structure (4-N-acetyl perosamine → fucose → glucose → GalNAc) (14). We have investigated the initiating reaction of E. coli O157 O-antigen subunit assembly to determine whether GalNAc-P-P-Und synthesis is catalyzed by WecA or by some previously undescribed mechanism. The evidence presented here shows that GalNAc-P-P-Und is not synthesized by GalNAc-P transfer from UDP-GalNAc catalyzed by WecA but rather by the reversible epimerization of the 4-OH of GlcNAc-P-P-Und catalyzed by a novel epimerase encoded by the Z3206 gene in E. coli O157.
Thus, this paper describes a novel biosynthetic pathway for the assembly of an important bacterial cell surface component as well as a new biosynthetic route for the synthesis of GalNAc-P-P-Und. The potential of the bacterial epimerase as a new target for antimicrobial agents is discussed.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmids
E. coli strains PR4019 (13) and PR21546 (15) were generous gifts from Dr. Paul Rick, Bethesda, MD, and E. coli O157:H45 (16) was a gift from Dr. Claudio Zweifel, Veterinary Institute, University of Zurich. E. coli DH5α (Invitrogen) was used as the host for cloning experiments and for protein glycosylation analysis. Plasmids used are listed in Table 1.
TABLE 1.
Plasmids used in this study
| Plasmid | Description | Refences |
|---|---|---|
| pMLBAD | Cloning vector, TmpR | 17 |
| pMLBAD:Z3206 | Z3206 in pMLBAD, TmpR, expression controlled by arabinose-inducible promoter | This study |
| pMLBAD:gne | gne in pMLBAD, TmpR, expression controlled by arabinose-inducible promoter | This study |
| pACYCpgl | C. jejuni pgl cluster CmR | 18 |
| pACYCgne::kan | C. jejuni pgl cluster containing a kan cassette in gne, CmR, KanR | 32 |
| pWA2 | Soluble periplasmic hexa-His-tagged AcrA under control of Tet promoter in pBR322, AmpR | 20 |
Materials
[1,6-3H]GlcNAc (30 Ci/mmol), UDP-[1-3H]GlcNAc (20 Ci/mmol), and UDP-[6-3H]GalNAc (20 Ci/mmol) were obtained from American Radiolabeled Chemicals (St. Louis, MO). Quantum 1 silica gel G thin layer plates are a product of Quantum Industries (Fairfield, NJ), and Baker Si250 Silica Gel G plates are manufactured by Mallinckrodt Chemical Works. Yeast extract and Bacto-peptone were products of BD Biosciences. All other chemicals were obtained from standard commercial sources. Trimethoprim (50 μg/ml), chloramphenicol (20 μg/ml), ampicillin (100 μg/ml), and kanamycin (50 μg/ml) were added to the media as needed.
Construction of Recombinant Plasmids
E. coli strain DH5α was used for DNA cloning experiments, and constructed plasmids were verified by DNA sequencing. The Z3206 gene was amplified from E. coli O157:H45 by PCR with oligonucleotides Z3206-Fw and Z3206-RvHA (AAACCCGGGATGAACGATAACGTTTTGCTC and AAATCTAGATTAAGCGTAATCTGGAACATCGTATGGGTACTCAGAAACAAACGTTATGTC; restriction sites are underlined). The PCR fragment was digested with SmaI and XbaI and ligated into SmaI-XbaI cleaved pMLBAD vector (17). This resulted in plasmid pMLBAD:Z3206 encoding Z3206 with a C-terminal hemagglutinin tag.
The gne gene was amplified from pACYCpgl (18), encoding Campylobacter jejuni pgl cluster, with oligonucleotides gne-Fw and gne-RV (AAACCATGGATGAAAATTCTTATTAGCGG and AAATCTAGATTAAGCGTAATCTGGAACATCGTATGGGTAGCACTGTTTTTCCCAATC; restriction sites are underlined). The PCR product was digested with NcoI and XbaI and ligated into the same sites of pMLBAD to generate plasmid pMLBAD:gne, which encodes Gne with a C-terminal hemagglutinin tag (Table 1).
Growth Conditions, Protein Expression, and Immunodetection
E. coli strains were cultured in Luria-Bertani medium (1% yeast extract, 2% Bacto-peptone, 0.6% NaCl) at 37 °C with vigorous shaking. Arabinose-inducible expression was achieved by adding arabinose at a final concentration of 0.02–0.2% (w/v) to E. coli cells grown up to an A600 of 0.05–0.4. The same amount of arabinose was added again 5 h post-induction, and incubation continued for 4–15 h.
Total E. coli cell extracts were prepared for immunodetection analysis using cells at a concentration equivalent to 1 A600 unit that were resuspended in 100 μl of SDS loading buffer (19). Aliquots of 10 μl were loaded on 10% SDS-PAGE. Periplasmic extracts of E. coli cells were prepared by lysozyme treatment (20), and 10 μl of the final sample (corresponding to 0.2 A600 units of cells) was analyzed by SDS-PAGE. After being blotted on nitrocellulose membrane, sample was immunostained with the specific antiserum (21). Anti-AcrA (18) antibodies were used. Anti-rabbit IgG-HRP (Bio-Rad) was used as secondary antibody. Detection was carried out with ECLTM Western blotting detection reagents (Amersham Biosciences).
For the preparation of membrane fractions, bacterial cells were collected by centrifugation at 1000 × g for 10 min, washed once in ice-cold phosphate-buffered saline, once with cold water, and once with 10 mm Tris-HCl, pH 7.4, 0.25 m sucrose. The cells were resuspended to a density of ∼200 A600 units/ml in 10 mm Tris-HCl, pH 7.4, 0.25 m sucrose, 10 mm EDTA containing 0.2 mg/ml lysozyme, and incubated at 30 °C for 30 min. Bacterial cells were recovered by centrifugation at 1000 × g for 10 min, quickly resuspended in 40 volumes of ice-cold 10 mm Tris-HCl, pH 7.4, and placed on ice. After 10 min the cells were homogenized with 15 strokes with a tight-fitting Dounce homogenizer and supplemented with 0.1 mm phenylmethylsulfonyl fluoride and sucrose to a final concentration of 0.25 m. Unbroken cells were removed by centrifugation at 1000 × g for 10 min, and cell envelopes were recovered by centrifugation at 40,000 × g for 20 min. The membrane fraction was resuspended in 10 mm Tris-HCl, pH 7.4, 0.25 m sucrose, 1 mm EDTA and again sedimented at 40,000 × g and resuspended in the same buffer to a protein concentration of ∼20 mg/ml. Membrane fractions were stored at −20 °C until needed.
Assay for the Biosynthesis of [3H]GlcNAc-P-P-Und and [3H]GalNAc-P-P-Und in E. coli Membranes in Vitro
Reaction mixtures for the synthesis of GlcNAc-P-P-Und and GalNAc-P-P-Und contained 50 mm Tris-HCl, pH 8, 40 mm MgCl2, 5 mm dithiothreitol, 5 mm 5′AMP, E. coli membrane fraction (50–200 μg of membrane protein, and either 5 μm UDP-[3H]GlcNAc/GalNAc (500–2500 dpm/pmol) or purified [3H]GlcNAc-P-P-Und/[3H]GalNAc-P-P-Und (2000 dpm/pmol, dispersed in 1% Triton X-100, final concentration 0.1%) in a total volume of 0.05 ml. After incubation at 37 °C, reactions were terminated by the addition of 40 volumes of CHCl3/CH3OH (2:1), and the total lipid extract containing [3H]HexNAc-P-P-undecaprenols was prepared as described previously (22). After partitioning, the organic phase was dried under a stream of nitrogen and redissolved in 1 ml CHCl3/CH3OH (2:1), and an aliquot (0.2 ml) was removed, dried in a scintillation vial, and analyzed for radioactivity by liquid scintillation spectrometry in a Packard Tri-Carb 2100 TR liquid scintillation spectrometer. To determine the rate of synthesis of [3H]GlcNAc-P-P-Und or [3H]GalNAc-P-P-Und, the lipid extract was dried under a stream of nitrogen, redissolved in a small volume of CHCl3/CH3OH (2:1), and spotted on a 10 × 20-cm borate-impregnated Baker Si250 silica gel plate, and the plate was developed with CHCl3, CH3OH, H2O, 0.2 m sodium borate (65:25:2:2). Individual glycolipids were detected with a Bioscan AR2000 Imaging Scanner (Bioscan, Washington, D. C.). The biosynthetic rates for each glycolipid were calculated by multiplying the total amount of radioactivity in [3H]GlcNAc/GalNAc-P-P-Und by the percentage of the individual [3H] glycolipids.
Metabolic Labeling of Bacterial Cells
E. coli cells were cultured with vigorous shaking in Luria-Bertani medium at 37 °C to an A600 of 0.5–1. [3H]GlcNAc was added to a final concentration of 1 μCi/ml, and the incubation was continued for 5 min at 37 °C. The incorporation of radiolabel into glycolipids was terminated by the addition of 0.5 gm/ml crushed ice, and the cultures were thoroughly mixed. The bacterial cells were recovered by centrifugation at 4000 × g for 10 min, and the supernatant was discarded. The cells were washed with ice-cold phosphate-buffered saline two times, resuspended by vigorous vortex mixing in 10 volumes (cell pellet) of methanol, and sonicated briefly with a probe sonicator at 40% full power. After sonication, 20 volumes of chloroform were added, and the extracts were mixed vigorously and allowed to stand at room temperature for 15 min. The insoluble material was sedimented by centrifugation, and the pellet was re-extracted with a small volume of CHCl3/CH3OH (2:1) twice. The combined organic extracts were then processed as described below.
Purification of GlcNAc-P-P-Und and GalNAc-P-P-Und
GlcNAc/GalNAc-P-P-Und was extracted with CHCl3/CH3OH (2:1) and freed of water-soluble material by partitioning as described elsewhere (22). The organic extract was then dried under a stream of nitrogen, and the bulk glycerophospholipids were destroyed by deacylation in toluene/methanol (1:3) containing 0.1 n KOH at 0 °C for 60 min. The deacylation reaction was neutralized with acetic acid, diluted with 4 volumes of CHCl3/CH3OH (2:1), and washed with 1/5 volume of 0.9% NaCl. The organic (lower) phase was washed with ⅓ volume of CHCl3, CH3OH, 0.9% NaCl (3:48:47), and the aqueous phase was discarded. The organic phase was diluted with sufficient methanol to accommodate the residual aqueous phase in the organic phase and applied to a DEAE-cellulose column (5 ml) equilibrated with CHCl3/CH3OH (2:1). The column was washed with 20 column volumes of CHCl3/CH3OH/H2O (10:10:3) and then eluted with CHCl3/CH3OH/H2O (10:10:3) containing 20 mm ammonium acetate. Fractions (2 ml) were collected and monitored for either radioactivity or GlcNAc/GalNAc-P-P-Und using an anisaldehyde spray reagent (23) after resolution by thin layer chromatography on borate-impregnated silica plates (as described earlier).
GlcNAc/GalNAc-P-P-Und elute in fractions 3–10. These fractions were combined, supplemented with sufficient CHCl3 and H2O to give a final composition of CHCl3/CH3OH/H2O (3:2:1), and partitioned. The aqueous layer was aspirated, and the organic layer was dried under a stream of nitrogen, spotted on a Quantum 1 thin layer plate (silica gel G, not borate-impregnated), and developed with CHCl3/CH3OH/H2O (65:25:4). GlcNAc/GalNAc-lipid zones were located by staining with iodine vapors and by comparison with radioactive marker lipids (if nonradioactive lipids were being purified) or detected by Bioscanning if radiolabeled GlcNAc/GalNAc-lipids were present. The desired zones were scraped from the plate, loaded into a Pasteur pipette, equipped with a glass wool plug, and washed sequentially with 2 column volumes of CH3OH and then with CHCl3/CH3OH/H2O (65:35:6) (∼10 column volumes). The eluate was adjusted to CHCl3/CH3OH/H2O (3:2:1) by the addition of CHCl3 and H2O, and the phases were separated by a brief centrifugation. The aqueous layer was discarded, and the lower phase was dried under a stream of nitrogen, spotted on a borate-impregnated Quantum 1 thin layer plate, and developed with CHCl3, CH3OH, H2O, 0.2 m sodium borate (65:25:2:2). GlcNAc/GalNAc-P-P-Und were located by exposure of the TLC plate to iodine vapors and by comparison to radioactive marker lipids and recovered from the silica gel as described above, dried under a stream of nitrogen, and stored at −20 °C until use.
Preparation of Borate-impregnated Thin Layer Plates and Whatman No. 1 Paper
Silica gel thin layer plates were impregnated with sodium borate by briefly immersing the plates in 2.5% Na2B4O7·10 H2O in 95% methanol as described by Kean (24). The borate-impregnated TLC plates were dried overnight at room temperature and stored in a vacuum desiccator over Drierite until use. Immediately before chromatography, the plates were activated by heating briefly (∼10–15 min) to 100 °C. Whatman No. 1 paper was impregnated with sodium borate by dipping 20 × 30-cm sheets of Whatman 1 paper in 0.2 m Na2B4O7·10 H2O. The Whatman No. 1 paper sheets were pressed firmly between two sheets of Whatman No. 3MM paper and allowed to dry at room temperature for several days, as described by Cardini and Leloir (25).
Characterization of Glycan Products Formed in in Vitro Reactions
The glycans of the individual glycolipids were characterized by descending paper chromatography after release by mild acid hydrolysis. The GlcNAc/GalNAc lipids were dried under a stream of nitrogen in a conical screw-cap tube and heated to 100 °C, 15 min in 0.2 ml of 0.01 m HCl. After hydrolysis the samples were applied to a 0.8-ml mixed-bed ion-exchange column containing 0.4 ml of AG50WX8 (H+) and 0.4 ml AG1X8 (acetate form) and eluted with 1.5 ml water. The eluate was dried under a stream of nitrogen, redissolved in a small volume of H2O (0.02 ml), spotted on a 30-cm strip of borate-impregnated Whatman No. 1 paper, and developed in descending mode with butanol/pyridine/water (6:4:3) for 40–50 h. After drying, the paper strips were cut into 1-cm zones and analyzed for radioactivity by scintillation spectrometry. GlcNAc and GalNAc standards were detected using an aniline-diphenylamine dip reagent (26).
Glycan products were converted to their corresponding alditols by reduction with 0.1 m NaBH4 in 0.1 m NaOH (final volume 0.1 ml) after mild acid hydrolysis as described above. After incubation at room temperature overnight, the reactions were quenched with several drops of glacial acetic acid and dried under a stream of nitrogen out of methanol containing 1 drop of acetic acid, several times. The alditols were dissolved in water, desalted by passage over 0.5-ml columns of AG50WX8 (H+) and AG1X8 (acetate), dried under nitrogen, and spotted on 30-cm strips of Whatman No. 3MM paper. The Whatman No. 3MM strips were developed overnight in descending mode with ethyl acetate, pyridine, 0.1 m boric acid (65:25:20), dried, cut into 1-cm zones, and analyzed for radioactivity by scintillation spectrometry. GlcNAcitol and GalNAcitol standards were visualized using a modification of the periodate-benzidine dip procedure (27). The paper strips were dipped in acetone, 0.1 m NaIO4 (95:5), allowed to air dry for 3 min, and then dipped in acetone/acetic acid/H2O/o-tolidine (96:0.6:4.4:0.2 gm). Alditols containing cis-diols stained as yellow spots on a blue background.
Mass Spectrometry of Glycolipids
Purified glycolipids were analyzed using an ABI/MDS Sciex 4000 Q-Trap hybrid triple quadrupole linear ion trap mass spectrometer with an ABI Turbo V electrospray ion source (ABI/MDS-Sciex, Toronto, Canada). In brief, samples were infused at 10 μl/min with ion source settings determined empirically, and MS/MS information was obtained by fragmentation of the molecular ion in linear ion trap mode.
Analytical Procedures
Protein concentrations were determined using the BCA protein assay (Pierce) after precipitation of membrane proteins with deoxycholate and trichloroacetic acid according to the Pierce Biotechnology bulletin “Eliminate Interfering Substances from Samples for BCA Protein Assay.” Samples were analyzed for radioactivity by scintillation spectrometry in a Packard Tri-Carb 2100TR liquid scintillation spectrometer after the addition of 0.5 ml of 1% SDS and 4 ml of Econosafe Economical Biodegradable Counting Mixture (Research Products International, Corp., Mount Prospect, IL).
RESULTS
UDP-GalNAc Is Not a Substrate for E. coli WecA (GlcNAc- phosphotransferase)
To determine whether E. coli WecA will utilize UDP-GalNAc as a GalNAc-P donor to form GalNAc-P-P-Und, membrane fractions from E. coli strains K12, PR4019, a WecA-overexpressing strain, and O157, which synthesize a tetrasaccharide O-antigen repeat unit with GalNAc at the reducing terminus presumably initiated by the synthesis of GalNAc-P-P-Und, were incubated with UDP-[3H]GalNAc. As seen in Table 2, no labeled glycolipids were detected after the incubation with UDP-[3H]GalNAc.
TABLE 2.
Synthesis of [3H]GlcNAc/GalNAc-P-P-undecaprenol in E. coli membrane fractions using either UDP-[3H]GlcNAc or UDP-[3H]GalNAc as substrate
Membrane fractions from the indicated E. coli strains were incubated with either UDP-[3H]GlcNAc or UDP-[3H]GalNAc for 10 min at 37 °C, and the incorporation into [3H]GlcNAc/GalNAc-P-P-Und was determined as described under “Experimental Procedures.”
| Source of membranes | Sugar nucleotide added | [3H]Glycolipid formed |
|
|---|---|---|---|
| GlcNAc-P-P-Und | GalNAc-P-P-Und | ||
| pmol/mg | |||
| K12 | UDP-[3H]GlcNAc | 6.4 | <0.01 |
| K12 | UDP-[3H]GalNAc | <0.01 | <0.01 |
| PR4019 | UDP-[3H]GlcNAc | 44 | <0.01 |
| PR4019 | UDP-[3H]GalNAc | <0.01 | <0.01 |
| O157 | UDP-[3H]GlcNAc | 1.5 | 0.5 |
| O157 | UDP-[3H]GalNAc | <0.01 | <0.01 |
Moreover, neither the addition of exogenous Und-P to incubations with membranes from PR4019, the WecA-overexpressing strain, or the addition of cytosolic fractions from O157 cells resulted in the formation of GalNAc-P-P-Und from UDP-GalNAc (data not shown). These results demonstrated that UDP-GalNAc is not a substrate for WecA and suggested that GalNAc-P-P-Und is formed by an alternative mechanism.
When membranes from strain K12 were incubated with UDP-[3H]GlcNAc, [3H]GlcNAc-P-P-Und was synthesized as expected (13). However, when membranes from strain O157 were incubated with UDP-[3H]GlcNAc, in addition to [3H]GlcNAc-P-P-Und, a second labeled lipid shown to be [3H]GalNAc-P-P-Und (see below) was observed. When the time course for the formation of the two glycolipids was examined, the incorporation of radioactivity into [3H]GlcNAc-P-P-Und (Fig. 1, O) occurred more quickly and to a higher extent than into [3H]GalNAc-P-P-Und (Fig. 1, ●), compatible with a precursor-product relationship (Fig. 2).
FIGURE 1.
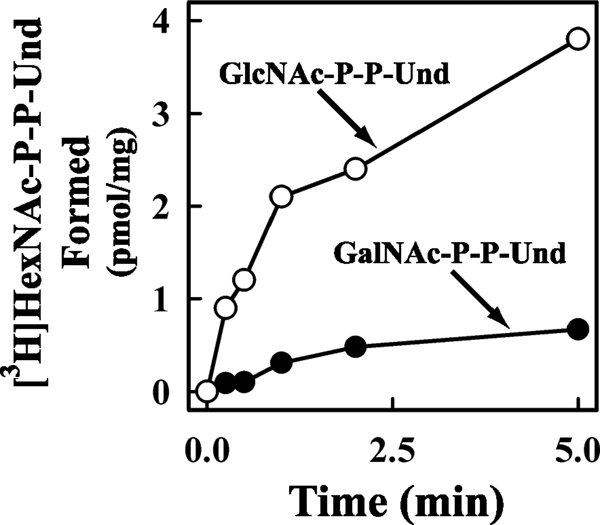
Time course of [3H]GlcNAc/GalNAc-P-P-Und synthesis by membrane fractions from E. coli O157. The membrane fraction from E. coli strain O157 was incubated with UDP-[3H]GlcNAc for the indicated times at 37 °C. The [3H]lipid products were extracted, and the incorporation of [3H]GlcNAc into [3H]GlcNAc-P-P-Und (○) and [3H]GalNAc-P-P-Und (●) was assayed as described under “Experimental Procedures.”
FIGURE 2.

Proposed biosynthetic pathway for the formation of GalNAc-P-P-Und from GlcNAc-P-P-Und.
The observation that E. coli O157 membranes do not utilize UDP-GalNAc as a GalNAc-P donor for the synthesis of GalNAc-P-P-Und prompted us to propose an alternative biosynthetic pathway for the formation of GalNAc-P-P-Und illustrated in Fig. 2. In this scheme GlcNAc-P-P-Und is formed by the transfer of GlcNAc-P from UDP-GlcNAc, catalyzed by WecA, and then GlcNAc-P-P-Und is epimerized by the action of a previously undescribed 4-epimerase to produce GalNAc-P-P-Und. The research described in this report was designed to test this hypothesis and to identify the novel GlcNAc 4-epimerase (E. coli O157 Z3206) catalyzing the formation of GalNAc-P-P-Und.
Characterization of [3H]GalNAc-P-P-Und Formed in Vitro with Membrane Fractions from E. coli Strain O157
Consistent with the additional O157-specific glycolipid product detected in Fig. 1, as GalNAc-P-P-Und, it was stable to mild alkaline methanolysis (toluene/methanol 1:3, containing 0.1 n KOH, 0 °C, 60 min), retained by DEAE-cellulose equilibrated in CHCl3/CH3OH/H2O (10:10:3), and eluted with CHCl3/CH3OH/H2O (10:10:3) containing 20 mm ammonium acetate as reported previously for [3H]GlcNAc1–2-P-P-Dol (28).
The putative [3H]GalNAc-P-P-Und was clearly resolved from [3H]GlcNAc-P-P-Und by thin layer chromatography on borate-impregnated silica gel G (24) and purified by preparative TLC as shown in Fig. 3, panel A and B. When the glycolipid was treated with mild acid (0.01 n HCl, 100 °C, 15 min), the water-soluble product co-chromatographed with [3H]GalNAc on descending paper chromatography with borate-impregnated Whatman No. 1 paper (Fig. 3, panel C). In addition, when the labeled sugar was reduced, it was converted to [3H]alditol, GalNAc-OH (Fig. 3, panel D). Moreover, negative-ion MS analysis yielded the [M-H]− ion of m/z = 1128, expected for GalNAc-P-P-Und, and the MS/MS daughter ion spectrum showed a prominent ion at m/z = 907, expected for a glycolipid containing P-P-Und (data not included) (29). The identification of the glycolipid product formed by strain O157 as GalNAc-P-P-Und is also supported by its formation from exogenous GlcNAc-P-P-Und (see below).
FIGURE 3.

Purification and Characterization of [3H]GalNAc-P-P-Und synthesized by membrane fractions from E. coli strain O157. Membrane fractions from E. coli O157 were incubated with UDP-[3H]GlcNAc, and the [3H]GalNAc lipids were purified as described under “Experimental Procedures.” Panel A, preparative thin layer chromatogram of [3H]HexNAc lipids on borate-impregnated silica gel G (Quantum 1) after purification on DEAE-cellulose is shown. Panel B, thin layer chromatography of purified [3H]GalNAc-P-P-Und on borate-impregnated silica gel G (Baker, Si250) after recovery from the preparative plate in panel A is shown. Panel C, descending paper chromatogram (borate-impregnated Whatman No. 1 paper) of the 3H amino sugar recovered after mild acid hydrolysis of [3H]GalNAc-P-P-Und purified in panel B is shown. Panel D, descending paper chromatogram (Whatman No. 3MM) of the [3H]HexNAc-alditol produced by reduction of the [3H] amino sugar from panel C with NaBH4.
Identification of an E. coli O157 Gene Encoding GlcNAc-P-P-Und 4-Epimerase
Genomic sequences of different bacteria encoding O antigen repeating units having a GalNAc at the reducing terminus were screened. One group with a repeating unit containing a GalNAc at the reducing terminus and a second group lacking a terminal GalNAc in the repeating unit were compared to identify potential epimerases. Using these criteria Z3206 was identified as a candidate GlcNAc-P-P-Und 4-epimerase (Table 3). The gene encoding a candidate for the GlcNAc-P-P-Und 4-epimerase was identified by a combination of genetic and bioinformatic approaches. The genomic location of the Z3206 gene is consistent with a role in this pathway, as it resides between galF of the O-antigen cluster and wcaM, which belongs to the colanic acid cluster.
TABLE 3.
Correlation of Z3206 gene in bacterial strains expressing O-antigen chains with GalNAc at the reducing termini
| % Identity with Z3206 | GalNAc at the reducing terminus of O-antigen repeat unit | |
|---|---|---|
| E .coli O55 gne | 100 | Yes |
| E. coli O86 gne1 | 100 | Yes |
| Shigella boydii O18 gne | 88 | Yes |
| Salmonella enterica O30 gne | 94 | Yes |
| C. jejuni gne | 21 | No |
| E. coli O16 galE | 27 | No |
| E. coli O86 gne2 | 18 | Yes |
The GlcNAc 4-epimerase genes present in E. coli strains with O-antigen repeat units containing GalNAc can be separated into two homology groups as shown in Table 3. One homology group (containing gne1) clearly is correlated with the presence of GalNAc as the initiating sugar on the O-antigen repeat unit. The second group (containing gne2) exhibits a high degree of similarity to the UDP-Glc epimerase, GalE, and is found in E. coli strains that do not initiate O-antigen repeat unit synthesis with GalNAc. Z3206 in E. coli O157, a gene with a high degree of homology to gne1, was selected for further study as a candidate GlcNAc-P-P-Und 4-epimerase.
Metabolic Labeling of [3H]GalNAc-P-P-Und with [3H]GlcNAc in E. coli Cells Expressing the Z3206 Gene
To investigate whether expression of the E. coli O157 Z3206 gene enabled cells to synthesize GalNAc-P-P-Und, E. coli strain 21546 (15) expressing the Z3206 gene was labeled metabolically with [3H]GlcNAc and analyzed for [3H]GlcNAc/GalNAc-P-P-Und formation. E. coli strain 21546 was selected as the host for the Z3206 expression studies because a mutation in UDP-ManNAcA synthesis results in a block in the utilization of GlcNAc-P-P-Und for the synthesis of the enterobacterial common antigen. Because E. coli 21546 is derived from E. coli K12, it does not synthesize an O-antigen repeat as well (30), and thus, larger amounts of GlcNAc-P-P-Und accumulate for the conversion to GalNAc-P-P-Und. When strain 21546 and the transformant expressing the Z3206 gene were labeled with [3H]GlcNAc and the radiolabeled lipids were analyzed by thin layer chromatography on borate-impregnated silica gel plates, the parental strain (Fig. 4, panel A) synthesized only one labeled lipid, GlcNAc-P-P-Und. However, 21546 cells expressing the Z3206 gene (Fig. 4, panel B) also synthesized an additional labeled lipid shown to be GalNAc-P-P-Und (see above).
FIGURE 4.
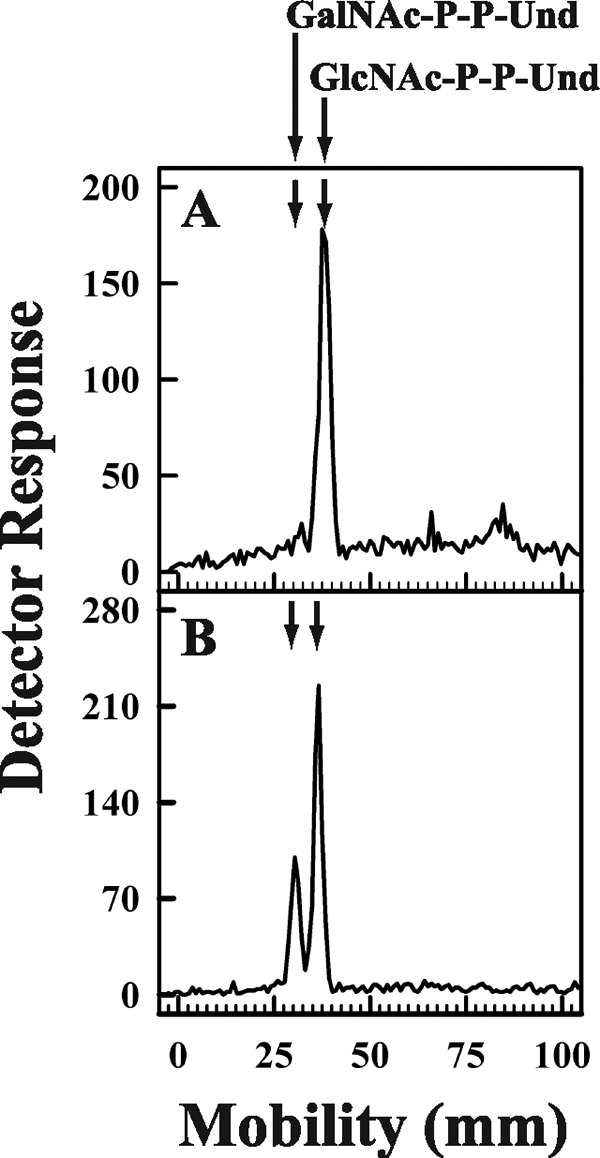
Metabolic labeling of E. coli 21546 cells and E. coli 21546 cells after transformation with pMLBAD:Z3206. E. coli 21546 (panel A) and E. coli 21546:pMLBAD/Z3206 (panel B) were labeled metabolically with [3H]GlcNAc for 5 min at 37 °C. [3H]GlcNAc/GalNAc-P-P-Und were extracted, freed of water soluble contaminants, and separated by thin layer chromatography on borate-impregnated silica gel plates (Baker Si250) as described under “Experimental Procedures.” Radioactive lipids were detected using a Bioscan chromatoscanner. The chromatographic positions of GalNAc-P-P-Und and GlcNAc-P-P-Und are indicated by arrows.
Membrane Fractions from E. coli Cells Expressing the Z3206 Gene Synthesize GalNAc-P-P-Und in Vitro
To corroborate that the protein encoded by the E. coli O157 Z3206 gene catalyzed the synthesis of GalNAc-P-P-Und, membrane fractions from E. coli cells expressing the Z3206 gene were incubated with [3H]UDP-GlcNAc, and the [3H]glycolipid products were analyzed by thin layer chromatography on borate-impregnated silica gel plates as shown in Fig. 5. When membrane fractions from E. coli K12 or the host strain E. coli 21546 cells were incubated with UDP-[3H]GlcNAc, only [3H]GlcNAc-P-P-Und was observed (Fig. 5, panels A and C). However, membrane fractions from E. coli O157 and E. coli 21546 expressing Z3206 formed GalNAc-P-P-Und as well (Fig. 5, panels B and D).
FIGURE 5.

Thin layer chromatography of [3H]GlcNAc/GalNAc-P-P-Und formed by incubation of membrane fractions from E. coli strains with UDP-[3H]GlcNAc. Membrane fractions from E. coli strains K12 (panel A), O157 (panel B), 21546 (panel C), and 21546:pMLBAD/Z3206 (panel D) were incubated with UDP-[3H]GlcNAc for 10 min at 37 °C, and the [3H]lipid products were extracted, freed of water-soluble contaminants by partitioning, and separated by thin layer chromatography on borate-impregnated silica gel plates (Baker Si250) as described under “Experimental Procedures.” The chromatographic positions of GalNAc-P-P-Und and GlcNAc-P-P-Und are indicated by arrows.
Formation of GlcNAc-P-P-Und, but Not GalNAc-P-P-Und, Is Reversed in the Presence of UMP
To provide additional evidence that GalNAc-P-P-Und is synthesized from GlcNAc-P-P-Und and not by the action of WecA using UDP-GalNAc as a glycosyl donor, the effect of discharging endogenous, pre-labeled [3H]GlcNAc-P-P-Und and [3H]GalNAc-P-P-Und with UMP was examined. The GlcNAc-phosphotransferase reaction catalyzed by WecA is freely reversible by the addition of excess UMP re-synthesizing UDP-GlcNAc and releasing Und-P.
In this experiment membrane fractions from the E. coli strain 21546 expressing Z3206 were pre-labeled for 10 min with UDP-[3H]GlcNAc followed by the addition of 1 mm UMP, and the amount of each labeled glycolipid remaining was determined. The results illustrated in Fig. 6, panel A, show the relative amounts of [3H]GlcNAc-P-P-Und and [3H]GalNAc-P-P-Und at the end of the 10-min labeling period. After incubation with 1 mm UMP for 1 min, it can be seen that there is a substantial loss of [3H]GlcNAc-P-P-Und, whereas the [3H]GalNAc-P-P-Und peak is relatively unchanged (Fig. 6, panel B). This observation is consistent with the results in Table 2 indicating that WecA does not catalyze the transfer of GalNAc-P into GalNAc-P-P-Und from UDP-GalNAc. It is noteworthy that during the second minute of incubation with UMP (Fig. 6, panel C), the loss of GlcNAc-P-P-Und slows, and there is a slight reduction in the peak of [3H]GalNAc-P-P-Und, suggesting that [3H]GalNAc-P-P-Und is re-equilibrating with the [3H]GlcNAc-P-P-Und pool by reversal of the epimerase reaction (see below).
FIGURE 6.
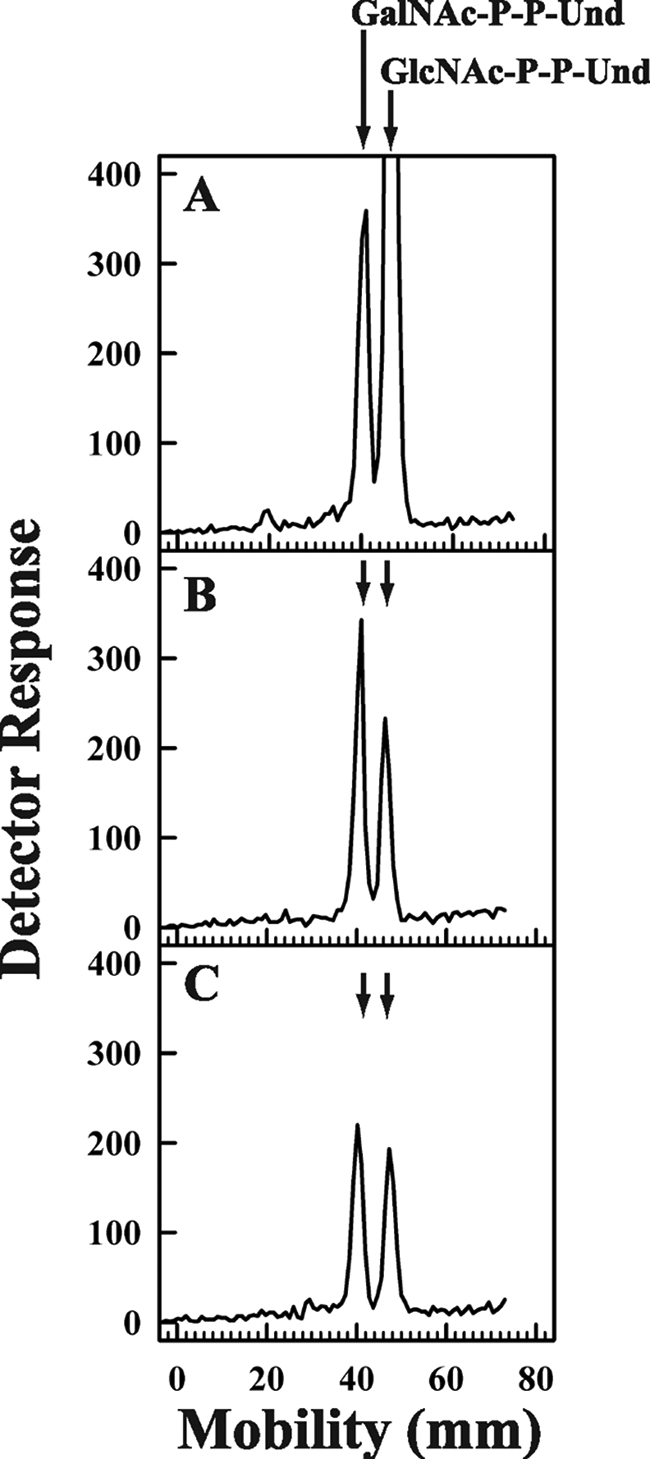
Discharge of GlcNAc-P by incubation with UMP. Membrane fractions from E. coli 21546:Z3206 were preincubated with UDP-[3H]GlcNAc to enzymatically label GlcNAc-P-P-Und for 10 min (panel A) at 37 °C followed by a second incubation period with 1 mm UMP included for either 1 min (panel B) or 2 min (panel C). After the indicated incubation periods [3H]GlcNAc/GalNAc-P-P-Und were extracted and resolved by thin layer chromatography on borate-impregnated silica gel plates (Baker Si250) as described under “Experimental Procedures.” The chromatographic positions of GalNAc-P-P-Und and GlcNAc-P-P-Und are indicated by arrows.
Interconversion of Exogenous, Purified [3H]GlcNAc-P-P-Und, and [3H]GalNAc-P-P-Und Catalyzed by Membranes from E. coli Cells Expressing Z3206
To provide direct evidence that GlcNAc-P-P-Und and GalNAc-P-P-Und can be directly interconverted by membrane fractions from E. coli cells expressing Z3260, purified [3H]GlcNAc-P-P-Und and [3H]GalNAc-P-P-Und were tested as exogenous substrates. Preliminary experiments showed that the epimerase was active when exogenous [3H]GlcNAc-P-P-Und was added to the reaction mixtures dispersed in Triton X-100, CHAPS, Nonidet P-40, or octylglucoside and exhibited a pH optimum in the range 7–8.5 (data not shown). The chromatographic mobility of the purified [3H]GlcNAc-P-P-Und and [3H]GalNAc-P-P-Und before incubation with membrane fractions is shown in Fig. 7, panels A and D. As seen in Fig. 7, panels B and E, the glycolipids are unaffected by incubation with membrane fractions from E. coli 21546. However, incubation of the purified glycolipids with membrane fractions from E. coli 21546 expressing Z3206 catalyzes the conversion of exogenous [3H]GlcNAc-P-P-Und to [3H]GalNAc-P-P-Und (Fig. 7, panel C) and the conversion of [3H]GalNAc-P-P-Und to [3H]GlcNAc-P-P-Und (Fig. 7, panel F). These results demonstrate directly that GlcNAc-P-P-Und and GalNAc-P-P-Und can be enzymatically interconverted in E. coli strains expressing the Z3206.
FIGURE 7.

Conversion of exogenous [3H]GlcNAc-P-P-Und and [3H]GalNAc-P-P-Und to the pertinent [3H]HexNAc-P-P-Und product catalyzed by membranes from strain 21546 expressing Z3206. Membrane fractions from E. coli strain 21546 (panels B and E) and 21546:pMLBAD/Z3206 (panels C and F) were incubated with purified [3H]GlcNAc-P-P-Und (panels A, B, and C) or [3H]GalNAc-P-P-Und (panels D, E, and F) (dispersed ultrasonically in 1% Triton X-100) for 1 min at 37 °C. [3H]GlcNAc/GalNAc-P-P-Und were extracted, resolved by thin layer chromatography on borate-impregnated silica gel plates (Baker Si250), and detected with a Bioscan AR2000 radiochromatoscanner as described under “Experimental Procedures.”
E. coli Z3206 Is Not a UDP-GlcNAc 4-Epimerase
To determine whether Z3206 can catalyze the formation of UDP-GalNAc, the N-glycosylation apparatus from C. jejuni was expressed in E. coli. In this reporter system, glycosylation of the target protein AcrA is dependent on the presence of the pgl locus (18), including a functional Gne UDP-Glc/UDP-GlcNAc epimerase (31). Glycosylation of AcrA is lost if the pgl cluster contains a deletion of gne (32). The ability of Z3206 to restore AcrA glycosylation in the presence of the pgl operon Δgne was investigated in vivo by expressing AcrA (pWA2) together with the pgl locus Δgne complemented by either Gne (pMLBAD:gne) or Z3206 (pMLBAD:Z3206). As shown in Fig. 8, the glycosylated protein, which migrates slower than the unglycosylated form, was formed only when cells expressing pgl locus Δgne were complemented by Gne (lane 2). Z3206 was unable to restore glycosylation of the reporter glycoprotein (Fig. 8, lane 1). Expression of Gne and membrane-associated Z3206 were confirmed by immunodetection (data not shown).
FIGURE 8.

Z3206 does not complement glycosylation of AcrA in a Gne-dependent glycosylation system. Periplasmic extracts prepared from E. coli DH5α cells carrying the AcrA expression plasmid and the pgl operon Δgne complemented with pMLBAD:Z3206 (lane 1), pMLBAD:gne (lane 2), or the vector control pMLBAD (lane 3) were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. AcrA and its glycosylated forms were detected with anti AcrA antisera. The position of bands corresponding to unglycosylated (AcrA) and glycosylated AcrA (gAcrA) is indicated.
DISCUSSION
E. coli O157 synthesizes an O-antigen with the repeating tetrasaccharide structure (4-N-acetyl perosamine → fucose → glucose → GalNAc). This study was conducted to determine whether the biosynthesis of the lipid-linked tetrasaccharide intermediate was initiated by the enzymatic transfer of GalNAc-P from UDP-GalNAc to Und-P catalyzed by WecA, as indicated by earlier genetic studies (12). The results described here obtained from genetic, enzymology, and metabolic labeling experiments indicate that WecA does not utilize UDP-GalNAc as a substrate but that WecA is required to synthesize GlcNAc-P-P-Und, which is then reversibly converted to GalNAc-P-P-Und by a novel epimerase encoded by the Z3206 gene in strain O157.
The Z3206 gene was selected as a candidate to encode the epimerase because it belongs to a family of genes present in several strains that produce surface O-antigen repeat units containing GalNAc residues at their reducing termini (Table 3). Previous reports identified two genes from E. coli O55 (33) and E. coli O86 (34), gne and gne1, respectively, that are 100% identical to Z3206 (Table 3). We conclude that these genes also encode an epimerase capable of converting GlcNAc-P-P-Und to GalNAc-P-P-Und in strains O55 and O86, which also produce O-antigen repeat units with GalNAc at the reducing termini (Table 3).
The gne and gne1 genes were previously proposed to encode a UDP-GlcNAc 4-epimerase (33, 34). The gne and gne1 proteins from strains O55 and O86 were not examined in this study. However, two experimental approaches in this study indicate that the Z3206 protein does not catalyze the epimerization of UDP-GlcNAc to UDP-GalNAc in strain O157. First, when membranes from strain O157 were incubated with [3H]UDP-GalNAc, neither [3H]GlcNAc-P-P-Und nor [3H]GalNAc-P-P-Und was detected (Table 2). If Z3206 catalyzed the conversion of [3H]UDP-GalNAc to [3H]UDP-GlcNAc, it would be expected that [3H]GlcNAc-P-P-Und should be observed. Second, we have shown that hemagglutinin-tagged Z3206 was incapable of complementing the UDP-GalNAc-dependent C. jejuni N-glycosylation reporter system (Fig. 8).
Although it cannot be excluded that accessory proteins in strains O55 and O86, not expressed in strain O157, might alter the specificity of the Gne and Gne1 proteins, it is also possible that the assays used to detect the epimerization of UDP-GlcNAc/GalNAc in the earlier studies (33, 34) produced misleading results. For example, Gne1 was assayed by incubating crude cell extracts from strain O86 (34), which could have contained membranes capable of forming GlcNAc/GalNAc-P-P-Und, with UDP-GlcNAc and following a decrease in the reaction with p-dimethylaminobenzaldehyde. Because the extracts were treated with 0.1 n HCl, the loss of reactivity with the reagent could plausibly be due to the conversion of GlcNAc-P-P-Und to GalNAc-P-P-Und and the subsequent release of GalNAc from the lipid intermediate during acid hydrolysis.
Gne from strain O55 (33) was also assayed for epimerase activity by incubating crude extracts with UDP-GalNAc and indirectly assaying the conversion to UDP-GlcNAc by measuring an increase in reactivity with p-dimethylaminobenzaldehyde after acid hydrolysis. In both studies the formation of the putative product was based on changes in reactivity with p-dimethylaminobenzaldehyde and not a definitive characterization of the sugar nucleotide end product. A 90% pure polyhistidine-tagged Gne1 was also shown to have a low level of UDP-glucose epimerase activity relative to Gne2 in a coupled assay. Z3206 from strain O157 was not assayed for UDP-glucose epimerase in the study described here.
It is significant that E. coli O86, which synthesizes an O-antigen containing two GalNAc residues, which would presumably require UDP-GalNAc as the glycosyl donor for the additional, non-reducing terminal GalNAc, also possesses an additional putative GlcNAc 4-epimerase gene, termed gne2, within the O-antigen gene cluster (34). This additional epimerase gene has high homology with the galE gene of the colanic acid gene cluster and appears to be a UDP-GlcNAc 4-epimerase capable of synthesizing UDP-GalNAc.
The Z3206 gene appears to be highly conserved in E. coli O-serotypes initiated with GalNAc. In a recent study, 62 E. coli strains with established O-antigen repeat unit structures were screened for expression of Z3206 by a polymerase chain reaction-based method using nucleotide primers designed to specifically detect the E. coli O157 Z3206 gene (33). In this study Z3206 was detected in 16 of the 22 E. coli strains that were known to contain GalNAc and in only 4 of the 40 strains lacking GalNAc. Moreover, a similar screen of the 22 GalNAc-containing strains with primers designed to detect an alternative epimerase with UDP-GlcNAc 4-epimerase activity (the GalE gene of E. coli O113) detected no strains carrying this gene, indicating that Z3206 is the GlcNAc 4-epimerase gene most commonly associated with the presence of a reducing-terminal GalNAc in O-antigen repeat units of E. coli.
Analysis of the Z3206 protein sequence by a variety of web-based topological prediction algorithms indicates that the Z3206 protein is not highly hydrophobic. The majority of the topological prediction algorithms indicate that Z3206 is a soluble 37-kDa protein, although TMPred (35) predicted a single weak N-terminal transmembrane helix. However, Western blotting after SDS-PAGE of cellular fractions from E. coli cells expressing hemagglutinin-tagged Z3206 clearly shows that the tagged protein is associated with the particulate fraction after hypotonic lysis of the cells (data not shown). Preliminary experiments show that the protein remains associated with the particulate fraction after incubation of the membrane fraction with 1 m KCl but is solubilized in an active form by incubation with 0.1% Triton X-100 (data not shown). Further studies will be required to determine whether the Z3206 protein associates with the membrane fraction via a transmembrane helix or by some other means, perhaps through association with a membrane-bound binding partner.
E. coli O157 Z3206 has significant sequence homology with the short-chain dehydrogenase/reductase family of oxidoreductases including the GXXGXXG motif (Rossman fold), consistent with the NAD(P) binding pocket (36) and the conserved S X24YX3K sequence, involved in proton abstraction and donation (37). Molecular modeling based on crystal structures of UDP-Glc 4-epimerase, another member of the short-chain dehydrogenase/reductase family, suggests that, after hydride abstraction, the 4-keto intermediate rotates around the β phosphate of UDP to present the opposite face of the keto intermediate and allow re-insertion of hydride from the opposite side, thus inverting the configuration of the hydroxyl at carbon 4. The presence of these conserved sequences suggests that Z3206 probably functions via a similar mechanism, but more experimentation will be required to verify this. Although the equilibrium distribution of the epimerase products, seen in Fig. 7, seems to favor the formation of GlcNAc-P-P-Und, the utilization of GalNAc-P-P-Und for O-antigen repeat unit assembly would drive the epimerization reaction in the direction of GalNAc-P-P-Und by mass action.
Epimerization of the glycosyl moieties of polyisoprenoid lipid intermediates has not been widely reported in nature. In one previous study the 2-epimerization of ribosyl-P-decaprenol to form arabinosyl-P-decaprenol, an arabinosyl donor in arabinogalactan biosynthesis in mycobacteria, was reported (38). Arabinosyl-P-decaprenol is formed via a two-step oxidation/reduction reaction requiring two mycobacterial proteins, Rv3790 and Rv3791. Although epimerization was modestly stimulated by the addition of NAD and NADP, neither Rv3790 nor Rv3791 contain either the Rossman fold or the SX24YXXXK motif, characteristic of the short-chain dehydrogenase/reductase family (36, 37).
In summary, a novel biosynthetic pathway for the formation of GalNAc-P-P-Und by the epimerization of GlcNAc-P-P-Und is described. Several antibiotics have been shown to inhibit the synthesis of GlcNAc-P-P-Und but are limited in their utility because they also block the synthesis of GlcNAc-P-P-dolichol, the initiating dolichol-linked intermediate of the protein N-glycosylation pathway. Although GlcNAc-P-P-dolichol is a structurally related mammalian counterpart of the bacterial glycolipid intermediate, GlcNAc-P-P-Und, there is no evidence for a similar epimerization reaction converting GlcNAc-P-P-dolichol to GalNAc-P-P-dolichol. Thus, this raises the possibility that in strains where the surface O-antigen containing GalNAc at the reducing termini are involved in a pathological process, O-antigen synthesis could potentially be blocked by inhibiting the bacterial epimerases.
Acknowledgments
We thank Dr. Andrew J. Morris (University of Kentucky) for assistance in mass spectrometric analysis of GalNAc-P-P-Und, Dr. Paul Rick (Bethesda, MD) for providing E. coli strains PR4019 and PR21546, and Michael Kowarik (GlycoVaxyn) for helpful discussions and careful editing of the manuscript. We also thank Miguel Valvano for providing the PMLBAD plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grant GM36065 (to C. J. W.).
- LPS
- lipopolysaccharide
- Und-P
- undecaprenyl monophosphate
- GlcNAc-P-P-Und
- GlcNAc-pyrophosphorylundecaprenol
- GalNAc-P-P-Und
- GalNAc-pyrophosphorylundecaprenol
- MS
- mass spectroscopy
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
REFERENCES
- 1.Raetz C. R., Whitfield C. (2002) Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitfield C. (2006) Annu. Rev. Biochem. 75, 39–68 [DOI] [PubMed] [Google Scholar]
- 3.Samuel G., Reeves P. R. (2003) Carbohydr. Res. 338, 2503–2519 [DOI] [PubMed] [Google Scholar]
- 4.Reeves P. R., Hobbs M., Valvano M. A., Skurnik M., Whitfield C., Coplin D., Kido N., Klena J., Maskell D., Raetz C. R., Rick P. D. (1996) Trends Microbiol. 4, 495–503 [DOI] [PubMed] [Google Scholar]
- 5.Law D. (2000) J. Appl. Microbiol. 88, 729–745 [DOI] [PubMed] [Google Scholar]
- 6.Spears K. J., Roe A. J., Gally D. L. (2006) FEMS Microbiol. Lett. 255, 187–202 [DOI] [PubMed] [Google Scholar]
- 7.Liu B., Knirel Y. A., Feng L., Perepelov A. V., Senchenkova S. N., Wang Q., Reeves P. R., Wang L. (2008) FEMS Microbiol. Rev. 32, 627–653 [DOI] [PubMed] [Google Scholar]
- 8.Stenutz R., Weintraub A., Widmalm G. (2006) FEMS Microbiol. Rev. 30, 382–403 [DOI] [PubMed] [Google Scholar]
- 9.Alexander D. C., Valvano M. A. (1994) J. Bacteriol. 176, 7079–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L., Radziejewska-Lebrecht J., Krajewska-Pietrasik D., Toivanen P., Skurnik M. (1997) Mol. Microbiol. 23, 63–76 [DOI] [PubMed] [Google Scholar]
- 11.Amor P. A., Whitfield C. (1997) Mol. Microbiol. 26, 145–161 [DOI] [PubMed] [Google Scholar]
- 12.Wang L., Reeves P. R. (1998) Infect. Immun. 66, 3545–3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rush J. S., Rick P. D., Waechter C. J. (1997) Glycobiology 7, 315–322 [DOI] [PubMed] [Google Scholar]
- 14.Perry M. B., MacLean L., Griffith D. W. (1986) Biochem. Cell Biol. 64, 21–28 [DOI] [PubMed] [Google Scholar]
- 15.Meier-Dieter U., Starman R., Barr K., Mayer H., Rick P. D. (1990) J. Biol. Chem. 265, 13490–13497 [PubMed] [Google Scholar]
- 16.Stephan R., Borel N., Zweifel C., Blanco M., Blanco J. E. (2004) BMC Microbiol. 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lefebre M. D., Valvano M. A. (2002) Appl. Environ. Microbiol. 68, 5956–5964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wacker M., Linton D., Hitchen P. G., Nita-Lazar M., Haslam S. M., North S. J., Panico M., Morris H. R., Dell A., Wren B. W., Aebi M. (2002) Science 298, 1790–1793 [DOI] [PubMed] [Google Scholar]
- 19.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 20.Feldman M. F., Wacker M., Hernandez M., Hitchen P. G., Marolda C. L., Kowarik M., Morris H. R., Dell A., Valvano M. A., Aebi M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 3016–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aebi M., Gassenhuber J., Domdey H., te Heesen S. (1996) Glycobiology 6, 439–444 [DOI] [PubMed] [Google Scholar]
- 22.Waechter C. J., Kennedy J. L., Harford J. B. (1976) Arch. Biochem. Biophys. 174, 726–737 [DOI] [PubMed] [Google Scholar]
- 23.Dunphy P. J., Kerr J. D., Pennock J. F., Whittle K. J., Feeney J. (1967) Biochim. Biophys. Acta 136, 136–147 [DOI] [PubMed] [Google Scholar]
- 24.Kean E. L. (1966) J. Lipid Res. 7, 449–452 [PubMed] [Google Scholar]
- 25.Cardini C. E., Leloir L. F. (1957) J. Biol. Chem. 225, 317–324 [PubMed] [Google Scholar]
- 26.Schwimmer S., Bevenue A. (1956) Science 123, 543–544 [DOI] [PubMed] [Google Scholar]
- 27.Gordon H. T., Thornburg W., Werum L. N. (1956) Anal. Chem. 28, 849–855 [Google Scholar]
- 28.Waechter C. J., Harford J. B. (1977) Arch. Biochem. Biophys. 181, 185–198 [DOI] [PubMed] [Google Scholar]
- 29.Guan Z., Breazeale S. D., Raetz C. R. (2005) Anal. Biochem. 345, 336–339 [DOI] [PubMed] [Google Scholar]
- 30.Stevenson G., Neal B., Liu D., Hobbs M., Packer N. H., Batley M., Redmond J. W., Lindquist L., Reeves P. (1994) J. Bacteriol. 176, 4144–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernatchez S., Szymanski C. M., Ishiyama N., Li J., Jarrell H. C., Lau P. C., Berghuis A. M., Young N. M., Wakarchuk W. W. (2005) J. Biol. Chem. 280, 4792–4802 [DOI] [PubMed] [Google Scholar]
- 32.Linton D., Dorrell N., Hitchen P. G., Amber S., Karlyshev A. V., Morris H. R., Dell A., Valvano M. A., Aebi M., Wren B. W. (2005) Mol. Microbiol. 55, 1695–1703 [DOI] [PubMed] [Google Scholar]
- 33.Wang L., Huskic S., Cisterne A., Rothemund D., Reeves P. R. (2002) J. Bacteriol. 184, 2620–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo H., Yi W., Li L., Wang P. G. (2007) Biochem. Biophys. Res. Commun. 356, 604–609 [DOI] [PubMed] [Google Scholar]
- 35.Hofmann K., Stoffel W. (1993) Biol. Chem. Hoppe-Seyler 374, 166 (abstr) [DOI] [PubMed] [Google Scholar]
- 36.Allard S. T., Giraud M. F., Naismith J. H. (2001) Cell. Mol. Life Sci. 58, 1650–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Field R. A., Naismith J. H. (2003) Biochemistry 42, 7637–7647 [DOI] [PubMed] [Google Scholar]
- 38.Mikusová K., Huang H., Yagi T., Holsters M., Vereecke D., D'Haeze W., Scherman M. S., Brennan P. J., McNeil M. R., Crick D. C. (2005) J. Bacteriol. 187, 8020–8025 [DOI] [PMC free article] [PubMed] [Google Scholar]