

Abstract
Mitochondrial biogenesis is a complex process. It necessitates the participation of both the nuclear and the mitochondrial genomes. This process is highly regulated, and mitochondrial content within a cell varies according to energy demand. In the yeast Saccharomyces cerevisiae, the cAMP pathway is involved in the regulation of mitochondrial biogenesis. An overactivation of this pathway leads to an increase in mitochondrial enzymatic content. Of the three yeast cAMP protein kinases, we have previously shown that Tpk3p is the one involved in the regulation of mitochondrial biogenesis. In this paper, we investigated the molecular mechanisms that govern this process. We show that in the absence of Tpk3p, mitochondria produce large amounts of reactive oxygen species that signal to the HAP2/3/4/5 nuclear transcription factors involved in mitochondrial biogenesis. We establish that an increase in mitochondrial reactive oxygen species production down-regulates mitochondrial biogenesis. It is the first time that a redox sensitivity of the transcription factors involved in yeast mitochondrial biogenesis is shown. Such a process could be seen as a mitochondria quality control process.
Keywords: Yeast, Mitochondrial Biogenesis, ROS, Signaling
Introduction
Cells adapt to their energy needs by adjusting their mitochondrial enzymatic content, resulting in a capacity to modulate ATP turnover (1). This has been shown to occur in a wide range of cell types, from yeast (2) and HeLa cells (3) to skeletal muscle (4). Mitochondria have their own genome that encodes a few (13 in mammals and 8 in yeast) of the 100 proteins necessary for oxidative phosphorylation (5). The remaining proteins necessary for the biogenesis of oxidative phosphorylation complexes are encoded by the nuclear genome. Mitochondrial biogenesis thus depends on the coordination of nuclear and mitochondrial events. Moreover, mitochondria to nucleus signaling has been a longstanding question.
In mammalian cells, it has been shown that the mitochondrial reactive oxygen species are involved in the activation of the serine/threonine protein kinase D, which in turn activates NF-κB, leading to induction of SOD2 (mitochondrial superoxide dismutase) and consequently efficient detoxification from mitochondrial reactive oxygen species (6). In HeLa cells, respiratory uncoupling, which is well known to decrease mitochondrial reactive oxygen species (ROS)2 production, activates NRF-1 (nuclear respiratory factor-1) (3). NRF-1 is a transcription factor encoded by nuclear DNA (7), and functional binding sites for NRF-1 have been described in several genes critical for mitochondrial biogenesis (8). Furthermore, during aging, it has been shown that while mitochondrial ROS production increases, there is a concomitant decrease in mitochondrial oxidative phosphorylation complex amount and activity (9). Altogether, these results point to a possible role of ROS as signaling molecules in the cross-talk between mitochondria and nucleus.
In living cells, growth is the result of coupling between substrate catabolism and multiple metabolic processes taking place during net biomass formation and cell maintenance. A crucial parameter for growth description is its yield (i.e. the efficiency of the transformation from substrate consumption to biomass formation). When yeast cells are grown on a purely respiratory substrate, biomass generation is entirely connected to substrate oxidation through oxidative phosphorylation and, hence, to oxygen consumption. In a previous paper (1), we have shown that, in non-fermentable media, the growth yield is identical regardless of the strain, growth phase, and respiratory substrate used. This homeostasis is the consequence of a strict linear relationship between growth and respiratory rates. Moreover, the oxygen consumption rate was strictly controlled by the cellular content in respiratory chains in such a way that, in vivo, the steady state of oxidative phosphorylation was kept constant. The cAMP signaling pathway is now well known to be involved in the regulation of mitochondrial biogenesis, both in mammalian cells and in yeast, although the molecular mechanisms of this process are not well defined. It has been shown that treatment of human preadipocytes with forskolin (which leads to an overactivation of the cAMP pathway) increased mitochondrial DNA copy number (10). In yeast, there is now a growing amount of evidence showing that overactivation of the Ras/cAMP pathway leads to an increase in the cell mitochondrial content (11–13). Yeast has three A kinase catalytic subunits, which have greater than 75% identity and are encoded by the TPK (TPK1, TPK2, and TPK3) genes (14). Although they are redundant for viability and functions such as glycogen storage regulation, the three A kinases are not redundant for other functions (15–18). We have shown that in the absence of the yeast protein kinase Tpk3p, there is a significant decrease in cellular mitochondrial content, when cells are grown in non-fermentable medium (19). This generates a drastic decrease in cell growth in the Δtpk3 cells versus the wild type cells, because when yeast cells are grown on respiratory substrate, energy transformation processes involve oxidative phosphorylation (1). Briefly, our previous study has allowed us to show that in Δtpk3 cells, (i) respiratory rates are decreased in these cells when compared with the wild type cells, (ii) cellular mitochondrial content that was assessed quantitatively by measuring the amount of mitochondrial cytochromes (namely aa3, b, and cc1) is decreased, and (iii) growth rate is decreased.
Here, we investigated the mechanisms involved in the regulation of mitochondrial biogenesis via the yeast protein kinase Tpk3p. We show that the decrease in mitochondrial content in the Δtpk3 cells originates in a decrease in mitochondrial biogenesis. Indeed, the activity of the transcription factors (HAP2/3/4/5 complex) involved in this process is decreased in the Δtpk3 cells. Moreover, we show that the activity of this complex is modulated by mitochondrial ROS production, which is increased in the mitochondria isolated from the Δtpk3 cells. This is the first report showing that the activity of the HAP2/3/4/5 complex is sensitive to ROS signaling, thus clearly involving ROS in mitochondria to nucleus signaling.
EXPERIMENTAL PROCEDURES
Yeast Strains, Culture Medium, and Growth Conditions
The following yeast strains were used in this study: BY4742, MATa ura3Δ0 lys2Δ0 leu2Δ0 his3Δ0 (EUROSCARF); Y15016, MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 tpk3:: kanMX4 (EUROSCARF).
Cells were grown aerobically at 28 °C in the following medium: 0.175% yeast nitrogen base (Difco), 0.2% casein hydrolysate (Merck), 0.5% (NH4)2SO4, 0.1% KH2PO4, 0.2% dl-lactate (w/v) (Prolabo), pH 5.5, 20 mg/liter l-tryptophan (Sigma), 40 mg/liter adenine hydrochloride (Sigma), and 20 mg/liter uracil (Sigma). When cells carried a plasmid, the relevant amino acid was taken out of the medium. Growth was measured at 600 nm in a Safas spectrophotometer (Monaco). Dry weight determinations were performed on samples of cells harvested throughout the growth period and washed twice in distilled water.
Oxygen Consumption Assays
The oxygen consumption was measured polarographically at 28 °C using a Clark oxygen electrode in a 1-ml thermostatically controlled chamber. Respiratory rates (JO2) were determined from the slope of a plot of O2 concentration versus time. Respiration assays of growing cells were performed in the growth medium except in the case of uncoupled respiration, which was performed after cells were harvested in the following buffer: 2 mm magnesium sulfate, 1.7 mm sodium chloride, 10 mm potassium sulfate, 10 mm glucose, and 100 mm ethanol, pH 6.8 (20).
Cytochrome Content Determination
The cellular and mitochondrial content of c + c1, b, and a + a3 hemes were calculated as described by Dejean et al. (2), taking into account the respective molar extinction coefficient values and the reduced minus oxidized spectra recorded using a dual beam spectrophotometer (Aminco DW2000).
Mitochondria Preparation
Cells were grown aerobically at 28 °C in the following medium: 0.175% yeast nitrogen base (Difco), 0.2% casein hydrolysate (Merck), 0.5% (NH4)2SO4, 0.1% KH2PO4, 0.2% lactate (w/v) (Prolabo) as carbon source, pH 5.5, 20 mg/liter l-tryptophan (Sigma), 40 mg/liter adenine hydrochloride (Sigma), and 20 mg/liter uracil (Sigma). Yeast cells were harvested in the exponential growth phase, and mitochondria were isolated from protoplasts as described (21). Protein concentration was measured by the biuret method using bovine serum albumin as a standard. Yeast mitochondria were suspended in the following medium: 0.65 m mannitol, 0.36 mm EGTA, 10 mm Tris-maleate, 5 mm Tris-phosphate, pH 6.8.
Catalase Activity
Cells were washed and then broken by vigorous shaking with an equal volume of glass beads in the following buffer: 0.65 m mannitol, 2 mm EDTA, 10 mm Tris-maleate (pH 6.8), and a mixture of protease inhibitors (Complete EDTA-freeTM, Roche Applied Science). Centrifugation (700 × g, 2 min) allowed the elimination of pelleted unbroken cells and glass beads. Cellular proteins were quantified by the biuret method. To assess catalase activity, proteins were suspended in 90 mm potassium phosphate buffer (pH 7.2) and dispatched in the cuvettes of a double beam Safas spectrophotometer; 0.03% hydrogen peroxide was added in the sample cuvette, and the absorbance difference between the cuvettes was measured at 240 nm. Catalase activity was determined from the slope of a plot of H2O2 concentration versus time.
Determination of H2O2 Production Rate
The rate of H2O2 production was determined by monitoring the oxidation of the fluorogenic indicator amplex red in the presence of horseradish peroxidase. The concentrations of horseradish peroxidase and amplex red in the incubation medium were 0.06 unit/ml and 1 μm, respectively. Fluorescence was recorded at the following wavelengths: excitation, 560 nm; emission, 584 nm. A standard curve was obtained by adding known amounts of H2O2 to the assay medium in the presence of the reactants. Mitochondria (0.3 mg of protein/ml) were incubated in the respiratory medium at 28 °C, and H2O2 production was initiated by substrate (glycerol 3-phosphate, 5 mm) addition. Phosphorylating conditions were obtained by the addition of 1 mm ADP. Antimycin A (0.6 μg/mg of protein) was added into the incubation medium in order to inhibit the activity of complex III. The H2O2 production rate was determined from the slope of a plot of the fluorogenic indicator versus time (22).
β-Galactosidase Assay
A standard permeabilization procedure was used as described (23). After the preincubation period, 0.4 mg/ml o-nitrophenyl-β-d-galactopyranoside was added, and the tube was briefly vortexed. The reaction was stopped by the addition of 0.5 m Na2CO3. The samples were centrifuged for 30 s at 14,000 × g, and the absorbance of the supernatant was read at 420 nm. Activity is given in arbitrary units.
Quinone Redox State
Frozen mitochondria were used to measure oxidized and reduced coenzyme Q6 contents. After dissolving and extraction in 2-propanol, CoQ6 was detected by reverse-phase high performance liquid chromatography with electrochemical detection as described (24).
Protein Carbonylation
The amount of proteins indicated in Fig. 4 was derivatized, using the Chemicon OxyBlot kit, to 2,4-dinitrophenolhydrazone by reaction with 2,4-dinitrophenylhydrazine (25). The 2,4-dinitrophenol-derivatized crude protein extracts were resolved by SDS-PAGE onto polyvinylidene difluoride (Millipore) membrane filters. The membranes were incubated with a primary antibody, specific to the 2,4-dinitrophenol moiety of the proteins, and subsequently incubated with a secondary (IgG goat anti-rabbit) horseradish peroxidase-antibody conjugate directed against the primary antibody. Filters were then treated with chemiluminescence blotting substrate (horseradish peroxidase) for detection. The filters were exposed to a blue-sensitive film that was subsequently developed.
FIGURE 4.

Protein carbonylation of mitochondria isolated from wild type and Δtpk3 cells. Detection of carbonylated proteins was performed using the Chemicon OxyBlot detection kit as described under “Experimental Procedures.” Lanes 1, 3, and 5, wild type isolated mitochondrial extracts; lanes 2, 4, and 6, Δtpk3 extracts. The three sets correspond to increasing amounts of mitochondrial proteins. The numbers on the quantification graph correspond to the ratio between carbonylation levels in the mutant versus the wild type. Quantification was assessed using the ImageJ software.
HAP4, SOD1, and SOD2 Subcloning
Yep352-SOD1 was kindly provided by P. Fabrizio (26). Yep351-URA-SOD2 was constructed by switching the original auxotrophic marker with an URA3-HpAI/KasI fragment. Both genes are driven by their natural promoters. The plasmid carrying the HAP4 gene expressed under control of the TET promoter was constructed by inserting a BamHI/PstI fragment amplified from the HAP4 locus with the oligonucleotides CCCGGATCCATCATGACCGCAAAGACT and CGCCTGCAGCTATTTCAAAATACTTGTACC, in pCM189 (27) linearized with BamHI/PstI.
Cellular Glycogen Content Determination
Glycogen was quantitated according to Ref. 28 on 4–10 mg of cell dry weight.
Proteins Extraction, Electrophoresis, and Blotting
Cells were suspended in 50 μl of a mixture of 3.5% β-mercaptoethanol in 2 m NaOH. After a 15-min incubation on ice, proteins were precipitated with 50 μl of 3 m trichloroacetic acid for 15 min on ice. After a rapid centrifugation, the pellet was resuspended in a 1:1 (v/v) mixture of 10% SDS and sample buffer (0.1 m Tris, 2% SDS, 2% β-mercaptoethanol, 25% glycerol, 0.002% bromphenol blue). After quantification with a Bio-Rad kit, proteins were analyzed by 12% SDS-PAGE performed according to the method of Laemmli. After electrotransfer onto polyvinylidene difluoride membranes (Amersham Biosciences), blots were probed with the desired antibodies. The proteins were visualized by ECL (Amersham Biosciences), according to the manufacturer's instructions.
Antibodies
Anti-green fluorescent protein antibody was purchased from Roche Applied Science, and anti-Ade4 antibody was a gift from Dr. B. Daignan-Fornier.
RESULTS
Δtpk3 Cells Exhibit Decreased Mitochondrial Content
Fig. 1 shows that (i) growth rate is decreased in Δtpk3 cells when compared with the wild type cells (A), (ii) cellular mitochondrial content that was assessed quantitatively by measuring the amount of mitochondrial cytochromes (namely aa3, b, and cc1) is decreased (B), and (iii) respiratory rates are decreased in these cells (C).
FIGURE 1.

A, growth of wild type (■) and Δtpk3 (□) cells. Cells were grown aerobically in minimal medium containing 0.2% (w/v) dl-lactate. Growth was measured at 600 nm as described under “Experimental Procedures.” B, cytochrome content of wild type (black bars) and Δtpk3 (white bars) cells. Cytochrome content was determined as described under “Experimental Procedures.” Results are mean ± S.D. of at least three measurements performed on three independent cell cultures. C, respiratory rates of wild type (black bars) and Δtpk3 (white bars) cells. Wild type and Δtpk3 respiratory rates were assessed on cells grown aerobically in minimal medium containing 0.2% (w/v) dl-lactate. Spontaneous respiratory rate was directly assessed on cells from the growth medium. The respiratory rate in the absence of phosphorylation (indicated by triethyltin (TET)) was assessed in the presence of 0.2 mm triethyltin, and the uncoupled respiratory rate was assessed in the presence of 40 μm carbonyl cyanide p-chlorophenylhydrazone (CCCP) as described under “Experimental Procedures.” Results are mean ± S.D. of at least three measurements performed on three independent cell cultures.
Coenzyme Q Is Highly Reduced, and ROS Production Is Drastically Increased in the Δtpk3 Mitochondria
In the Δtpk3 cells, we have shown (Fig. 1B) that cytochrome b (and thus the bc1 complex, assuming a constant b/c1 stoichiometry in this complex) is decreased by 20%, and cytochrome c is drastically decreased (50%), whereas cytochrome aa3 is slightly decreased (Fig. 1B). We hypothesized that the decreased amount of cytochrome c in the Δtpk3 cells would represent a bottleneck for electron transfer. If this were the case, one would expect the quinones (which are upstream of the cytochrome c) to be in a more reduced state. We thus determined the quinone redox states on isolated mitochondria under different conditions: non-phosphorylating (substrate oxidation), phosphorylating (oxidation coupled to phosphorylation), or fully inhibited (respiratory chain inhibition). Fig. 2 clearly shows that in the mitochondria isolated from the wild type cells, under non-phosphorylating (substrate oxidation) conditions, the quinones were about 50% reduced, and as expected, this redox state decreased under phosphorylating conditions. In the presence of antimycin A, a bc1 complex inhibitor, 80% of the quinone pool was reduced due to the well known site of inhibition of this inhibitor. Indeed, antimycin A acts in the Q-cycle directly downstream of the ubisemiquinone, thus not allowing the whole pool of quinones to be reduced. In the presence of potassium cyanide, a well known cytochrome oxidase inhibitor, as expected, over 90% of the quinone pool was reduced. In the mitochondria isolated from the Δtpk3 cells, the quinone redox state under non-phosphorylating conditions was comparable with the one determined in the presence of potassium cyanide, indicating that in this condition, the quinones were fully reduced. This redox state was only slightly decreased under phosphorylating conditions. Moreover, these quinones are involved in one-electron transfer processes that are well known to participate in reactive oxygen species production (29–31). Since yeast mitochondria do not harbor any complex I, ubisemiquinone is the most likely candidate as a superoxide generator at the level of complex III. Because the quinone redox state was very much increased in the Δtpk3 isolated mitochondria, we investigated ROS production in these mitochondria. Table 1 shows that the rate of H2O2 production in these mitochondria was 4–5 times higher than the rate in the wild type mitochondria, establishing that the mutant mitochondria produce high levels of ROS. It is noteworthy that whereas in the wild type mitochondria, coupling oxidation to phosphorylation decreased the ROS production rate by 40%, in the mutant mitochondria, there was no significant decrease when substrate oxidation was coupled to ATP synthesis.
FIGURE 2.

Quinone redox state of wild type (black bars) and Δtpk3 (white bars) isolated mitochondria. Quinone redox state was determined as described (46) on mitochondria isolated from the wild type and Δtpk3 cells. Results are means ± S.D. of at least three measurements performed on two independent mitochondrial preparations. Respiratory substrate was glycerol 3-phosphate (10 mm), phosphorylating respiratory rate was determined in the presence of 1 mm ADP, and fully inhibited state was determined in the presence of either 0.6 μg/mg protein antimycin A or 1 mm KCN.
TABLE 1.
H2O2 production in isolated mitochondria
The rate of H2O2 production in mitochondria was determined as described under “Experimental Procedures.” Respiratory substrate was glycerol 3-phosphate (10 mm), phosphorylating respiratory rate was determined in the presence of 1 mm ADP, and fully inhibited state was determined in the presence of 0.6 μg/mg protein antimycin A. Results are mean ± S.D. of at least three measurements performed on two independent mitochondrial preparations.
| Rate of H2O2 production |
||
|---|---|---|
| Wild type | Δtpk3 | |
| pmol/min/mg protein | ||
| Non-phosphorylating | 59 ± 5 | 238 ± 38 |
| Phosphorylating | 42 ± 4 | 230 ± 47 |
| Antimycin A | 283 ± 65 | 417 ± 34 |
cAMP-induced Decrease in ROS Generation Is Dependent on Tpk3p on Isolated Mitochondria
One of the questions raised by this study is how Tpk3p deficiency led to an increase in mitochondrial ROS production. The activity of some of the respiratory chain complexes has been shown to be regulated by cAMP-dependent phosphorylation of one of their subunits (32). This process leads to a regulation of the flux through the respiratory chain and might have an impact on the rate of ROS production. In order to decipher whether mitochondrial ROS generation was regulated by Tpk3p, their production rate was assessed in the presence of cAMP. Fig. 3 clearly shows that cAMP induces a decrease in ROS production rate on isolated wild type mitochondria, whereas there is no significant decrease of this rate on isolated Δtpk3 mitochondria.
FIGURE 3.

Tpk3p-dependent decrease in mitochondrial ROS production of wild type (■) and Δtpk3 (□) isolated mitochondria. The rate of H2O2 production in mitochondria was determined as described under “Experimental Procedures.” Respiratory substrate was glycerol 3-phosphate (G3P; 10 mm), and cAMP was added at the indicated concentration in the presence of 5 mm ATP and 5 mm Mg2+. Results are mean ± S.D. of at least three measurements performed on two independent mitochondrial preparations.
The Activity of the ROS Protection Enzymes Is Increased in the Δtpk3 Cells
Because the Δtpk3 isolated mitochondria produced more ROS, we investigated the status of the ROS protection enzymes that allow the cells to avoid the deleterious effects of these species. Superoxide (O2˙̄) produced by the mitochondrial respiratory chain can be dismutated to hydrogen peroxide by superoxide dismutase. There are two superoxide dismutases, SOD1 (copper-zinc superoxide dismutase), which is cytosolic and present in the mitochondrial intermembrane space, and SOD2 (manganese superoxide dismutase), which is located within the mitochondrial matrix. Further, mitochondrial and cytosolic catalases convert the hydrogen peroxide generated by the activity of these enzymes in water and oxygen. The activity of these enzymes was investigated on whole cells. There was only a slight increase in mitochondrial SOD activity, whereas the cytosolic SOD was significantly increased in the Δtpk3 cells (see Fig. 6B), and catalase activity was doubled in the mutant cells (35 × 106 units/mg proteins for the wild type and 72 × 106 units/mg proteins for the Δtpk3 cells), indicating an oxidative stress in these cells. However, data relative to ROS production at the mitochondrial level (see above) indicate that this increase is not sufficient to prevent the increase in ROS production in the Δtpk3 mitochondria. The oxidative stress was further confirmed by the level of carbonylation of the mitochondrial proteins from both strains (Fig. 4). Indeed, due to its irreversible nature, the level of protein carbonylation is considered a good indicator of oxidative stress. Fig. 4 further strengthens the finding that the Δtpk3 cells are subjected to an oxidative stress.
FIGURE 6.

A, respiratory rates in wild type-, Δtpk3-, and Δtpk3-overexpressing SOD cells. Wild type (WT; black), Δtpk3 (white), Δtpk3-SOD1 (gray), and Δtpk3-SOD2 (light gray) respiratory rates were assessed on cells grown aerobically in minimal medium containing 0.2% (w/v) dl-lactate. Spontaneous respiratory rate was directly assessed on cells from the growth medium. Results are means of at least three measurements performed on three independent cell cultures. B, SOD1 and -2 in gel activities in wild type-, Δtpk3-, and Δtpk3-overexpressing SOD cells. SOD activity was assessed according to Ref. 53 on cell extracts. C, cytochrome content in wild type-, Δtpk3-, and Δtpk3-overexpressing SOD cells. Wild type (black), Δtpk3 (white), Δtpk3-SOD1 (gray), and Δtpk3-SOD2 (light gray) cytochromes were assessed on cells grown aerobically in minimal medium containing 0.2% (w/v) dl-lactate. Results are the means of at least three measurements performed on three independent cell cultures.
The Decrease in Mitochondrial Enzymatic Equipment in Δtpk3 Cells Originates in ROS Production
Oxidative stress has long been considered an “accident” of aerobic metabolism, a stochastic process of free radical production and nonspecific tissue damage that is fundamentally unregulated aside from the antioxidant defense mechanisms. In recent years, a paradigm shift has been occurring, wherein certain ROS have become appreciated as signaling molecules whose production may be regulated as a part of routine cellular signal transduction (33). Because ROS production was largely increased in the mutant mitochondria, we hypothesized that these species might be involved in the signaling leading to a decrease in the mitochondrial enzymatic content. We thus monitored the growth of both wild type and Δtpk3 cells in the presence of a potent antioxidant: N-acetylcysteine. Fig. 5A clearly shows that growth was fully restored in the mutant cells in the presence of an antioxidant. Cellular mitochondrial content was assessed quantitatively by measuring the amount of mitochondrial cytochromes, namely aa3, b, and cc1. Fig. 5B shows that there was a full restoration in the mitochondrial cytochrome content of the Δtpk3 cells when grown in the presence of N-acetylcysteine. Furthermore, metabolic activity of these cells grown on respiratory substrate was assessed through respiratory rate measurement. Fig. 5C unequivocally shows that any respiratory rate in the mutant cells in the presence of N-acetylcysteine was comparable with the wild type cells with N-acetylcysteine. This clearly indicates that the decrease in mitochondrial enzymatic content of the Δtpk3 cells is due to an increase in ROS production.
FIGURE 5.

A, growth of wild type (●) and Δtpk3 (○) cells in the presence of an antioxidant. Cells were grown aerobically in minimal medium containing 0.2% (w/v) dl-lactate and 5 mm N-acetylcysteine. Growth was measured at 600 nm as described under “Experimental Procedures.” B, cytochrome content of wild type (black bars) and Δtpk3 (white bars) cells grown in the presence of an antioxidant. Cytochrome content was determined as described under “Experimental Procedures.” Results are mean ± S.D. of at least three measurements performed on three independent cell cultures. C, respiratory rates of wild type and Δtpk3 cells grown in the presence of an antioxidant. Wild type (black) and Δtpk3 (white) respiratory rates were assessed on cells grown aerobically in minimal medium containing 0.2% (w/v) dl-lactate and 5 mm N-acetylcysteine. Spontaneous respiratory rate was directly assessed on cells from the growth medium. The respiratory rate in the absence of phosphorylation (indicated by triethyltin (TET)) was assessed in the presence of 0.2 mm triethyltin, and the uncoupled respiratory rate was assessed in the presence of 40 μm carbonyl cyanide p-chlorophenylhydrazone (CCCP), as described under “Experimental Procedures.” Results are mean ± S.D. of at least three measurements performed on three independent cell cultures.
Moreover, the bc1 complex can produce reactive oxygen species both on the matrix side of the membrane and on the intermembranal side (34). Because the diffusible and thus signaling species of ROS is thought to be hydrogen peroxide, either matricial and/or inner membrane ROS could be involved in the signaling process. To sort out whether the localization of the ROS produced was of importance in the signaling process, we overexpressed either the matricial SOD (SOD2) or the cytosolic SOD (SOD1) in the Δtpk3 strain. Fig. 6A clearly shows that overexpression of the cytosolic SOD (which is mostly located in the mitochondrial inner membrane space) led to an almost complete abrogation of the respiratory defect in Δtpk3 cells. Matricial SOD overexpression had little effect on the Δtpk3 cell respiratory rate. Fig. 6B shows that both SOD1 and -2 are indeed overexpressed under our experimental conditions. Because SOD1 is overexpressed 2.5 times in Δtpk3-SOD1 cells versus Δtpk3 cells and SOD2 is overexpressed about 3 times in Δtpk3-SOD2 cells versus SOD2 cells, their overexpression level is comparable (see Fig. 6B). Fig. 6C shows that cellular cytochrome content is almost completely restored in the Δtpk3-SOD1 cells.
In order to ensure that the ROS-induced decrease of mitochondrial content and its sensitivity to an antioxidant in the Δtpk3 cells was indeed specific to the mitochondrial compartment, we assessed cellular glycogen content. It is well known that glycogen storage is impaired in mutants of the yeast Ras/cAMP pathway (35), a decrease in the activity of this pathway being associated with an increase in cellular glycogen content. Table 2 shows that there is an important increase in cellular glycogen content in the Δtpk3 cells versus the wild type cells and that this increase is not affected by an antioxidant, such as N-acetylcysteine. This strengthens the fact that the ROS signaling here is indeed specific to the mitochondrial compartment.
TABLE 2.
Cellular glycogen content in wild type and Δtpk3 cells
Glycogen content in whole cells was determined as described under “Experimental Procedures.” Results are mean ± S.D. of at least four measurements.
| Glycogen |
||
|---|---|---|
| Non-treated | N-Acetylcysteine | |
| μg/mg protein | ||
| Wild type | 3.5 ± 1.3 | 3.2 ± 1.1 |
| Δtpk3 | 26 ± 3.4 | 24 ± 3.8 |
ROS Regulate the Activity of the Transcription Factors HAP2, -3, -4, and -5
Microorganisms adapt their metabolism to environmental conditions and, particularly, to nutrient availability. The yeast has a clear preference for glucose as a carbon source, with subsequent conversion to ethanol by fermentation. During this shift from fermentation to respiration, gene expression is largely reprogrammed (36). This is achieved by derepression of glucose-repressed genes and specific induction of genes involved in gluconeogenesis, metabolism of alternate carbon sources, respiration, and mitochondrial development (37). The Hap2/3/4/5p transcription factor has been shown to be involved in these transcriptional changes. Indeed, disruption of any subunits of this complex renders the cells unable to grow on non-fermentable carbon sources (38–41). Moreover, many genes involved in energy metabolism have been shown to be regulated by this complex (42, 43). In order to determine whether the decrease in mitochondrial enzymatic content in the Δtpk3 cells was linked to a decrease in mitochondrial biogenesis in these cells, we assessed the activity of the Hap2/3/4/5p transcription factor with a widely used reporter gene, pCYC1-lacZ (pLG669Z) (44). Fig. 7A shows that there was an 80% decrease in the activity of this complex (as assessed by βlacZ reporter gene activity) in the Δtpk3 cells, indicating a potent defect in mitochondrial biogenesis in these cells. Moreover, whereas an antioxidant had no effect on this activity in the wild type cells, there was a full restoration of Hap2/3/4/5p transcription factor activity in the mutant cells grown in the presence of an antioxidant, indicating that (i) the decrease in Hap2/3/4/5p transcription factor activity in the Δtpk3 cells goes through ROS, and (ii) in the signaling pathway leading to the decrease in mitochondrial enzymatic content, ROS act downstream of Tpk3p. Sensitivity of the Hap2/3/4/5p transcription factor to cytosolic ROS was further confirmed by the decrease in its activity in the presence of menadione, a pro-oxidant (Fig. 7A). Because the CYC1 promoter has been shown to contain two upstream activation sequences (UAS1 and UAS2) and to ensure that we were indeed assessing the activity of the Hap2/3/4/5p transcription factor (that has been shown to bind to UAS2), we performed a similar experiment with a truncated version of pCYC1 that only contains the UAS2 (45). Fig. 7B shows that the results obtained with the UAS2 of pCYC1 are comparable with the ones obtained with the full promoter, further confirming the sensitivity of the Hap2/3/4/5p transcription factor to oxidative stress. To ensure that this regulation was not specific to pCYC1, similar experiments were realized using the ACO1-lacZ reporter gene (46) that has been shown to be HAP-responsive. Similar results were obtained using such a lacZ reporter gene (data not shown).
FIGURE 7.

Activity of the transcription factors HAP2/3/4/5. A, the activity of the transcription factors HAP2, -3, -4, and -5 was assessed with a widely used reporter gene, pCYC1-lacZ (pLG669Z) as described under “Experimental Procedures.” N-acetylcysteine, when used, was 5 mm, and menadione was 20 μm. Results are mean ± S.D. of at least three measurements performed on three independent cell cultures. B, the activity of the transcription factors HAP2, -3, -4, and -5 was assessed with pLG CYC1-UAS2. Results are mean ± S.D. of at least three measurements performed on three independent cell cultures. WT, wild type.
HAP4p Amount Plays a Key Role in ROS-induced Decrease in Mitochondrial Biogenesis
In order to confirm that ROS signaling in the Δtpk3 cells did indeed go through the HAP2/3/4/5 complex, we determined the influence of HAP4p (the only subunit of the complex whose expression is transcriptionally regulated (36, 40)) overexpression in Δtpk3 cells. Fig. 8A shows that HAP4p overexpression in the Δtpk3 cells suppressed their respiratory rate defect (as well as the growth defect; data not shown), confirming that ROS signaling to mitochondrial biogenesis goes through the HAP2/3/4/5 complex. Fig. 8B shows that the cellular cytochrome content is in accordance with the respiratory rates and that its decrease in the Δtpk3 cells is abrogated by HAP4p overexpression.
FIGURE 8.

Effect of HAP4p overexpression in wild type and Δtpk3 cells. A, wild type (Wt; black), Δtpk3 (white), wild type HAP4p (dark gray), and Δtpk3-HPA4p (light gray) respiratory rates were assessed on cells grown aerobically in minimal medium containing 0.2% HAP4p (w/v) dl-lactate. Spontaneous respiratory rate was directly assessed on cells from the growth medium. Results are mean ± S.D. of at least three measurements performed on three independent cell cultures. B, wild type (black), Δtpk3 (white), wild type HAP4p (dark gray), and Δtpk3-HAP4p (light gray) cytochrome content were assessed on cells grown aerobically in minimal medium containing 0.2% (w/v) dl-lactate. Results are mean ± S.D. of at least three measurements performed on three independent cell cultures.
Furthermore, because HAP4p overexpression was able to suppress the respiratory defect in the Δtpk3 cells, we hypothesized that HAP4p might be the target of ROS signaling. Fig. 9 shows that when wild type cells are subjected to H2O2 treatment, there is an important decrease in the cellular amount of HAP4p.
FIGURE 9.
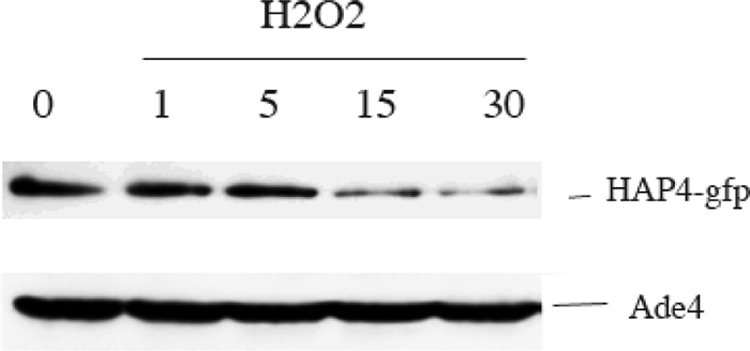
Green fluorescent protein-HAP4p cellular amount is decreased in the presence of H2O2. Cells were treated with 0.4 mm H2O2 for the time indicated (min). Proteins were extracted and resolved as described under “Experimental Procedures.” Ade4 was probed as a loading control.
DISCUSSION
In yeast, the Ras/cAMP pathway cascades as follows (47). Cdc25p catalyzes the conversion of GDP-Ras1p and Ras2p into GTP-Ras1p and Ras2p, which are the activators of Cyr1p, the adenylate cyclase. Cyr1p catalyzes cAMP synthesis. The intracellular concentration of cAMP thus depends on the respective activities of Cyr1p and the phosphodiesterases Pde1p and -2p. High cAMP concentrations promote the dissociation of the regulatory subunit (Bcy1p) from the catalytic subunits (Tpk1p, -2p, and 3p), activating the catalytic subunits of the protein kinase A, which phosphorylates a variety of substrates (32). We have shown that, from the top of the pathway down to Bcy1p, any mutation leading to an overactivation of this pathway leads to an increase in the mitochondrial content (11). This shows that the signaling of this pathway to mitochondrial content regulation goes through the PKA (i.e. Tpk1p, 2p, and 3p in yeast). These three catalytic subunits of the PKA have redundant functions. However, specificity in their respective signaling has recently been proposed (15–18). In a previous paper (19), we have shown that the yeast Tpk3p was specifically involved in the regulation of mitochondrial content in the transition phase. Here, we investigate the molecular mechanisms of this process in a medium where the amount of carbon substrate controls growth. We show that the absence of the cAMP protein kinase Tpk3p leads to a growth defect due to a decrease in the mitochondrial enzyme content. This originates in an increased mitochondrial ROS production that is due to a deficiency in Tpk3p-induced phosphorylation at the mitochondrial level. These reactive oxygen species are involved in mitochondria-to-nucleus signaling and induce a decrease in the activity of the transcription factor complex HAP2/3/4/5 that is involved in mitochondrial biogenesis. Moreover, we show that the ROS involved in this signaling are the ones produced on the external side of the mitochondrial inner membrane. Thus, a deficiency in the activity of Tpk3p leads to a decreased mitochondrial biogenesis induced by ROS signaling. Although the cells sense the oxidative stress and respond to it by increasing the amount of antioxidant enzymes (SOD and catalase), this increase is not sufficient to suppress the overflow of ROS.
The activity of some of the respiratory chain complexes has been shown to be regulated by cAMP-dependent phosphorylation of one of their subunits (32, 48). Tpk3p is most likely involved in the direct or indirect regulation of one (or more) of the subunits of one of the respiratory chain complexes that leads to an increase in the electron flux through this complex and thus a decrease in ROS production rate. Consequently, in the absence of Tpk3p, there is an increase in ROS production.
ROS are generated as by-products of cellular metabolism, primarily in the mitochondria. When cellular production of ROS overwhelms its antioxidant capacity, damages to cellular macromolecules, such as lipids, protein, and DNA, may ensue. Such a state of “oxidative stress” is thought to contribute to the pathogenesis of a number of human diseases. Recent studies have also implicated ROS in normal physiological signaling (49). In yeast, the Yap1p transcription factor regulates hydroperoxide homeostasis, and it has been shown that the thiol peroxidase Gpx3 is the hydroperoxide sensor that promotes the oxidation of Yap1 to its intramolecular disulfide bond, the activated form (50, 51). In mammalian cells, mitochondrial ROS have been shown to be involved in the regulation of the activity of the HIF-1α transcription factor (52). Here, we show that in the yeast Saccharomyces cerevisiae, one of the subunits of the nuclear transcription factor complex responsible for the transcription of most of the genes involved in mitochondria biogenesis is most probably down-regulated by an increase in mitochondrial ROS production. Such an increase can be deleterious to the cell and is often associated with a mitochondrial malfunction. Through this signaling pathway, the cell protects itself by decreasing mitochondrial biogenesis and thus the amount of dysfunctional mitochondria. This work indicates that ROS act as a sensor of the mitochondrial functional state and that over a threshold, they signal to the nucleus through regulation of the activity of transcription factor(s). In addition, it is clear that the site of ROS production (compartmentalization) and the concentration of ROS generation are important factors in determining the physiological actions and effects of ROS in the regulation of mitochondrial biogenesis. Such a down-regulation of mitochondrial biogenesis when mitochondrial alterations lead to increased ROS production could be seen as a mitochondria quality control process.
Acknowledgments
We thank P. Fabrizio for the gift of Yep352-SOD1 and SOD2, Prof. G. Lauquin for the gift of the aconitase promoter reporter gene, Dr. B Guiard for the gift of pLG CYC1-UAS2, and Dr. G. Dujardin for the gift of the plasmid encoding green fluorescent protein-HAP4p.
This work was supported in part by Agence Nationale de la Recherche Grant NT05-2_42268 and the Conseil Regional D'Aquitaine.
- ROS
- reactive oxygen species.
REFERENCES
- 1.Devin A., Dejean L., Beauvoit B., Chevtzoff C., Avéret N., Bunoust O., Rigoulet M. (2006) J. Biol. Chem. 281, 26779–26784 [DOI] [PubMed] [Google Scholar]
- 2.Dejean L., Beauvoit B., Guérin B., Rigoulet M. (2000) Biochim. Biophys. Acta 1457, 45–56 [DOI] [PubMed] [Google Scholar]
- 3.Li B., Holloszy J. O., Semenkovich C. F. (1999) J. Biol. Chem. 274, 17534–17540 [DOI] [PubMed] [Google Scholar]
- 4.Oscai L. B., Holloszy J. O. (1971) J. Biol. Chem. 246, 6968–6972 [PubMed] [Google Scholar]
- 5.Grivell L. A. (1989) Nature 341, 569–571 [DOI] [PubMed] [Google Scholar]
- 6.Storz P., Döppler H., Toker A. (2005) Mol. Cell. Biol. 25, 8520–8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans M. J., Scarpulla R. C. (1989) J. Biol. Chem. 264, 14361–14368 [PubMed] [Google Scholar]
- 8.Larsson N. G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G. S., Clayton D. A. (1998) Nat. Genet. 18, 231–236 [DOI] [PubMed] [Google Scholar]
- 9.Shigenaga M. K., Hagen T. M., Ames B. N. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 10771–10778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogacka I., Ukropcova B., McNeil M., Gimble J. M., Smith S. R. (2005) J. Clin. Endocrinol. Metab. 90, 6650–6656 [DOI] [PubMed] [Google Scholar]
- 11.Dejean L., Beauvoit B., Bunoust O., Guérin B., Rigoulet M. (2002) Biochem. Biophys. Res. Commun. 293, 1383–1388 [DOI] [PubMed] [Google Scholar]
- 12.Dejean L., Beauvoit B., Alonso A. P., Bunoust O., Guérin B., Rigoulet M. (2002) Biochim. Biophys. Acta 1554, 159–169 [DOI] [PubMed] [Google Scholar]
- 13.Noubhani A., Bunoust O., Bonini B. M., Thevelein J. M., Devin A., Rigoulet M. (2009) J. Biol. Chem. 284, 27229–27234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toda T., Cameron S., Sass P., Zoller M., Wigler M. (1987) Cell 50, 277–287 [DOI] [PubMed] [Google Scholar]
- 15.Robertson L. S., Fink G. R. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13783–13787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan X., Heitman J. (1999) Mol. Cell. Biol. 19, 4874–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan X., Heitman J. (2002) Mol. Cell. Biol. 22, 3981–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson L. S., Causton H. C., Young R. A., Fink G. R. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5984–5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chevtzoff C., Vallortigara J., Avéret N., Rigoulet M., Devin A. (2005) Biochim. Biophys. Acta 1706, 117–125 [DOI] [PubMed] [Google Scholar]
- 20.Beauvoit B., Rigoulet M., Bunoust O., Raffard G., Canioni P., Guérin B. (1993) Eur. J. Biochem. 214, 163–172 [DOI] [PubMed] [Google Scholar]
- 21.Guérin B., Labbe P., Somlo M. (1979) Methods Enzymol. 55, 149–159 [DOI] [PubMed] [Google Scholar]
- 22.Votyakova T. V., Reynolds I. J. (2001) J. Neurochem. 79, 266–277 [DOI] [PubMed] [Google Scholar]
- 23.Kippert F. (1995) FEMS Microbiol. Lett. 128, 201–206 [DOI] [PubMed] [Google Scholar]
- 24.Galinier A., Carrière A., Fernandez Y., Bessac A. M., Caspar-Bauguil S., Periquet B., Comtat M., Thouvenot J. P., Casteilla L. (2004) FEBS Lett. 578, 53–57 [DOI] [PubMed] [Google Scholar]
- 25.Levine R. L., Williams J. A., Stadtman E. R., Shacter E. (1994) Methods Enzymol. 233, 346–357 [DOI] [PubMed] [Google Scholar]
- 26.Fabrizio P., Liou L. L., Moy V. N., Diaspro A., Valentine J. S., Gralla E. B., Longo V. D. (2003) Genetics 163, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garí E., Piedrafita L., Aldea M., Herrero E. (1997) Yeast 13, 837–848 [DOI] [PubMed] [Google Scholar]
- 28.Parrou J. L., François J. (1997) Anal. Biochem. 248, 186–188 [DOI] [PubMed] [Google Scholar]
- 29.Chance B., Sies H., Boveris A. (1979) Physiol. Rev. 59, 527–605 [DOI] [PubMed] [Google Scholar]
- 30.Turrens J. F., Alexandre A., Lehninger A. L. (1985) Arch. Biochem. Biophys. 237, 408–414 [DOI] [PubMed] [Google Scholar]
- 31.Casteilla L., Rigoulet M., Pénicaud L. (2001) IUBMB Life 52, 181–188 [DOI] [PubMed] [Google Scholar]
- 32.Bender E., Kadenbach B. (2000) FEBS Lett. 466, 130–134 [DOI] [PubMed] [Google Scholar]
- 33.Suzuki Y. J., Forman H. J., Sevanian A. (1997) Free Radic. Biol. Med. 22, 269–285 [DOI] [PubMed] [Google Scholar]
- 34.Sun J., Trumpower B. L. (2003) Arch. Biochem. Biophys. 419, 198–206 [DOI] [PubMed] [Google Scholar]
- 35.Smith A., Ward M. P., Garrett S. (1998) EMBO J. 17, 3556–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeRisi J. L., Iyer V. R., Brown P. O. (1997) Science 278, 680–686 [DOI] [PubMed] [Google Scholar]
- 37.Ronne H. (1995) Trends Genet. 11, 12–17 [DOI] [PubMed] [Google Scholar]
- 38.Olesen J., Hahn S., Guarente L. (1987) Cell 51, 953–961 [DOI] [PubMed] [Google Scholar]
- 39.Hahn S., Pinkham J., Wei R., Miller R., Guarente L. (1988) Mol. Cell. Biol. 8, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forsburg S. L., Guarente L. (1989) Genes Dev. 3, 1166–1178 [DOI] [PubMed] [Google Scholar]
- 41.McNabb D. S., Xing Y., Guarente L. (1995) Genes Dev. 9, 47–58 [DOI] [PubMed] [Google Scholar]
- 42.Dang V. D., Valens M., Bolotin-Fukuhara M., Daignan-Fornier B. (1994) Yeast 10, 1273–1283 [DOI] [PubMed] [Google Scholar]
- 43.Fondrat C., Kalogeropoulos A. (1996) Comput. Appl. Biosci. 12, 363–374 [DOI] [PubMed] [Google Scholar]
- 44.Guarente L., Ptashne M. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 2199–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guarente L., Lalonde B., Gifford P., Alani E. (1984) Cell 36, 503–511 [DOI] [PubMed] [Google Scholar]
- 46.Vélot C., Haviernik P., Lauquin G. J. (1996) Genetics 144, 893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devin A., Rigoulet M. (2007) Am. J. Physiol. Cell Physiol. 292, C52–C58 [DOI] [PubMed] [Google Scholar]
- 48.Chen R., Fearnley I. M., Peak-Chew S. Y., Walker J. E. (2004) J. Biol. Chem. 279, 26036–26045 [DOI] [PubMed] [Google Scholar]
- 49.Thannickal V. J., Fanburg B. L. (2000) Am. J. Physiol. Lung Cell Mol. Physiol. 279, L1005–L1028 [DOI] [PubMed] [Google Scholar]
- 50.Delaunay A., Pflieger D., Barrault M. B., Vinh J., Toledano M. B. (2002) Cell 111, 471–481 [DOI] [PubMed] [Google Scholar]
- 51.Wood M. J., Storz G., Tjandra N. (2004) Nature 430, 917–921 [DOI] [PubMed] [Google Scholar]
- 52.Chandel N. S., Maltepe E., Goldwasser E., Mathieu C. E., Simon M. C., Schumacker P. T. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 11715–11720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beauchamp C., Fridovich I. (1971) Anal. Biochem. 44, 276–287 [DOI] [PubMed] [Google Scholar]