

Abstract
Previously we have identified the lipid mediator sphingosylphosphorylcholine (SPC) as the first potentially endogenous inhibitor of the ubiquitous Ca2+ sensor calmodulin (CaM) (Kovacs, E., and Liliom, K. (2008) Biochem. J. 410, 427–437). Here we give mechanistic insight into CaM inhibition by SPC, based on fluorescence stopped-flow studies with the model CaM-binding domain melittin. We demonstrate that both the peptide and SPC micelles bind to CaM in a rapid and reversible manner with comparable affinities. Furthermore, we present kinetic evidence that both species compete for the same target site on CaM, and thus SPC can be considered as a competitive inhibitor of CaM-target peptide interactions. We also show that SPC disrupts the complex of CaM and the CaM-binding domain of ryanodine receptor type 1, inositol 1,4,5-trisphosphate receptor type 1, and the plasma membrane Ca2+ pump. By interfering with these interactions, thus inhibiting the negative feedback that CaM has on Ca2+ signaling, we hypothesize that SPC could lead to Ca2+ mobilization in vivo. Hence, we suggest that the action of the sphingolipid on CaM might explain the previously recognized phenomenon that SPC liberates Ca2+ from intracellular stores. Moreover, we demonstrate that unlike traditional synthetic CaM inhibitors, SPC disrupts the complex between not only the Ca2+-saturated but also the apo form of the protein and the target peptide, suggesting a completely novel regulation for target proteins that constitutively bind CaM, such as ryanodine receptors.
Keywords: Calcium Signaling, Calmodulin, Calmodulin-binding Mechanism, Competitive Inhibition, Fluorescence, Sphingolipid, Sphingosylphosphorylcholine, Stopped-flow
Introduction
Calmodulin (CaM)3 is the ubiquitous Ca2+ sensor of eukaryotic cells (1). It plays a central role in cellular signaling, regulating the activity of numerous proteins, including kinases, phosphatases, ion channels, and pumps. Vertebrate CaM is a small (148 residue), acidic (pI 4.1), highly conserved protein comprised of four Ca2+-binding α-helical EF-hand motifs. The first two EF-hands combine to form a globular N-terminal domain separated by a short flexible linker from a highly homologous C-terminal domain consisting of the remaining EF-hands (2). The binding of Ca2+ leads to structural rearrangements that result in exposure of hydrophobic groups in a methionine-rich crevice of each domain (3). In the classical mode of target binding, Ca2+-saturated CaM wraps around its targets, largely driven by interactions between hydrophobic residues of the target sequence with the hydrophobic surface cavities of CaM (4–6). The CaM-binding domains of target proteins share no sequence homology, but fulfill minimal structural characteristics: they are ∼20 residues long and have the potential to fold into a basic amphiphilic α-helix (7).
Widely used CaM antagonists such as trifluoperazine, W7, and calmidazolium are all synthetic and bind only to the Ca2+-saturated form of the protein but not to the apo form (8). Although anti-CaM drugs can interact with CaM in a versatile manner (9, 10), the binding to apoCaM has only been demonstrated in case of KAR-2. Natural products with anti-CaM properties also exist, but can mainly be found in certain plant species and animal venoms (11). We have shown that the putative lipid second messenger sphingosylphosphorylcholine (SPC) (12, 13) binds specifically to both apo- and Ca2+-saturated CaM, and inhibits the action of the protein on the target enzymes phosphodiesterase and calcineurin (14). Data on SPC metabolism is still scarce, but its presence has been demonstrated under several physiologic and pathologic conditions (15–18). Our findings suggest a novel endogenous regulation for CaM and also proposes that CaM might be an intracellular receptor for the sphingolipid. SPC has been shown to affect several cellular processes (19–22), and interestingly, it can act as a first and second messenger as well (23, 24). The best studied intracellular effect of SPC is the liberation of Ca2+ from the endoplasmic reticulum (25). Suggestions have been made on how the sphingolipid elicits Ca2+ release (15, 26–28), but its site of action is still unclear.
Here we study the mechanism and the putative consequences of CaM inhibition by SPC, using fluorescence stopped-flow and equilibrium techniques with the model CaM-binding domain melittin, and several CaM-binding peptides of target proteins involved in Ca2+ signaling. We provide a detailed kinetic model for Ca2+-saturated CaM binding to its targets, and we show that SPC is a strong competitive inhibitor of CaM-target peptide complexes. Our in vitro results give a plausible explanation to how SPC can lead to intracellular Ca2+ mobilization in vivo.
EXPERIMENTAL PROCEDURES
Preparation of Dansyl-labeled CaM
CaM was purified from bovine brain using phenyl-Sepharose affinity chromatography and dansylated according to Kovacs and Liliom (14).
Peptides Representing CaM-binding Domains
Melittin was purchased from Sigma (M2272). The CaM-binding domain of the skeletal muscle Ca2+ release channel (RyR1) (29) and two putative CaM-binding domains from the type 1 inositol 1,4,5-trisphosphate receptor (IP3R1) (30, 31) were synthesized by Bio-Science Trading Ltd. The CaM-binding domain of the plasma membrane Ca2+-ATPase (PMCA) (32) was a kind gift of Dr. Agnes Enyedi. For the exact sequence of peptides used in the study refer to Table 1.
TABLE 1.
Sequence of peptides used in the study
Lipids
d-Erythro-sphingosylphosphoryl-choline (SPC, catalog number 860600), d-erythro-sphingosine-1-phosphate (S1P, catalog number 860492), oleoyl-lysophosphatidylcholine (LPC, catalog number 845875), and oleoyl-lysophosphatidic acid (LPA, catalog number 857130) was purchased from Avanti Polar Lipids. l-Threo-sphingosylphosphorylcholine (LT-SPC, catalog number 1319) was from Matreya. Lipids were delivered from 10 mm methanolic stock solutions.
Stopped-flow
All measurements were carried out in a buffer comprised of 10 mm HEPES, pH 7.4, 100 mm KCl, and 1 mm CaCl2 at 25 °C. Fluorescence time courses were recorded using an SX-20 (Applied Photophysics, UK) stopped-flow apparatus having 2-ms dead time. Dansyl fluorescence was excited at 340 nm and emission was selected with a 455-nm long-pass filter. Time courses were analyzed using the curve fitting software provided with the stopped-flow apparatus or by Origin 7 (OriginLab Corp., Northampton, MA). At least 5 individual curves were collected and averaged for each data point. Each experiment was repeated 2–5 times. Error bars represent the sample standard deviation of the average of data points obtained from different experiments. In most amplitude versus concentration graphs no error bars are present due to the fact that signal amplitudes of different curves collected at different detector gains are not in the same range. In these cases, amplitudes of the actually shown set of curves are presented. Amplitude titration curves are fitted with the following quadratic equation (derived in the supplemental equations) to extract dissociation constants,
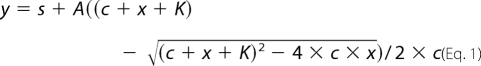 |
s = y at x = 0, A = amplitude, c = concentration of the constant component, and K = dissociation constant. Errors reported on the fitted parameters comprise not only the fitting error, but the standard deviation of the individual data points as well. Kinetic simulation was performed using the Gepasi software (33) and the kinetic parameters given in Table 2.
TABLE 2.
Measured kinetic and thermodynamic parameters of the interaction of Ca2+-saturated CaM with melittin or with SPC
| Experiment | Parametera | Value | Dimension | Fig. |
|---|---|---|---|---|
| CaM-ME binding | k+1,M | 1004 ± 366 | μm−1 s−1 | 1B |
| k−2,M + k+2,M | 49 ± 13 | s−1 | 1B | |
| Kd1,M | 0.3 ± 0.15 | μm | 1C | |
| Kd1–2,M | 0.067 ± 0.044 | μm | 1C | |
| Kdeq,M | 0.054 ± 0.016 | μm | 1D | |
| CaM-SPC binding | k+1,S | >40 | μm−1 s−1 | |
| SPC micelle formation | CMC | 33 ± 2 | μm | Ref. 14 |
| SPC chase | Kd1–2,SC | 32 ± 0.7 | μm | 2B |
| Kd1,SC | 51 ± 3 | μm | 2B | |
| k+1,SC | 2.4 ± 0.2 | μm−1 s−1 | 2C | |
| k1,sat,SC | 348 ± 30 | s−1 | 2C | |
| k2,obs,SC | 14 ± 6 | s−1 | 2C | |
| k3,obs,SC | 0.9 ± 0.3 | s−1 | 2C | |
| ME chase | Kd1–2,MC | 1.0 ± 0.3 | μm | 3B |
| k1,sat,MC | 59 ± 13 | s−1 | 3C | |
| K1(app),MC | 5.4 ± 3.4 | μm | 3C | |
| K2,obs,MC | 4–8 | s−1 | 3C |
Equilibrium Fluorescence Peptide-binding Assays
Fluorescence of dansyl-labeled CaM and the Trp residue of the RyR peptide was monitored on a Jobin Yvon Fluoromax-3 spectrofluorimeter at 25 °C in 10 mm HEPES, pH 7.4, 100 mm KCl, and 1 mm CaCl2. Bandwidths were set to 5 nm. Dansyl was excited at 340 nm, emission was monitored from 400 to 600 nm. Dansyl-CaM titration with melittin was carried out at 0.2 μm dansyl-CaM and the resulting curve was fitted with the above quadratic equation. When screening with lipids SPC, S1P, LPC, LPA, and LT-SPC, dansyl-CaM, RyR peptide, and lipid concentrations were 0.2, 0.5, and 100 μm, respectively. When measuring dose response for SPC, dansyl-CaM and RyR peptide concentrations were 0.2 and 0.5 μm, respectively, and the SPC concentration varied between 10 and 100 μm. In the complimentary set of experiments, the Trp residue of the RyR peptide was excited at 295 nm, and spectra were recorded from 310 to 400 nm. RyR peptide and CaM (unlabeled) concentrations were both 1 μm. In screening experiments, lipid concentrations were 100 μm, while measuring the dose response, SPC concentration varied between 10 and 100 μm. Experiments with dansyl-labeled apoCaM were carried out similarly to measurements with Ca2+-saturated CaM, only in buffer containing 1 mm EGTA instead of 1 mm CaCl2. Measurements with peptides derived from the IP3R1 and the PMCA were conducted as in the case of the RyR peptide. Mixed micelles were prepared by mixing the methanolic stock solutions of the two lipids, and then adding them to the appropriate assay buffer. Each spectrum was corrected for corresponding lipid, protein, peptide, and buffer effects by subtracting a matching buffer scan.
RESULTS
The Model Peptide Melittin Binds to Ca2+-saturated CaM in a Two-step Reversible Manner
The CaM-melittin complex is a widely used model to study the interaction between CaM and the effector proteins it regulates (34). The details of the CaM-melittin binding mechanism, nevertheless, have not been revealed before to the degree we needed to study a composite system with both putative CaM binding partners, SPC and melittin, present. Previous kinetic studies focused on the mutual effect of Ca2+ and target peptide binding to CaM (35, 36) and did not aim at characterizing the CaM-peptide interaction at saturating Ca2+ concentration. Therefore, we performed melittin binding experiments both by equilibrium and transient kinetic methods using the fluorescence of dansyl-CaM. Dansyl labeling was performed in conditions to produce a 1:1 homogeneous labeling to avoid artifacts in the transient kinetics experiments.
Time courses of fluorescence change after mixing dansyl-CaM with melittin are biphasic (Fig. 1A) indicating at least two biochemical transitions both characterized by the expected fluorescence increase (based upon previously determined spectral changes (37, 38)) upon binding. Time courses were analyzed by double exponential fitting. The concentration dependence of the observed rate constants of the two phases (Fig. 1B) suggests that the fast phase is a second-order reaction followed by a slow first-order reaction reflecting some conformational reorganization of the CaM-melittin complex (k2,obs,M = 49 ± 13 s−1, Table 2). The association rate constant of the second-order reaction was determined from the linear phase of the kobs versus concentration curve, in which range the pseudo first-order approximation applies (k+1,M = 1004 ± 366 μm−1 s−1, Table 2). The dissociation rate constant could not be reliably extracted from the linear fit because of the large uncertainty of the y intercept. We could extract the dissociation constant of the first process of the binding from the concentration dependence of the fast phase amplitude (Fig. 1C, Kd1,M = 0.3 ± 0.15 μm, Table 2). The total amplitude describing the entire binding process is analogous to equilibrium binding data and yielded Kd1–2,M = 0.067 ± 0.044 μm. Consistently, equilibrium fluorescence titration of dansyl-CaM with melittin in a fluorimeter yielded Kd,eq,M = 0.054 ± 0.016 μm (Fig. 1D, Table 2) close to the previous Kd1–2,M value within error. Taking Equations 2 and 3 (derived in the supplemental equations) and Kd1–2,M into consideration, we calculated all remaining parameters of the two-step binding process summarized in Scheme 1.
 |
Using the kinetic parameters in Scheme 1, we ran numerical simulations for the time courses shown in Fig. 1A to test the validity of our model. To model the experimentally observed time courses, we assumed that the two high fluorescence states have similar intensities and thus the observed fluorescence reflects the sum of the concentrations of the two populations (** in Scheme 1 and Fig. 8). The simulated time courses were subjected to the same analysis procedures as the experimental data. As a result, kobs values showed good agreement with the experimentally obtained ones (Fig. 1B) indicating that the established CaM-ME binding model is consistent with our experimental data.
FIGURE 1.

Binding of Ca2+-saturated dansyl-CaM to the model peptide melittin. A, fluorescence time courses on the reaction of 0.1 μm (final concentration) dansyl-CaM with buffer or with 0.15–0.6 μm melittin. Stopped-flow traces are shown in gray, black lines through the data represent the best double exponential fits to the curves. The x axis is shown from t = 0.002 s (the dead time of the stopped-flow apparatus), exponential fits converge to the fluorescence intensity level of the “buffer” curve. B, melittin concentration dependence of the observed binding rate constants from exponential fits to the stopped-flow traces (fast phase, ■; and slow phase, ●) or from kinetic simulation of the same time courses (fast phase, □; slow phase, ○). A linear fit to the pseudo-first order part of the curve yielded k+1,M = 1004 ± 366 μm−1 s−1. The rate constants of the slow phase (inset, ●) did not exhibit concentration dependence and had a first-order k2,obs,M = 49 ± 13 s−1. C, amplitude titration extracted from the exponential fits to the stopped-flow traces. The quadratic fit (smooth line through the data) to the concentration-dependent fast phase (■) comprising 88% of the total amplitude at the highest measured concentration yielded an apparent Kd of 0.3 ± 0.15 μm. The amplitude of the slow phase (●) exhibited a tendency to decrease with concentration. Fitting the total amplitude data (○) yielded a Kd of 0.067 ± 0.044 μm. D, equilibrium fluorescence titration of 0.2 μm dansyl-CaM with melittin. The Kd from the quadratic fit is 0.054 ± 0.016 μm. Error bars represent the sample standard deviation of the average of data points obtained from different experiments.
SCHEME 1.

FIGURE 8.

Schematic representation of the model by which SPC competes for CaM with target peptides. Protein Data Bank codes 1CLL and 1CDM were used to prepare the structures representing CaM and the CaM·peptide** complex, respectively. The CaM (blue) and peptide (magenta) molecules are shown as schematics. Bound Ca2+ ions are shown as light blue spheres. The CaM·peptide* and the CaM·SPC species are purely hypothetical, and are depicted by the Ca2+-saturated CaM structure PDB 1CLL bound to the peptide or the micelle. The SPC micelle is depicted as a gray sphere with a diameter comparable with the width of a lipid bilayer. The figure was prepared using PyMOL (DeLano Scientific). Rate constants shown in brackets are estimated values and were used for global simulations.
The Binding of SPC to CaM Is Rapid and Saturating above the Critical Micelle Concentration
We wished to characterize the kinetics of the interaction between dansyl-CaM and SPC as we did for the CaM-peptide interaction. Fluorescence time courses upon mixing dansyl-CaM with various concentrations of SPC proved to be extremely fast and yielded only small fluorescence changes. Even at the lowest SPC concentration the course of fluorescence change was lost in the 2-ms dead time of the stopped-flow apparatus and is therefore not shown. We can only put a lower estimate on the association rate constant (k+1,S > 40 μm−1 s−1, Table 2). Our previous observations suggested that the species interacting with CaM is the micelle not the monomer (14). Above the critical micelle concentration (CMC = 33 ± 2 μm (14)) the fluorescence intensity change upon binding became completely saturated. As a result of these SPC binding experiments we conclude that Ca2+-saturated CaM binding to SPC is fast and saturated above the CMC, which is relevant for designing the competition experiments.
SPC Competes for CaM with the Target Peptide
We reacted pre-mixed dansyl-CaM-melittin complexes with various concentrations of SPC to investigate whether these two ligands compete with each other for the same CaM target site. Luckily, formation of the two different dansyl-CaM complexes display opposite fluorescence changes and thus transformation of one complex to the other is expected to be accompanied by a large signal change (CaM·ME** → CaM·SPC). The large fluorescence decrease upon mixing indicated that SPC replaced the previously bound melittin on dansyl-CaM (Fig. 2A). Time courses could be fitted with one (1st data point), two (2nd data point), or three exponentials. The slower kinetic phases have smaller amplitudes that may explain why they go unseen when the total amplitude is relatively small (first data points). The first, concentration-dependent, fast phase corresponds to SPC binding to dansyl-CaM and is characterized by a cooperative amplitude saturation curve (Fig. 2B), which probably reflects micelle formation (parameters in Table 2). As previously observed, above the CMC the fluorescence change becomes saturated. The kobs of the fast phase saturates at about 350 s−1 (Fig. 2C, k1,sat,SC in Table 2), close to the value of the rate constant estimated for CaM-melittin dissociation (k−1,M = 301 s−1, see Scheme 1). The first-order rate constant observed for the second phase (k2,obs,SC = 14 ± 6 s−1) is also almost equal to k−2,M (11 s−1, Scheme 1) in the melittin binding mechanism. These observations imply that SPC binding to CaM is limited by the dissociation of melittin. The initially melittin-saturated dansyl-CaM-melittin complex (mostly populated in the dCaM·ME** state in Scheme 1) must go through the kinetic steps characterized by k−2,M and k−1,M before SPC can associate with CaM. The apparent SPC-CaM association (k+1,SC = 2.4 ± 0.2 μm−1 s−1) is not as fast as in the case of SPC binding to pure dansyl-CaM because melittin re-binding occurs and the observed rates are set as a function of the concentration ratios and kinetic parameters of the two ligands. A third kinetic phase with a slow observed rate constant of k3,obs,SC = 0.9 ± 0.3 s−1 and an 18% relative amplitude appears at [SPC] > CMC possibly due to a conformational change in the dansyl-CaM-micelle complex.
FIGURE 2.

Kinetics of the interaction of SPC with the Ca2+-saturated dansyl-CaM-melittin complex. A, time courses of melittin dissociation from dansyl-CaM after mixing an equilibrated sample of 0.4 μm dansyl-CaM and 0.8 μm melittin with 0–500 μm SPC (pre-mix concentrations). The first trace was best fitted with single, the second with double, and further curves with triple exponentials represented by the smooth lines through the data. B, concentration dependence of the amplitudes (inset, A1 (■), A2 (○), and A3 (△)) and maximal fluorescence changes derived from the exponential fits to the stopped-flow traces shown in panel A. Maximal fluorescence change was calculated from the ymax value of the exponential fits. A Hill equation having n = 4 ± 0.4 and a half-maximal signal change at [SPC] = 32 ± 0.7 μm, close to the previously determined CMC for SPC, provided the best fit to the curve. Similar fit to the amplitude of the fast phase yielded n = 3 ± 0.5 and A1,max/2 = 51 ± 3 μm. At saturation, the three observed kinetic phases A1, A2, and A3 take 78, 4, and 18% shares of the total amplitude, respectively. C, SPC concentration dependence of the observed rate constants of the fast (main panel, ■) and slow (inset, k2, ○; k3, △) phases. The fast phase data showed linear concentration dependence and reached saturation at about 150 μm SPC with k1,sat,SC = 348 ± 30 s−1. The two slower phases did not depend on SPC concentration in the measured range and varied in the range of k2,obs,SC = 14 ± 6 s−1 and k3,obs,SC = 0.9 ± 0.3 s−1. Error bars represent the sample standard deviation of the average of data points obtained from different experiments.
We also carried out the reverse chasing experiment in which dansyl-CaM saturated with SPC was mixed with various concentrations of melittin (Fig. 3). We again expected large fluorescence changes, a fluorescence increase this time, because SPC exchanges to melittin on dansyl-CaM (CaM·SPC → CaM·ME**). Time courses followed double exponentials (Fig. 3A) and the fluorescence change exhibited hyperbolic saturation with an apparent dissociation constant of 1 μm (Fig. 3B, Kd1–2,MC = 1.0 ± 0.3 μm, Table 2). The observed rate constants of both phases were dependent on melittin concentration in the measured concentration range (Fig. 3C). The fast phase exhibited a saturating character and could be fitted with a hyperbole that saturates at k1,sat,MC = 59 ± 13 s−1. This rate constant likely originates from the one observed for the second process in melittin binding that is in the same range within error (k+2, M + k−2,M = 49 ± 13 s−1). At infinite melittin concentration, the initial binding of melittin (characterized by k+1,M × [CaM]) will be fast and saturated, thus, the entire binding process will be limited by the second, slower kinetic step. The apparent half-maximal saturation of the fast phase (K1(app)MC = 5.4 ± 3.4 μm) is the result of an interplay between the three reversible processes shown in Fig. 8 and it appears to be close to the equilibrium constant calculated for the second CaM-melittin binding step. The slow phase (4–8 s−1) represents the smaller portion of the total amplitude. On the basis of our parallel binding model (Fig. 8), it should originate from the dissociation of the CaM-micelle complex (consistently with the relatively small signal change observed upon the CaM-SPC interaction) and is expected to reach saturation (kobs,sat = k−1,S). Because micelle binding to CaM was too fast to be measured, these observed rate constants are the only accessible parameters to indicate that k−1,S is relatively slow.
FIGURE 3.

Dissociation of the Ca2+-saturated dansyl-CaM-SPC complex by melittin. A, time courses of the chasing experiment. Equilibrated 0.4 μm dansyl-CaM and 100 μm SPC were mixed with 0–20 μm melittin (pre-mix concentrations). Smooth lines represent double exponential fits to the data. B, concentration dependence of the amplitudes (fast phase, circo; slow phase, □; and total amplitude, ■) derived from exponential fits to the stopped-flow traces. The best quadratic fit to the total amplitude curve yielded an apparent Kd1–2,MC of 1 ± 0.3 μm. C, concentration dependence of the observed rate constants of the fast (○) and slow (□) phases. The fast phase was best fitted with a quadratic equation having an y intercept at 10 ± 4 s−1, a rate constant of 59 ± 13 s−1 at saturation, and an apparent Kd = 5.4 ± 3.4 μm. The slow phase exhibited a weak dependence on melittin concentration and the kobs varied between 4 and 8 s−1. D, analysis of simulated time courses. Kinetic parameters used for the simulation are shown in Fig. 8. Concentration dependence of the amplitudes of exponential fits to the simulated curves (fast phase, ○; slow phase, □, and total amplitude, ■). The best quadratic fit to the total amplitude curve yielded an apparent Kd of 1.5 ± 0.12 μm. Error bars represent the sample standard deviation of the average of data points obtained from different experiments.
We formulated a relationship between the thermodynamic parameters of CaM binding to either SPC or melittin and the apparent Kd-s of the chasing experiments (Equation 4; for details, see the supplemental equations).
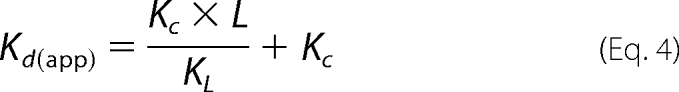 |
C represents the chaser, L represents the prebound ligand, and KL and KC indicate the dissociation constants for the ligand and the chaser. Substituting the previously determined values for KC (∼60 nm, Fig. 1, C and D, Kd1–2,M and Kd,eq,M in Table 2), Kd(app) (1 μm, Fig. 3B, and Kd1–2,MC in Table 2), and L (50 μm) into Equation 4, we calculate the experimentally inaccessible parameter, KL, to be 3 μm (in SPC monomer concentration). This corresponds to 3/n μm in SPC micelle concentration, where n is the number of monomers per micelle. We estimate the molecular ratio to be 100–200 monomers per micelle,4 which would imply that the Kd for the dansyl-CaM-micelle complex is 0.015–0.03 μm.
We performed kinetic simulations to test if our kinetic model is plausible taking an average size of 150 monomers/micelle into account. We used the herein defined kinetic parameters and relative fluorescence levels of 2 and 0.8 for the dansyl-CaM-melittin and dansyl-CaM-micelle complexes, respectively, compared with free dansyl-CaM. By simulating the experiment shown in Fig. 3A, we obtained double exponential curves similar to the measured ones. The amplitude analysis of the simulated curves (Fig. 3D) resulted in an apparent Kd of 1.5 ± 0.12 μm, close to the experimentally determined 1 μm. Simulated kobs values were also in the range of the measured ones. As a summary of our results on the SPC-melittin competition experiments we suggest the model shown in Fig. 8.
SPC Dissociates the Complex between Ca2+-saturated CaM and the CaM-binding domain of RyR1
To investigate the possible functional consequences of the CaM inhibitory effect of SPC, we examined the impact of SPC on interactions between CaM and CaM-binding domains of proteins involved in Ca2+ homeostasis, because the best described function of SPC as a putative second messenger is the liberation of Ca2+ from intracellular stores (25). As SPC has been suggested to be involved in activation of ryanodine receptors (RyRs) (15, 26), we started working with the CaM-binding domain (amino acids 3614–3643) of the skeletal muscle Ca2+ release channel (RyR1) (29). By monitoring the fluorescence of both the dansyl-labeled protein and Trp of the RyR1 peptide, we were able to examine their interaction from the aspect of both CaM and the Ca2+ channel. As both fluorescence signals undergo large changes upon complexation, they provide a convenient method to distinguish whether a third compound dissociates the complex or not.
The fluorescence intensity of dansyl-labeled Ca2+-saturated CaM increases ∼2-fold accompanied by an ∼30-nm blue-shift upon binding to its target peptide on the RyR. After the addition of saturating amounts of SPC to the peptide-CaM complex, the spectrum resembles the SPC-bound form of dansyl-labeled CaM, implying that all peptide was replaced by SPC on CaM (Fig. 4A). We have demonstrated that this complex-dissociating effect of SPC is selective compared with structurally and functionally related lysophospholipids S1P, LPC, and LPA (Fig. 4C), and occurs with a half-effective concentration of 19.4 ± 1.4 μm and a cooperativity coefficient of 2.6 ± 0.4 (Fig. 4E).
FIGURE 4.

Dissociation of the complex between Ca2+-saturated CaM and the CaM-binding domain of RyR1 by SPC, revealed by the fluorescence of dansyl-labeled CaM and the Trp of the RyR peptide. A, spectra of 0.2 μm Ca2+-saturated dansyl-CaM (○), 0.2 μm Ca2+-saturated dansyl-CaM with 0.5 μm RyR peptide (●), 0.2 μm Ca2+-saturated dansyl-CaM in the presence of 100 μm SPC (△), and 0.2 μm Ca2+-saturated dansyl-CaM in the presence of 0.5 μm RyR peptide and 100 μm SPC (▲). B, spectra of 1 μm RyR peptide (□), 1 μm RyR peptide with 1 μm Ca2+-saturated CaM (■), 1 μm RyR peptide in the presence of 100 μm SPC (▿), and 1 μm RyR peptide in the presence of 1 μm Ca2+-saturated CaM and 100 μm SPC (▼). Spectra in panels A and B are averaged from three independent measurements. C, effect of related lysophospholipids on the interaction between Ca2+-saturated CaM and the CaM-binding domain of RyR1. Bars depict the fluorescence intensity of 0.2 μm Ca2+-saturated dansyl-CaM in the presence of 0.5 μm RyR peptide and 100 μm lipids. D, the same as in panel C, but measuring the Trp fluorescence of the RyR peptide. Bars depict the fluorescence intensity of 1 μm RyR peptide in the presence of 1 μm Ca2+-saturated CaM and 100 μm lipids. Mean ± S.E. values in panels C and D were calculated from three independent experiments. The addition of SPC and LT-SPC brought forth a significant decrease in intensity (p < 0.05, see asterisks), based on Student's t test. E, concentration dependence of the complex dissociating ability of SPC. Ca2+-saturated dansyl-CaM and RyR peptide concentrations were 0.2 and 0.5 μm, respectively. F, the same as in panel E, but measuring the Trp fluorescence of the RyR peptide. RyR peptide and Ca2+-saturated CaM concentrations were both 1 μm. Data points in panels E and F represent the mean ± S.E. values of three independent determinations. Fitting a sigmoidal dose-response function yielded an EC50 value of 19.4 ± 1.4 μm with a cooperativity coefficient of 2.6 ± 0.4 for dansyl-CaM, and an EC50 value of 19.3 ± 3.4 μm with a cooperativity coefficient of 2.2 ± 0.8 for Trp fluorescence of the peptide.
In the complementary experiment, when monitoring Trp fluorescence of the RyR peptide, the acquired results correspond exactly to the ones obtained using dansyl-CaM fluorescence as a reporter. Binding of Ca2+-saturated CaM brought forth an ∼2.5-fold increase in Trp fluorescence intensity and an ∼20 nm blueshift. The addition of SPC resulted in a spectrum similar to the spectrum of the free RyR peptide (Fig. 4B). This effect was again specific (Fig. 4D), and gave an EC50 value of 19.3 ± 3.4 μm and a cooperativity coefficient of 2.2 ± 0.8 (Fig. 4F), very similar to the ones obtained from dansyl-CaM fluorescence.
l-Threo-SPC, a synthetic stereoisomer of the naturally occurring d-erythro-SPC, can also potently dissociate the complex between Ca2+-saturated CaM and the CaM-binding domain of RyR1 (Fig. 4, C and D). Note that by SPC, we refer to d-erythro-SPC throughout this report.
SPC Dissociates the Complex between ApoCaM and the CaM-binding Domain of RyR1
Calcium binding to CaM leads to an N-terminal shift in its binding site on the RyR, hence this region of the channel possesses the unique feature of containing a distinct binding site for both apo- and Ca2+-saturated CaM (29). Because traditional CaM inhibitors only interact with Ca2+-saturated CaM (8), whereas SPC binds to both forms of the protein (14), we investigated the effect of SPC on the apoCaM-RyR peptide interaction. Although the complex formation between apoCaM and the RyR peptide yielded significantly smaller changes in fluorescence than in the case of Ca2+-saturated CaM, the complex dissociating ability of SPC could still be demonstrated. The spectra of dansyl-labeled apoCaM revealed that if saturating amounts of SPC are present, the protein is predominantly bound to the sphingolipid (Fig. 5).
FIGURE 5.

Dissociation of the complex between apoCaM and the CaM-binding domain of RyR1 by SPC. Spectra of 0.2 μm dansyl-labeled apoCaM (○), 0.2 μm dansyl-labeled apoCaM with 0.5 μm RyR peptide (●), 0.2 μm dansyl-labeled apoCaM in the presence of 100 μm SPC (△), and 0.2 μm dansyl-labeled apoCaM in the presence of 0.5 μm RyR peptide and 100 μm SPC (▲). Spectra are averaged from three independent measurements.
SPC Dissociates the Complex between Ca2+-saturated CaM and the CaM-binding Domain of Several Proteins Involved in Ca2+ Homeostasis
The effect of SPC on the interaction of Ca2+-saturated CaM with further CaM-binding proteins involved in Ca2+ homeostasis was also explored. Besides the RyR1 peptide, two peptides corresponding to residues 1564–1585 and 106–128 of the IP3R1 and a peptide corresponding to residues 2–21 of the human erythrocyte PMCA were examined. For details on these peptides refer to Table 1. We found that SPC disrupted the complex between each of these peptides and Ca2+-saturated CaM, as the fluorescence of the dansyl-labeled protein in the presence of both the peptide and SPC resembled the SPC-bound form (Fig. 6). Other lysophospholipids such as S1P, LPC, and LPA did not significantly affect the fluorescence of the CaM-target complex.
FIGURE 6.

SPC-induced dissociation of complexes between Ca2+-saturated CaM and peptides derived from proteins involved in Ca2+ homeostasis. Bars depict the fluorescence intensity of 0.2 μm Ca2+-saturated dansyl-CaM with peptides from RyR1, IP3R1, and PMCA (see Table 1 for details) at a concentration of 0.5 μm, in the absence (white bars) and presence (gray bars) of 100 μm SPC, S1P, LPC, and LPA, respectively. Mean ± S.E. values were calculated from three independent experiments, and the asterisks represent a significant decrease (p < 0.05), based on Student's t test.
SPC Exerts Its Effects in Mixed Micelles, More Relevant to in Vivo Conditions
To assess whether SPC can displace CaM from its targets under conditions more resembling the in vivo situation, experiments with mixed micelles were carried out. In these experiments, varying amounts of SPC were incorporated into micelles consisting of lipids that did not have any significant effect on the CaM-target peptide system, such as S1P, LPC, and LPA. Fig. 7A clearly demonstrates that SPC dissociates the CaM-peptide complex in the presence of other lipids just as potently as pure SPC. To comprehend the effect of “dilution” caused by other lipids, we measured the dose response of complex dissociation in SPC/LPC mixed micelles keeping the SPC content at a constant 20% (Fig. 7B). Comparing these results with the dose response for pure SPC revealed that the fluorescence of dansyl-CaM changes less steeply in the case of mixed micelles. A possible explanation for this phenomenon might be that at lower concentrations additional lipids aid the effect of SPC by forming micelles at lower SPC concentrations. While at higher concentrations, the presence of other lipids seems to have a minor negative diluting effect on the ability of SPC to interfere with CaM function. These observations point to the potential of SPC to displace CaM under in vivo conditions near membrane surfaces enriched in the signaling sphingolipid.
FIGURE 7.

Effect of SPC on the Ca2+-saturated CaM-RyR1 peptide complex in mixed micelles. A, bars depict the fluorescence intensity of 0.2 μm Ca2+-saturated dansyl-CaM with 0.5 μm RyR1 peptide in the absence (white bars) and presence of pure SPC (lightest gray bar) or mixed micelles of various amounts of SPC incorporated into S1P, LPC, or LPA micelles, respectively (darker gray bars). Total lipid concentration was held constant at 100 μm, and mixed micelles contained either 20 or 50 μm SPC. Error bars depict an average experimental error of 5%. B, concentration dependence of the complex dissociating ability of 20% SPC containing LPC micelles (▿) compared with pure SPC (○) taken from Fig. 4E. Ca2+-saturated dansyl-CaM and RyR peptide concentrations were 0.2 and 0.5 μm, respectively. In contrast to panel A, the total lipid concentration varied, and the SPC content was held constant at 20%. Data points represent the mean ± S.E. values of three independent measurements.
DISCUSSION
In our previous studies (14) we have shown that the putative lipid second messenger SPC can bind to CaM selectively and inhibit its activity in in vitro assay systems. We believe that this finding is of particular importance for the following reasons: 1) it suggests an intracellular target site for SPC, the mechanism of action of which is yet unclear; 2) it proposes a novel type of endogenous regulation for CaM, since classical CaM inhibitors are all synthetic; 3) the feature of SPC binding to both apo and Ca2+-saturated CaM is unique even among the synthetic CaM inhibitors; and 4) SPC is a lipid, and the fact that a lipid would be able to specifically regulate the well known Ca2+ sensor is a novel concept. Due to these reasons, we find it highly important to decipher the mechanism and also the possible functional consequences of this novel regulation of CaM by SPC. To do so, we turned to the simplest model system for CaM-target interactions, and studied the impact of SPC on interaction between CaM and its target peptides using fluorescence methods.
Having recognized that relevant kinetics data are missing on the mechanism of CaM binding to its target peptides, we first aimed to characterize the CaM-melittin interaction at saturating Ca2+ concentrations (see Fig. 1). Details of the model we established are shown in Scheme 1 and Fig. 8 and its main features are: 1) rapid, reversible binding of the peptide to CaM accompanied by a fluorescence increase of the dansyl label and 2) a slower, reversible conformational change with a further apparent fluorescence increase. The overall process is shifted to the right implying that the predominant conformation is the compact dCaM·ME** species. Our two-step sequential binding model agrees with literature data proposing that an initial binding of target proteins occurs on the N-terminal domain of CaM followed by formation of the compact structure shown in Fig. 8 (36).
We demonstrated that SPC competes for CaM with the model CaM-binding domain melittin, using ligand chasing experiments for both ligands (see Figs. 2 and 3). The kinetic parameters we obtained in the SPC chasing assay (see Fig. 2) clearly indicate that SPC competes with melittin for the same binding site. SPC could only bind to CaM upon melittin dissociation as its binding rate constant was limited by the dissociation rate constant of the CaM-melittin complex (see Table 2). No sign of a trimeric complex comprising all three interacting partners has been observed. Structural characterization of the CaM-SPC complex also revealed that the peptide and the sphingolipid occupy the same binding site on CaM.4 The previous observation that SPC binds to CaM as a micelle, not as a monomer (14), was reinforced in the stopped-flow measurements as well (Fig. 2B).
The reverse chasing experiment, in which melittin served as a competitor of the pre-bound SPC (Fig. 3), also indicated the concentration-dependent replacement of the pre-bound ligand for melittin on CaM. In addition, this experiment yielded information relevant to the size of the SPC micelle that interacts with CaM. Using the relationship described in Equation 4 and kinetic simulations, we can estimate that an SPC micelle is composed of about 150 monomers.
Next, to investigate the specificity of the complex-dissociating effect of SPC regarding the peptide, and also to assess its possible functional consequences, we studied the interaction between the CaM-binding domain of RyR1 and CaM. We chose this peptide because SPC has previously been suggested to be involved in regulation of RyRs (15, 26), and also because calcium binding to CaM leads to an N-terminal shift in its binding site on the peptide (29). Thus, this peptide is unique in a way that it binds constitutively to either apo- or Ca2+-saturated CaM, so it is a convenient tool to study the effect of SPC on the apoCaM-target interaction as well. Here we clearly demonstrate that SPC dissociates the complex between Ca2+-saturated CaM and the CaM-binding domain of RyR1 (Fig. 4). This effect is selective compared with structurally related signaling lipids such as S1P, LPC, and LPA, and occurs with an EC50 of ∼20 μm. This is in a similar low micromolar range as the observed Ca2+ mobilizing action of SPC in previous reports (15, 24–26). Moreover, Meyer zu Heringdorf et al. (24) also showed that l-threo-SPC, a synthetic stereoisomer, liberated Ca2+ only if administered intracellullarly, but was ineffective extracellularly. The fact that in our measurements l-threo-SPC gave similar results as the naturally occurring d-erythro-SPC implies that the effect we observed and the findings by Meyer zu Heringdorf et al. (24) may share a common underlying mechanism.
Furthermore, we also show that SPC dissociates the complex between apoCaM and the CaM-binding domain of RyR1 (Fig. 5). This finding suggests an entirely novel endogenous regulation for RyRs and other proteins that constitutively bind CaM regardless of Ca2+. SPC is the first compound having the potential to completely free these proteins from the Ca2+ sensor.
As we have mentioned before, the best characterized intracellular action of SPC is that it can liberate Ca2+ from the endoplasmic reticulum. CaM is known to regulate several proteins involved in modulating intracellullar Ca2+ levels, providing a negative feedback mechanism for the Ca2+ signal. Ca2+-saturated CaM has been shown to inhibit the two most abundant Ca2+ channels, RyRs and IP3Rs (39), and to activate the PMCA (40). Our hypothesis is that if SPC interferes with this function of CaM, it would have exactly the opposite effect. That is, to activate Ca2+ channels and inhibit Ca2+ pumps, which would eventually lead to the elevation of intracellullar Ca2+ levels. In this report, we show that SPC disrupts the complex between CaM and the CaM-binding domain of RyR1, IP3R1, and PMCA (Fig. 6). We excluded sarcoplasmic reticulum Ca2+-APTases from our study because they are not directly regulated by CaM. We demonstrated that SPC does not differentiate between CaM-target complexes in vitro. The interactions it can actually modify in vivo are probably selected by their cellular localization. Currently our knowledge of SPC metabolism is scarce (15–18), so to accurately address this question, further study into the mechanism and location of in vivo SPC production is necessary. Nevertheless, experiments carried out with micelles containing other lipids besides SPC confirmed that SPC can displace CaM from its targets even when incorporated into a mixed lipid environment. This finding argues for the plausibility of the same phenomenon to occur under in vivo conditions near a membrane surface enriched in SPC.
To conclude the possible physiological relevance of our study, we propose that SPC interference with any of the above mentioned interactions will lead to elevated intracellular Ca2+-levels. Thus, we suggest a mechanism by which the putative second messenger SPC might perform its previously reported intracellular function.
Supplementary Material
Acknowledgments
We thank Dr. Agnes Enyedi (National Blood Center, Dept. of Molecular Cell Biology, Budapest, Hungary) for the kind gift of the PMCA peptide and Dr. Mihaly Kovacs (Eotvos Lorand University, Budapest, Hungary) for help in interpretation of the kinetics data.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01TW008130-01 (to J. T.), Hungarian Scientific Research Fund OTKA Grants 61501 (to K. L.), 72008 (to J. T.), K68229 and CK-78646 (to B. G. V.), National Development Agency Grant KMOP-1.1.2-07/1-2008-0003, Howard Hughes Medical Institutes Grants 55005628 and 55000342 (to B. G. V.), the Alexander von Humboldt Foundation, National Office for Research and Technology, Hungary, Grant JÁP_TSZ_071128_TB_INTER, and European Union Grants FP6 SPINE2c LSHG-CT-2006-031220 and TEACH-SG LSSG-CT-2007-037198 (to B. G. V.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental equations.
E. Kovacs, V. Harmat, J. Tóth, B. G. Vértessy, K. Módos, J. Kardos, and K. Liliom, submitted for publication.
- CaM
- calmodulin
- RyR1
- ryanodine receptor type 1
- IP3R1
- inositol 1,4,5-trisphosphate receptor type 1
- PMCA
- plama membrane Ca2+ pump
- SPC
- sphingosylphosphorylcholine
- S1P
- sphingosine-1-phosphate
- LPC
- lysophosphatidylcholine
- LPA
- lysophosphatidic acid
- LT-SPC
- l-threo-sphingosylphosphorylcholine
- CMC
- critical micelle concentration
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl.
REFERENCES
- 1.Chin D., Means A. R. (2000) Trends Cell Biol. 10, 322–328 [DOI] [PubMed] [Google Scholar]
- 2.Chattopadhyaya R., Meador W. E., Means A. R., Quiocho F. A. (1992) J. Mol. Biol. 228, 1177–1192 [DOI] [PubMed] [Google Scholar]
- 3.LaPorte D. C., Wierman B. M., Storm D. R. (1980) Biochemistry 19, 3814–3819 [DOI] [PubMed] [Google Scholar]
- 4.Meador W. E., Means A. R., Quiocho F. A. (1992) Science 257, 1251–1255 [DOI] [PubMed] [Google Scholar]
- 5.Meador W. E., Means A. R., Quiocho F. A. (1993) Science 262, 1718–1721 [DOI] [PubMed] [Google Scholar]
- 6.Maximciuc A. A., Putkey J. A., Shamoo Y., Mackenzie K. R. (2006) Structure 14, 1547–1556 [DOI] [PubMed] [Google Scholar]
- 7.Rhoads A. R., Friedberg F. (1997) FASEB J. 11, 331–340 [DOI] [PubMed] [Google Scholar]
- 8.Massom L., Lee H., Jarrett H. W. (1990) Biochemistry 29, 671–681 [DOI] [PubMed] [Google Scholar]
- 9.Vertessy B. G., Harmat V., Böcskei Z., Náray-Szabó G., Orosz F., Ovádi J. (1998) Biochemistry 37, 15300–15310 [DOI] [PubMed] [Google Scholar]
- 10.Molnár A., Liliom K., Orosz F., Vértessy B. G., Ovádi J. (1995) Eur. J. Pharmacol. 291, 73–82 [DOI] [PubMed] [Google Scholar]
- 11.Martínez-Luis S., Pérez-Vásquez A., Mata R. (2007) Phytochemistry 68, 1882–1903 [DOI] [PubMed] [Google Scholar]
- 12.Meyer zu Heringdorf D., Himmel H. M., Jakobs K. H. (2002) Biochim. Biophys. Acta 1582, 178–189 [DOI] [PubMed] [Google Scholar]
- 13.Nixon G. F., Mathieson F. A., Hunter I. (2008) Prog. Lipid Res. 47, 62–75 [DOI] [PubMed] [Google Scholar]
- 14.Kovacs E., Liliom K. (2008) Biochem. J. 410, 427–437 [DOI] [PubMed] [Google Scholar]
- 15.Betto R., Teresi A., Turcato F., Salviati G., Sabbadini R. A., Krown K., Glembotski C. C., Kindman L. A., Dettbarn C., Pereon Y., Yasui K., Palade P. T. (1997) Biochem. J. 322, 327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liliom K., Sun G., Bünemann M., Virág T., Nusser N., Baker D. L., Wang D. A., Fabian M. J., Brandts B., Bender K., Eickel A., Malik K. U., Miller D. D., Desiderio D. M., Tigyi G., Pott L. (2001) Biochem. J. 355, 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okamoto R., Arikawa J., Ishibashi M., Kawashima M., Takagi Y., Imokawa G. (2003) J. Lipid Res. 44, 93–102 [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Lafrasse C., Vanier M. T. (1999) Neurochem. Res. 24, 199–205 [DOI] [PubMed] [Google Scholar]
- 19.Desai N. N., Spiegel S. (1991) Biochem. Biophys. Res. Commun. 181, 361–366 [DOI] [PubMed] [Google Scholar]
- 20.Bünemann M., Liliom K., Brandts B. K., Pott L., Tseng J. L., Desiderio D. M., Sun G., Miller D., Tigyi G. (1996) EMBO J. 15, 5527–5534 [PMC free article] [PubMed] [Google Scholar]
- 21.Beil M., Micoulet A., von Wichert G., Paschke S., Walther P., Omary M. B., Van Veldhoven P. P., Gern U., Wolff-Hieber E., Eggermann J., Waltenberger J., Adler G., Spatz J., Seufferlein T. (2003) Nat. Cell Biol. 5, 803–811 [DOI] [PubMed] [Google Scholar]
- 22.Chiulli N., Codazzi F., Di Cesare A., Gravaghi C., Zacchetti D., Grohovaz F. (2007) Eur. J. Neurosci. 26, 875–881 [DOI] [PubMed] [Google Scholar]
- 23.Meyer Zu Heringdorf D. (2004) J. Cell. Biochem. 92, 937–948 [DOI] [PubMed] [Google Scholar]
- 24.Meyer zu Heringdorf D., Niederdräing N., Neumann E., Fröde R., Lass H., Van Koppen C. J., Jakobs K. H. (1998) Eur. J. Pharmacol. 354, 113–122 [DOI] [PubMed] [Google Scholar]
- 25.Ghosh T. K., Bian J., Gill D. L. (1990) Science 248, 1653–1656 [DOI] [PubMed] [Google Scholar]
- 26.Dettbarn C., Betto R., Salviati G., Sabbadini R., Palade P. (1995) Brain Res. 669, 79–85 [DOI] [PubMed] [Google Scholar]
- 27.Mao C., Kim S. H., Almenoff J. S., Rudner X. L., Kearney D. M., Kindman L. A. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 1993–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schnurbus R., de Pietri Tonelli D., Grohovaz F., Zacchetti D. (2002) Biochem. J. 362, 183–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodney G. G., Moore C. P., Williams B. Y., Zhang J. Z., Krol J., Pedersen S. E., Hamilton S. L. (2001) J. Biol. Chem. 276, 2069–2074 [DOI] [PubMed] [Google Scholar]
- 30.Yamada M., Miyawaki A., Saito K., Nakajima T., Yamamoto-Hino M., Ryo Y., Furuichi T., Mikoshiba K. (1995) Biochem. J. 308, 83–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sienaert I., Nadif Kasri N., Vanlingen S., Parys J. B., Callewaert G., Missiaen L., de Smedt H. (2002) Biochem. J. 365, 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Enyedi A., Vorherr T., James P., McCormick D. J., Filoteo A. G., Carafoli E., Penniston J. T. (1989) J. Biol. Chem. 264, 12313–12321 [PubMed] [Google Scholar]
- 33.Mendes P. (1997) Trends Biochem. Sci. 22, 361–363 [DOI] [PubMed] [Google Scholar]
- 34.Comte M., Maulet Y., Cox J. A. (1983) Biochem. J. 209, 269–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown S. E., Martin S. R., Bayley P. M. (1997) J. Biol. Chem. 272, 3389–3397 [DOI] [PubMed] [Google Scholar]
- 36.Murase T., Iio T. (2002) Biochemistry 41, 1618–1629 [DOI] [PubMed] [Google Scholar]
- 37.Lucas J. L., Wang D., Sadée W. (2006) Pharm. Res. 23, 647–653 [DOI] [PubMed] [Google Scholar]
- 38.Turner J. H., Raymond J. R. (2005) J. Biol. Chem. 280, 30741–30750 [DOI] [PubMed] [Google Scholar]
- 39.Balshaw D. M., Yamaguchi N., Meissner G. (2002) J. Membr. Biol. 185, 1–8 [DOI] [PubMed] [Google Scholar]
- 40.Di Leva F., Domi T., Fedrizzi L., Lim D., Carafoli E. (2008) Arch. Biochem. Biophys. 476, 65–74 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.