

Abstract
During apoptosis, cells acquire new activities that enable them to modulate the fate and function of interacting phagocytes, particularly macrophages (mϕ). Although the best known of these activities is anti-inflammatory, apoptotic targets also influence mϕ survival and proliferation by modulating proximal signaling events, such as MAPK modules and Akt. We asked whether modulation of these same signaling events extends to epithelial cells, a minimally phagocytic cell type. We used BU.MPT cells, a mouse kidney epithelial cell line, as our primary model, but we also evaluated several epithelial cell lines of distinct tissue origins. Like mϕ, mouse kidney epithelial cells recognized apoptotic and necrotic targets through distinct non-competing receptors, albeit with lower binding capacity and markedly reduced phagocytosis. Also, modulation of inflammatory activity and MAPK-dependent signaling by apoptotic and necrotic targets was indistinguishable in kidney epithelial cells and mϕ. In contrast, modulation of Akt-dependent signaling differed dramatically between kidney epithelial cells and mϕ. In kidney epithelial cells, modulation of Akt was linked to target cell recognition, independently of phagocytosis, whereas in mϕ, modulation was linked to phagocytosis. Moreover, recognition of apoptotic and necrotic targets by kidney epithelial cells elicited opposite responses; apoptotic targets inhibited whereas necrotic targets stimulated Akt activity. These data confirm that nonprofessional phagocytes recognize and respond to dying cells, albeit in a manner partially distinct from mϕ. By acting as sentinels of environmental change, apoptotic and necrotic targets may permit neighboring viable cells, especially non-migratory epithelial cells, to monitor and adapt to local stresses.
Keywords: Apoptosis, Cell/Epithelial, Cell/Phagocytosis, Immunology/Innate Immunity, Tissue/Organ Systems/Kidney, Akt, MAP Kinase, Necrosis
Introduction
The defining property of an apoptotic cell is its ability to be removed without inflammation. As documented by several groups, apoptotic cells actively inhibit the secretion of proinflammatory cytokines by macrophages (mϕ)2 that engulf them (1–6). Necrotic cells, in contrast, tend to enhance proinflammatory responses, although alone they are often weak or insufficient triggers of inflammatory responses (6–9). The contrasting effects of apoptotic and necrotic cells on inflammation (10) imply that mϕ and other phagocytes can discriminate between cells undergoing these two forms of target cell death. Recognition of apoptotic or necrotic targets occurs via a saturable, receptor-mediated process (6). Notably, the receptors that mediate binding of apoptotic targets do not compete with those for necrotic targets, and vice versa (6). The existence of separate receptors is consistent with the distinct effects elicited by apoptotic and necrotic cells and implies that these two forms of cell death provide independent information to responding mϕ.
The mechanism(s) by which apoptotic targets exert their anti-inflammatory effects probably varies with time. Early inhibition entails direct receptor-initiated signaling events, leading to the inhibition of NFκB-dependent transcription (6, 11). Later inhibition occurs indirectly via the release of soluble mediators, such as transforming growth factor-β and interleukin 10, that act in a paracrine or autocrine fashion to block the expression of proinflammatory cytokines (1–5). Our focus has been on the early signaling events induced in mϕ by apoptotic and necrotic targets. We have shown that, in addition to inhibition of inflammation, apoptotic and necrotic targets potently modulate the survival, proliferation, and other transcriptional responses of mϕ with which they interact (6, 7, 11–14). Linked to these outcomes, dead target cells trigger a characteristic set of early signaling events in responding mϕ, especially those involving MAPK modules and the prosurvival kinase Akt (7, 12). These signaling events occur within minutes of the interaction between mϕ and apoptotic or necrotic targets (7, 12). Notably, we have distinguished between signaling events induced by receptor-mediated recognition and those induced by phagocytosis (6, 7, 11, 12, 15). We reasoned that signaling events for which apoptotic and necrotic targets elicit opposite responses (e.g. modulation of MAPK modules and NFκB-dependent transcription) must be triggered by distinct receptor-mediated recognition, whereas signaling events for which apoptotic and necrotic targets elicit similar responses (e.g. activation of Akt) may be triggered by the shared machinery of phagocytosis.
Importantly, recognition-dependent inhibition of proinflammatory responses by apoptotic targets is not restricted to professional phagocytes (14, 16). Indeed, apoptotic cells inhibit proinflammatory responses in all cell types examined, including non-professional phagocytes, such as epithelial, neuronal, and lymphoid cells (14). Here, we ask whether the ability of apoptotic and necrotic targets to modulate proximal signaling events involving MAPK- and Akt-dependent modules also extends to non-professional phagocytes. We used BU.MPT (Boston University mouse proximal tubule) cells, a conditionally immortalized mouse kidney epithelial cell line, as our primary model (17, 18).
We provide evidence that BU.MPT cells, like mϕ, discriminate between apoptotic and necrotic targets via distinct receptors. BU.MPT cells also evince the same set of recognition- dependent responses as mϕ with respect to inhibition of inflammation and modulation of MAPK modules. Remarkably, however, BU.MPT cells differ from mϕ in two important ways with respect to modulation of Akt; 1) modulation is triggered not by engulfment, but instead by recognition, and 2) apoptotic and necrotic targets have divergent effects. Specifically, apoptotic targets inhibit Akt in BU.MPT cells but not in mϕ, whereas necrotic targets activate Akt in both cell types. To determine whether the response of BU.MPT cells is characteristic of epithelial cells, we evaluated several distinct epithelial cell lines of different tissue origins. All epithelial cell lines evaluated showed a recognition-dependent response to apoptotic targets, but the direction of their Akt response was organ-specific, with activation of Akt occurring in some cell lines and inhibition in others.
Taken together, these data demonstrate that nonprofessional phagocytes, such as epithelial cells, recognize and respond to dying cells in a manner distinct from mϕ. We hypothesize that these early signaling responses to dead cells play an important role in the normal homeostasis of tissues. By acting as sentinels of environmental change, apoptotic and necrotic targets may permit neighboring viable cells, especially non-migratory cells like epithelial cells, to monitor and potentially adapt to local stresses.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise stated, all chemicals were obtained from Sigma, Invitrogen, or Fisher. Cell culture medium was obtained from Mediatech (Herndon, VA).
Antibodies
Affinity-purified polyclonal rabbit antibodies detecting the active phosphorylated or total forms of Akt (Akt1, Akt2, and Akt3), the active phosphorylated form of p90RSK, and the inactive phosphorylated forms of Bad and GSK-3β were obtained from Cell Signaling Technology (Beverly, MA). Affinity-purified polyclonal rabbit antibodies detecting the active phosphorylated or total forms of ERK (ERK1 and ERK2), JNK (JNK1 and JNK2), and p38 (p38α, p38β, and p38γ) were obtained from Promega (Madison, WI). Affinity-purified polyclonal goat antibodies to murine interleukin 1β (IL-1β) (31-kDa pro-IL-1β and 17-kDa mature IL-1β) were obtained from R & D Systems, Inc. (Minneapolis, MN). Anti-active Akt detects Akt1 when phosphorylated at Ser473 and detects Akt2 and Akt3 when phosphorylated at the corresponding residues. This antibody does not recognize Akt phosphorylated at other sites; nor does it recognize phosphorylated forms of related kinases, such as protein kinase C or p70 S6 kinase. Anti-phospho-Bad detects endogenous levels of Bad only when phosphorylated at Ser112 but does not detect unphosphorylated Bad or related family members. Anti-phospho-GSK-3β detects endogenous levels of GSK-3β only when phosphorylated at Ser9 and may cross-react weakly with the phosphorylated form of GSK-3α. Anti-phospho-p90RSK detects the dually phosphorylated Thr359-X-X-X-Ser363 region (pT359XXXpS363; where pT and pS represent phosphothreonine and phosphoserine, respectively) of p90RSK1 and cross-reacts with p90RSK3. Anti-active ERK1/2 detects the dually phosphorylated Thr183-Glu-Tyr185 region (pT183EpY185; where pY represents phosphotyrosine) derived from the catalytic core of the active ERK kinase. Anti-active JNK1/2 detects the dually phosphorylated Thr183-Pro-Tyr185 region (pT183PpY185) derived from the catalytic core of the active form of the JNK kinase. Anti-active p38 detects the dually phosphorylated Thr180-Gly-Tyr182 region (pT180GpY182) derived from the catalytic core of the active form of the p38 kinase. Horseradish peroxidase-linked donkey anti-rabbit F(ab′)2 from GE Healthcare was used as a secondary antibody for detection of Western blots by enhanced chemiluminescence.
Cell Culture
All cells were grown at 37 °C in a humidified 5% (v/v) CO2 atmosphere unless otherwise stated. The conditionally immortalized mouse kidney proximal tubule epithelial cell line (BU.MPT) was maintained in high glucose Dulbecco's modified Eagle's medium-F-12 medium containing 10% (v/v) heat-inactivated fetal bovine serum (FBS), 2 mm l-glutamine, 10 mm HEPES, 100 units/ml penicillin-streptomycin, and 10 units/ml interferon-γ. BU.MPT cells were derived from a transgenic mouse bearing a temperature-sensitive mutation (tsA58) of the SV40 large tumor antigen under the control of the mouse major histocompatibility complex H-2Kb class I promoter (17–19). Under permissive conditions, defined as growth at 33–37 °C in the presence of interferon-γ, the tsA58 large tumor antigen transgene is expressed. Under non-permissive temperatures, defined as growth at 39.5 °C in the absence of interferon-γ, expression of the transgene is inhibited (by >95%), and BU.MPT cells behave like primary cultures of mouse kidney proximal tubular cells. Prior to all experiments, BU.MPT cells are serum-starved and cultured under non-permissive conditions for 24 h.
Chinese hamster ovary (CHO) cells were grown in α-minimum Eagle's medium supplemented with 10% (v/v) heat-inactivated FBS and 2 mm l-glutamine. HeLa cells, a human cervical cancer epithelial cell line, were maintained in Dulbecco's modified Eagle's medium containing 10% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, and 2 mm l-glutamine. MCF-7 K1 cells, a human breast adenocarcinoma cell line, were maintained in Dulbecco's modified Eagle's medium containing 5% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, and 2 mm l-glutamine. OK cells, which were derived from the kidney of an adult female North American opossum and display many characteristics of kidney proximal tubular epithelial cells, were maintained in low glucose Dulbecco's modified Eagle's medium containing 10% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, and 2 mm l-glutamine. J774A.1 (J774) cells, a mouse monocyte-derived mϕ cell line, were grown in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, 2 mm l-glutamine, and 50 μm 2-mercaptoethanol. DO11.10 cells, a T cell hybridoma line, were grown in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, and 50 μm 2-mercaptoethanol.
Preparation of Apoptotic Cells, Necrotic Cells, and Latex Beads
Physiological cell death, or apoptosis, of BU.MPT, CHO, HeLa, MCF-7, and OK cells was induced by incubating cells with FBS-free medium containing the non-selective protein kinase inhibitor staurosporine (1 μg/ml, 3 h); by incubating cells with FBS-containing medium containing the macromolecular synthesis inhibitor actinomycin D (200 ng/ml, overnight); or by irradiating cells with UV-B irradiation (20–50 mJ/cm2) and incubation for 24 h. After induction of apoptosis, the remaining adherent cells were detached by the addition of 5 mm EDTA and pooled with floating cells, followed by three washes and resuspension in fresh FBS-free medium before use in experiments. Pathological cell death, or necrosis, was induced after detachment of the cells with 5 mm EDTA and suspension in the appropriate FBS-free medium by heating cells to 65 °C for 40 min, followed by incubation for 2 h. Latex beads (1.15 μm), obtained from Seradyne (Indianapolis, IN), were used as a neutral phagocytic stimulus.
Induction of apoptosis or necrosis was confirmed by flow cytometry. Viable cells were defined as cells that were both propidium iodide (PI)- and annexin V-negative. Early apoptotic cells (intact cell membranes) were defined as PI-negative cells with annexin V staining and decreased cell size. Necrotic cells were defined as PI-positive cells of normal or increased cell size. Late apoptotic cells (non-intact cell membranes) were defined as PI-positive cells with annexin V staining and decreased cell size. Loss of membrane integrity by necrotic cells was confirmed by trypan blue staining. By these criteria, apoptotic cell preparations contained ∼85% early apoptotic and ∼15% late apoptotic cells. Necrotic cell preparations contained ∼95% necrotic cells. In all preparations, viable cells, defined as PI-negative cells of normal size without annexin V staining, comprised <5% of the total cell population.
Western Blot Analysis
After stimulation of responder cells with apoptotic targets, necrotic targets, and/or latex beads, in the presence or absence of epidermal growth factor (EGF; 10 nm) (Calbiochem), responder cells were washed with ice-cold PBS and then lysed in ice-cold cell lysis buffer (PBS containing 1% (v/v) Nonidet P-40, 0.5% (w/v) deoxycholate, 0.1% (w/v) SDS, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride, and 200 μm orthovanadate). Lysates were centrifuged at 10,000 × g for 10 min at 4 °C, and the supernatants were stored at −70 °C.
Protein samples (20 μg each, as determined by the bicinchoninic acid protein assay (Pierce)) were boiled in 6× reducing sample buffer, electrophoresed on 12% SDS-polyacrylamide gels, and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). Membranes were blocked with either 2.5% bovine serum albumin or 5% dry milk in Tris-buffered saline before probing with one of the primary antibodies described above. Following incubation with secondary antibody, immunoreactive bands were visualized by the luminol reaction (GE Healthcare). Equivalent loading of protein samples was monitored by staining with Ponceau S (0.25% (w/v)) in 0.1% acetic acid for 5 min.
Phagocyte Interaction Assays
Binding and phagocytosis of apoptotic targets by BU.MPT and J774 cells were assessed, as previously described (6). In brief, after green prelabeling with 5,6-carboxyfluorescein diacetate succinimidyl ester (CFDA; 4 μm; excitation wavelength (λEx) = 488 nm; emission wavelength (λEm) = 525 nm; Molecular Probes, Inc. (Eugene, OR)), BU.MPT or DO11.10 target cells were induced to undergo apoptotic cell death as described above. BU.MPT and J774 engulfing responder cells were red-prelabeled with 5,6-(((4-chloromethyl)-benzoyl)-amino)-tetramethylrhodamine (CMTMR; 8 μm; λEx = 488 nm, λEm = 610 nm; Molecular Probes, Inc.). Both target and responder cells were labeled at 37 °C for 30 min while still adherent on the day preceding the experiment and cultured overnight in medium containing 10% FBS to eliminate unbound label.
For binding assays, green-labeled apoptotic or necrotic targets, either alone or in mixtures with unlabeled targets, were added to unlabeled BU.MPT or J774 monolayers (105 cells/well) and allowed to interact for 2–6 h. Wells were washed with ice-cold saline three times, and plate-bound fluorescence was analyzed on a Cytofluor 2350 fluorescence plate reader (Millipore, Marlborough, MA). For quantitation, a standard curve was prepared with a graded number of labeled target cells. The fluorescent labeling of target cells, which varied minimally (<10%) between experiments, yielded specific fluorescence intensities of ∼6.25 × 103 fluorescence units/105 cells (above a background of <50 fluorescence units). This degree of labeling permitted reliable quantitation of as few as 0.01 target cells per responder cell (based on 105 responder cells analyzed per well). Individual experiments represent the means of triplicate determinations, and all data points are the means ± S.E. of at least three experiments.
For phagoytosis assays, red-labeled responder cells were co-cultured with green-labeled apoptotic targets for 30 min at 37 °C. Cells were then harvested with PBS supplemented with 0.4 mm EDTA and analyzed cytofluorimetrically on a FACSCaliber instrument (BD Biosciences). Engulfing cells were identified by double-positive staining, and fractional engulfment was defined as the percentage of responder cells that contained at least one apoptotic target (percentage of red-labeled cells that are also green-labeled). Apoptotic targets bound to responder cells at the end of the 30-min co-incubation but not yet engulfed were detached by the treatment with 0.4 mm EDTA and did not remain bound during analysis.
Microscopy
Phase-contrast and corresponding fluorescent microscopic images were acquired with a SenSys CCD Camera (Photometrics, Tucson, AZ) and analyzed using Image-Pro Plus software (Media Cybernetics, Silver Spring, MD).
For confocal microscopy, BU.MPT responder cells were grown overnight on LabTek II chambered coverglass slides and then incubated in the absence or presence of CMTMR-labeled apoptotic or necrotic targets (prepared as described above) for 1 h. The cells were washed with PBS and then fixed with 4% paraformaldehyde for 10 min. After washing with PBS, cells were permeabilized by incubation in 1% Triton X-100 in PBS for 5 min at room temperature, washed, and blocked with 1% bovine serum albumin and 0.01% sodium azide in PBS (blocking solution) for 30 min. F-actin was detected by incubation of cells for 30 min in fluorescein isothiocyanate (FITC)-phalloidin in blocking solution. After three washes with PBS, the cells were mounted in Vectashield with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA) to stain the nuclei of responder and target cells and visualized by confocal fluorescence microscopy (LSM510, Zeiss) under the green, blue, and red channels. In control experiments, cells were incubated with PBS without FITC-phalloidin.
EGF Receptor (EGFR) Detection
After incubation of BU.MPT responder cells for 30 min in the absence or presence of apoptotic, necrotic, or latex targets, direct staining of BU.MPT responders for EGFR was performed either immediately or 18 h after exposure to targets. Cells were harvested and washed and, following centrifugation, were suspended in ice-cold suspension buffer (PBS with 10% FBS and 0.01% sodium azide) at a concentration of 1 to 5 × 106 cells/ml. Cells were then incubated with 0.5 μg of FITC-conjugated EGFR antibody (ICR10 clone, Novus Biologicals, Littleton, CO) or irrelevant control antibody (FITC-conjugated, rat IgG2a, BD Biosciences) for 1 h at room temperature. Cells were subsequently washed, resuspended in 500 μl of ice-cold suspension buffer, and analyzed cytofluorimetrically on a FACSCaliber instrument.
Phosphatase Inhibitors
The following phosphatase inhibitors were obtained from Calbiochem: calyculin A, protein-tyrosine phosphatase inhibitor (PTPI) IV, phenylarsine oxide, potassium bisperoxo(1,10-phenanthroline)oxovanadate, dipotassium bisperoxo(picolinato)oxovanadate V, and PTPI I.
RESULTS
Apoptotic Target Effects on MAPK Modular Signaling and Inflammatory Suppression Are Conserved between mϕ and Epithelial Cells
Apoptotic targets influence the proliferation, survival, and inflammatory responses of interacting mϕ through receptor-mediated modulation of a number of proximal signaling events, involving MAPK modules, Akt-dependent signaling, and NFκB-dependent transcription (6, 7, 11–14). Previous studies have suggested that many of these responses are elicited ubiquitously in a variety of cell types (13, 14, 16, 20, 21). We asked whether apoptotic modulation of these signaling events extends to epithelial cells, a minimally phagocytic cell type, not traditionally thought to play a major role in the response to dead cells. As our primary cell model, we used BU.MPT cells, a conditionally immortalized line of differentiated mouse kidney proximal tubular epithelial cells.
BU.MPT cells demonstrated the same set of signature signaling events involving MAPK modules (ERK1/2, p38, and JNK1/2) in response to apoptotic and necrotic targets as observed in mϕ (Fig. 1) (7, 12). Quiescent BU.MPT cells showed minimal levels of active phosphorylated ERK1/2. Stimulation with EGF or high dose insulin for 15 min induced strong phosphorylation of ERK1/2. In the absence of EGF, exposure to apoptotic targets induced no phosphorylated ERK1/2 and, in fact, in most cases, inhibited basal ERK1/2 phosphorylation. Prior exposure to apoptotic cells strongly inhibited EGF and insulin-induced phosphorylation of ERK1/2 (Fig. 1A). Inhibition of ERK1/2 phosphorylation correlated with inhibition of ERK1/2 activity, as detected by the parallel inhibition of the phosphorylation of p90RSK, a downstream target of ERK1/2 (Fig. 1A). Inhibition of ERK1/2 by apoptotic cells occurred in a dose-dependent manner (Fig. 1B). The ability of apoptotic targets to inhibit the phosphorylation of ERK1/2, either basal or induced by, for example, EGF and insulin, implies that apoptotic modulation in BU.MPT responders is generalized and independent of the factor or cytokine used to stimulate BU.MPT cells.
FIGURE 1.

MAPK signaling in epithelial cells in response to apoptotic, necrotic, or latex bead targets. A, serum-starved BU.MPT cells were stimulated with apoptotic cells (Apo), necrotic cells (Nec), or cell-sized latex beads (Lat) alone for 15 min, or serum-starred BU. MPT cells were stimulated with EGF (50 nm) or high dose insulin (50 μg/ml) for 15 min following a 30-min pre-exposure to apoptotic cell, necrotic cell, or latex bead targets. B, serum-starved BU.MPT cells were exposed to apoptotic cell targets at a target/responder cell ratio ranging from 1:32 to 1:1 in 2-fold increments, as indicated by the schematic, for 30 min, followed by a 15-min stimulation with EGF (50 nm). C, serum-starved BU.MPT cells were stimulated with apoptotic targets at a target/responder cell ratio ranging from 1:32 to 1:1 in 2-fold increments, as indicated by the schematic, for 15 min. In all cases, non-adherent cells were removed by washing, and epithelial cell lysates were probed with anti-active ERK1/2, anti-active p90RSK, anti-active p38, and anti-active JNK1/2 antibodies, as shown. Equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total ERK1/2, total p90RSK, total p38, and total JNK (supplemental Fig. 1).
The effect of necrotic targets on ERK1/2 phosphorylation and activity was opposite to that of apoptotic targets. Exposure to necrotic targets alone for 15 min, in the absence of EGF or insulin, activated ERK1/2. Prior exposure to necrotic targets also moderately up-regulated the subsequent response to EGF. In contrast, cell-sized latex beads, a neutral phagocytic stimulus, had no effect on ERK1/2 phosphorylation in either the absence or the presence of EGF (Fig. 1A).
The response of BU.MPT cells to apoptotic and necrotic targets also mirrored that of mϕ with respect to p38 and JNK1/2, two additional MAPK modules (7, 12). Quiescent BU.MPT cells had undetectable levels of phosphorylated JNK1/2 and p38. EGF induced weak phosphorylation of JNK1/2 and had no effect on p38. In contrast to their inhibitory effect on ERK1/2 phosphorylation, apoptotic targets induced phosphorylation of both p38 and JNK1/2 (Fig. 1A). Phosphorylation of p38 in response to apoptotic targets occurred in a dose-dependent manner (Fig. 1C). Although p38 and JNK1/2 are activated by a wide variety of stresses and inflammatory stimuli, necrotic targets had no detectable effect on these two pathways, whether added alone or in the presence of EGF (Fig. 1A). Similarly, cell-sized latex beads had no effect on the levels of phosphorylated p38 or JNK1/2 (Fig. 1A).
Apoptotic targets also modulated the inflammatory capacity of responding BU.MPT cells, as assessed by the expression of cell-associated IL-1β (22). Exposure to apoptotic targets inhibited both basal and lipopolysaccharide (LPS)- or phorbol 12-myristate 13-acetate (PMA)-induced IL-1β expression (Fig. 2). In contrast, necrotic targets and latex beads had no detectable effect on IL-1β expression (data not shown). Levels of secreted IL-1 in response to lipopolysaccharide and phorbol 12-myristate 13-acetate were low and indistinguishable from background by ELISA (data not shown).
FIGURE 2.
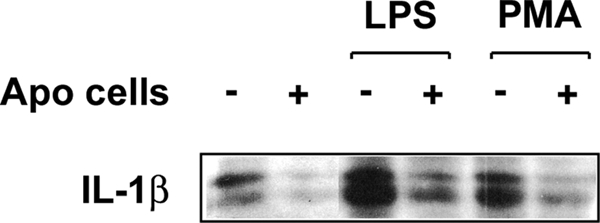
Anti-inflammatory response by epithelial cells in response to apoptotic targets. Serum-starved BU.MPT cells were stimulated with lipopolysaccharide (LPS; 10 μg/ml) or phorbol 12-myristate 13-acetate (PMA; 20 ng/ml) for 16 h in the absence or continuous presence of apoptotic targets at a target/responder cell ratio of 1:1. In all cases, non-adherent cells were removed by washing, and epithelial cell lysates were probed for cell-associated IL-1β, as shown. Equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total Akt (supplemental Fig. 2).
In these and subsequent experiments, apoptotic or necrotic targets alone contributed neither detectable IL-1β nor any phosphorylated or unphosphorylated ERK1/2, p90RSK, p38, JNK1/2, or Akt (7, 12) (data not shown). Although BU.MPT cells were the primary source of targets in these studies, we have observed similar modulation of MAPK activity with murine DO11.10 T cells (data not shown). The ability of apoptotic targets to modulate MAPK activity was also observed with several distinct apoptotic stimuli (e.g. staurosporine, actinomycin D, and UV-B irradiation) (data not shown).
Specific Recognition of Apoptotic and Necrotic Targets Is Conserved in mϕ and Epithelial Cells
The similarity in the response to dead targets by BU.MPT cells and mϕ in terms of MAPK modular activity and inflammatory cytokine expression suggested that BU.MPT cells, like mϕ, may recognize apoptotic and necrotic targets through distinct mechanisms. We therefore assessed the ability of unlabeled dead target cells (apoptotic or necrotic) to interfere with the binding of fluorescently labeled target cells (Fig. 3). Although unlabeled apoptotic targets inhibited the binding of labeled apoptotic targets to BU.MPT cells in a dose-dependent manner, unlabeled necrotic targets had no effect on this binding (Fig. 3A). Correspondingly, unlabeled necrotic, but not unlabeled apoptotic, targets inhibited the binding of labeled necrotic targets to BU.MPT cells (Fig. 3B). Of note, microscopic examination revealed that apoptotic and necrotic targets bound to the same BU.MPT cells (data not shown). The specificity of target binding exhibited in these competition studies implies the existence of independent mechanisms for the recognition of apoptotic and necrotic targets.
FIGURE 3.

Specific recognition of apoptotic and necrotic targets by epithelial cells versus macrophages. A and B, recognition and binding of CFDA-labeled apoptotic (A) and necrotic (B) DO11.10 target cells by BU.MPT or J774 monolayers (supplemental Fig. 3, A and B) (∼105 cells/well) was assessed in the presence or absence of unlabeled dead target competitors after incubation for 2 h at 37 °C. Labeled targets (10 × 105 cells/well) were mixed with unlabeled apoptotic or necrotic competitors at the indicated ratios. Unbound cells were removed by washing, and the number of target cells bound was determined by quantifying CFDA fluorescence (λEx = 490 nm; λEm = 525 nm), as described under “Experimental Procedures.” C, apoptotic and necrotic CMTMR-labeled DO11.10 target cells were added to subconfluent monolayers of unlabeled responder cells (BU.MPT or J774) at a target/responder cell ratio of 10:1 and allowed to interact for 1 h at 37 °C. Phase-contrast and corresponding fluorescent microscopic images reveal surface binding by both responder cells but uptake only by J774 cells. For BU.MPT responders, bound CMTMR-labeled apoptotic or necrotic targets can be seen to lie outside the cytoplasm at the cell surface (arrows). For J774 responders, CMTMR-labeled apoptotic or necrotic targets lie within the cytoplasm and, in many cases, show evidence of fragmentation and degradation (arrows). All panels were photographed at an equivalent magnification (×40) and scaled to the same size. D, apoptotic and necrotic CMTMR-labeled (red) DO11.10 target cells were added to subconfluent monolayers of BU.MPT cells at a target/responder cell ratio of 5:1 and allowed to interact for 1 h at 37 °C. The nuclei and cytoskeletons of fixed responders and bound targets were stained with 4′,6-diamidino-2-phenylindole (blue) and FITC-phalloidin (green), respectively. The white arrows in composite images indicate bound apoptotic and necrotic target cell fragments, which in most cases appear to contain exclusively nuclear or cytoplasmic material. The red arrows indicate largely intact bound targets containing both nuclear and cytoplasmic material. Interaction with apoptotic targets led to altered BU.MPT cell morphology, characterized by development of multiple cellular processes resembling pseudopodia and filopodia. Interaction with necrotic targets had essentially no effect on BU.MPT cell morphology. All panels were photographed at an equivalent magnification (×60). A high resolution version of D is given in the supplemental material.
In keeping with their status as non-professional phagocytes, BU.MPT cells have fewer binding sites for dead target cells than do J774 cells, a mϕ cell line. Under identical experimental conditions, BU.MPT cells bind less than one-tenth as many apoptotic (0.29 ± 0.07 versus 4.0 ± 0.9) or necrotic (0.27 ± 0.05 versus 2.9 ± 0.5) targets per responder as do J774 cells (Fig. 3, A and B) (supplemental Fig. 3). This difference is not the result of slower kinetics, because the binding of dead targets by BU.MPT cells was maximal by 2 h and did not increase between 2 and 6 h (data not shown). BU.MPT cells also have reduced phagocytic capacity compared with J774 mϕ. Microscopic examination of responder and target cells incubated together for 1 h revealed surface binding of dead target cells to BU.MPT responders but no uptake (Fig. 3C; see also Fig. 7). In contrast, at this same time point, J774 mϕ demonstrated significant uptake of dead target cells.
FIGURE 7.

Flow cytometric analysis of the capacity of epithelial cells to phagocytose apoptotic targets. A, responder cells, either BU.MPT epithelial cells or J774 macrophages, were prelabeled with CMTMR (FL2, y axis), following which they were incubated for 2 h in the presence or absence of cytochalasin D (2 μm). Apoptotic targets, BU.MPT cells induced to undergo apoptosis by overnight exposure to actinomycin D (200 ng/ml), were prelabeled with CFDA (FL1, x axis). Apoptotic targets were added to responder cells at a ratio of 10:1. After incubation together for 30 min, flow cytometric analysis was performed on the total cell population. Apoptotic targets bound to responder cells but not yet engulfed were detached by treatment with 0.4 mm EDTA and did not remain bound during analysis. CMTMR-positive CFDA-positive double-labeled cells in the upper right quadrant represent responder cells that have engulfed at least one apoptotic target. CMTMR-positive single-labeled cells in the upper left quadrant represent responder cells that have not ingested an apoptotic target. The percentage of the total CMTMR-positive responder cell population falling in each of these two quadrants is indicated. CFDA-positive single-labeled cells in the lower right quadrant represent non-engulfed apoptotic targets. B, the graph depicts the mean and S.E. from three separate flow cytometric analyses of the percentage of responder cells engulfing at least one apoptotic target.
Confocal microscopy revealed diffuse binding of dead targets (apoptotic or necrotic), with a predominance of cellular fragments over intact target cells (Fig. 3D). Many of the target cell fragments recognized by BU.MPT responders seemed to contain exclusively cytoplasmic or nuclear material. Interaction with apoptotic targets altered the morphology of BU.MPT responders, leading to the development of multiple projections and delicate extensions, resembling pseudopodia and filopodia. In contrast, interaction with necrotic targets had essentially no effect on the morphology of BU.MPT responders.
Apoptotic Targets Elicit Divergent Akt-dependent Signaling Events in Epithelial Cells and mϕ
In mϕ, modulation of the activity of Akt, a major regulator of cell survival, is linked to engulfment of cell corpses (7, 12, 15). Both apoptotic and necrotic targets activate Akt in a dose-dependent manner, an effect that is blocked by inhibition of phagocytosis. Consistent with the notion that Akt activation is dependent on phagocytosis, rather than specific receptor-mediated recognition of apoptotic or necrotic targets, exposure to latex beads, a neutral phagocytic stimulus, also activates Akt (12).
We were therefore interested in evaluating the effect of apoptotic and necrotic targets on Akt activity in BU.MPT cells, which are minimally phagocytic. As opposed to the response of MAPK modules, the Akt response of BU.MPT cells to dead cells diverged from that of mϕ in several important ways. Most remarkably, exposure to apoptotic targets induced a decrease, rather than an increase, in the phosphorylation of Akt (Fig. 4A). Inhibition of Akt phosphorylation by apoptotic targets was observed for basal as well as stimulated levels, whether by EGF or high dose insulin. A dose-dependent decrease was observed both for basal phosphorylation (Fig. 4B) and for induced phosphorylation, as assessed in the presence of EGF (Fig. 4C). Inhibition of Akt phosphorylation correlated with inhibition of Akt activity, as indicated by the parallel decrease of the phosphorylation of GSK3β and Bad, two downstream targets of Akt (Fig. 4, B and C). Inhibition of Akt phosphorylation following exposure to apoptotic targets was never complete, and maximal inhibition, for both basal and induced phosphorylation, was ∼50%. Increasing the ratio of apoptotic targets to BU.MPT responders did not yield further inhibition (data not shown).
FIGURE 4.

Akt signaling in epithelial cells in response to apoptotic, necrotic, or latex bead targets. A, serum-starved BU.MPT cells were stimulated with apoptotic cells (Apo), necrotic cells (Nec), or cell-sized latex beads (Lat) alone for 15 min, or serum-starred BU. MPT cells were stimulated with EGF (50 nm) or high dose insulin (50 μg/ml) for 15 min following a 30-min pre-exposure to apoptotic cell, necrotic cell, or cell-sized latex bead targets. B, serum-starved BU.MPT cells were stimulated with Apo targets at a target/responder cell ratio ranging from 1:32 to 1:1 in 2-fold increments, as indicated by the schematic, for 15 min. C, serum-starved BU.MPT cells were exposed to Apo targets at a target/responder cell ratio ranging from 1:32 to 1:1 in 2-fold increments, as indicated by the schematic, for 30 min, followed by a 15-min stimulation with EGF (50 nm). D, serum-starved BU.MPT cells were stimulated with Nec targets at a target/responder cell ratio ranging from 1:32 to 1:1 in 2-fold increments, as indicated by the schematic, for 15 min. In all cases, non-adherent cells were removed by washing, and BU.MPT cell lysates were probed with anti-active Akt, anti-inactive GSK3β, and anti-inactive Bad antibodies, as shown. Apparent differences in base-line Akt activity between panels reflect differences in exposure time for optimal presentation of data. Equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total Akt (supplemental Fig. 4).
In contrast to the effect of apoptotic targets, necrotic targets induced a dose-dependent increase in the phosphorylation of Akt and its downstream substrates (Fig. 4D). Cell-sized latex beads had no effect on Akt signaling (Fig. 4A). Thus, as opposed to mϕ, for which the Akt response is consonant for all three targets (12, 15), the response of BU.MPT cells differs for apoptotic targets, necrotic targets, and latex beads. Apoptotic targets inhibit Akt, necrotic targets activate Akt, and latex beads have no effect.
A second divergence between BU.MPT cells and mϕ emerged when we exposed BU.MPT cells to apoptotic and necrotic targets simultaneously (Fig. 5). In the case of mϕ, the signaling events induced by apoptotic targets are strongly dominant over those induced by necrotic targets, with one apoptotic cell able to overcome the effect of four necrotic cells on MAPK modular activity (7). In contrast, for BU.MPT cells, neither apoptotic nor necrotic targets dominate over the other. We observed this in two complementary sets of experiments. In the first (Fig. 5, left grouping), BU.MPT cells were incubated with a fixed number of necrotic cells (inducing maximal phosphorylation of Akt; lane 1, left grouping), together with a graded number of apoptotic cells, the ratio of apoptotic to necrotic cells ranging from 1:16 to 16:16. In the second (Fig. 5, right grouping), we added a fixed number of apoptotic cells (inducing maximal inhibition of Akt phosphorylation; lane 1, right grouping), together with a graded number of necrotic cells, the ratio of apoptotic to necrotic cells ranging from 16:1 to 16:16. When the numbers of apoptotic and necrotic targets were equal (Fig. 5, last lane, left and right groupings), Akt phosphorylation did not differ substantially from base-line levels in the absence of dead targets (rightmost lane, 0:0). However, when the number of necrotic targets exceeded that of apoptotic targets (Fig. 5, left), the level of phosphorylation of Akt was greater than base-line control levels. Conversely, when the number of apoptotic targets exceeded that of necrotic targets (Fig. 5, right), Akt phosphorylation was lower than base-line levels. Phosphorylation of GSK3β and Bad, two downstream targets of Akt, paralleled that of Akt.
FIGURE 5.

Akt signaling induced by simultaneous exposure to apoptotic and necrotic cell targets. Serum-starved BU.MPT cells were exposed to mixtures of apoptotic cell (Apo) and necrotic cell (Nec) targets for 30 min. In the left grouping, the number of apoptotic targets was kept constant at a target/responder ratio of 1:1, and the number of necrotic targets was increased in 2-fold increments, as shown, until the number of necrotic targets equaled that of apoptotic targets. In the right grouping, the number of necrotic targets was kept constant at a target/responder ratio of 1:1, and the number of apoptotic targets was increased in 2-fold increments, as shown, until the number of apoptotic targets equaled that of necrotic targets. The rightmost lane shows the BU.MPT response in the absence of apoptotic or necrotic targets (0:0). In all cases, non-adherent cells were removed by washing, and BU.MPT cell lysates were probed with anti-active Akt, anti-active GSK3β, and anti-active Bad antibodies, as shown. Equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total Akt (supplemental Fig. 5).
Further Divergence between Epithelial Cells and mϕ; Dependence of Akt Response on Recognition Rather than Phagocytosis
The dramatically different Akt responses elicited by apoptotic and necrotic targets in BU.MPT cells and mϕ suggested that the Akt response in these epithelial cells depends not upon a shared phagocytic machinery common to most cells but upon recognition-dependent signaling events in BU.MPT cells. This notion is reinforced by two other observations. First, latex beads, which are not specifically recognized, had no effect on Akt signaling; second, BU.MPT cells are essentially non-phagocytic, especially within the short duration in which these signaling events occur.
To explore this question more rigorously, we inhibited phagocytosis with cytochalasin D, a pharmacologic inhibitor of actin polymerization. As shown in Fig. 6, cytochalasin D at 2 μg/ml had no effect on the inhibition of phosphorylation of Akt or its downstream substrates in response to apoptotic targets. Similarly, cytochalasin D had no effect on Akt signaling induced by necrotic targets. Together, these data imply that modulation of Akt activity by dead targets, whether apoptotic or necrotic, is dependent on recognition and independent of phagocytosis. As expected, cytochalasin D had no effect on the p38 response of BU.MPT cells to dead cells, confirming that modulation of p38 activity is also dependent on recognition. The Akt response remained unchanged at higher concentrations of cytochalasin D (4 μg/ml), although this concentration of cytochalasin proved slightly toxic to BU.MPT cells (not shown).
FIGURE 6.

Akt response in epithelial cells following pharmacologic inhibition of phagocytosis. Serum-starved BU.MPT cells were stimulated with apoptotic cells (Apo), necrotic cells (Nec), or cell-sized latex beads for 15 min, following a 2-h incubation in the presence or absence of cytochalasin D (2 μm). In all cases, non-adherent cells were removed by washing, and BU.MPT cell lysates were probed with anti-active Akt, anti-active GSK3β, anti-active Bad, and anti-active p38 antibodies as shown. Equal loading was confirmed by probing for total Akt (supplemental Fig. 6).
To confirm the effectiveness of this dose of cytochalasin D as an inhibitor of phagocytosis, BU.MPT cells (or J774 mϕ) and apoptotic targets were individually labeled and incubated together for 30 min, the duration of exposure used in our signaling studies. Apoptotic targets that were bound to responder cells but not yet engulfed were detached by treatment with 0.4 mm EDTA. Responder cells that have ingested apoptotic targets are easily detected as dually stained cells by flow cytometry (Fig. 7A) or immunofluorescence microscopy (not shown). Cytochalasin D (2 μg/ml) significantly inhibited the percentage of double-positive J774 mϕ (35.4 ± 3.4% versus 12.3 ± 0.6%, p < 0.02), whereas it had no effect on the percentage of double-positive BU.MPT cells (8.4 ± 0.2% versus 10.2 ± 0.6%, p = not significant) (Fig. 7B), consistent with the limited phagocytic capacity of these cells. Notably, the percentage of double-positive BU.MPT cells, in the presence or absence of cytochalasin D, did not differ from that of J774 mϕ in the presence of cytochalasin D (p = not significant). Finally, microscopic examination also failed to detect any double-positive BU.MPT cells (data not shown).
Tissue-specific Variation of the Akt Response by Epithelial Cells to Apoptotic Targets
To determine whether the Akt response of BU.MPT cells to apoptotic targets is a general feature of epithelial cells or specific to these cells, we evaluated the Akt response to apoptotic targets in a panel of epithelial cell lines derived from different tissues. As shown in Fig. 8, epithelial cells could be divided into two types: those that resembled BU.MPT cells and showed inhibition of Akt in response to apoptotic targets (OK kidney and CHO ovarian epithelial cells) and those that differed from BU.MPT cells and showed activation of Akt in response to apoptotic targets (HeLa cervical and MCF-7 breast epithelial cells) (Fig. 8). The Akt response by these cell lines was independent of the cell of origin for the apoptotic targets, because cells from each line showed a similar Akt response, irrespective of whether apoptotic targets were derived from BU.MPT cells (heterotypic targets) or from the same line as responder cells (homotypic targets). Notably, in each case, the heterotypic targets were species-disparate from the responders. These data indicate that the specific recognition of apoptotic targets is species-independent, consistent with our earlier findings (11, 14). They also suggest that the response of epithelial cells to apoptotic targets may be tissue-specific, with activation of Akt occurring in some epithelial cells, especially those derived from kidney, and inhibition of Akt occurring in others.
FIGURE 8.

Tissue-specific Akt response of epithelial cells to apoptotic targets. Serum-starved epithelial cells were incubated for 15 min with heterotypic (hetero) or homotypic (homo) apoptotic target cells. Heterotypic targets were BU.MPT cells, and homotypic targets were identical in origin to the responding cell line. Responder cell lines segregated into two types: those showing inhibition of Akt (OK opossum kidney and CHO Chinese hamster ovary epithelial cells) (A) and those showing activation of Akt (MCF-7 breast and HeLa cervical epithelial cells) (B). In all cases, non-adherent cells were removed by washing, and cell lysates were probed with anti-active Akt. Equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total Akt (supplemental Fig. 7).
Inhibition of ERK1/2- and Akt-dependent Signaling following Exposure to Apoptotic Targets Is Not the Result of EGFR Down-modulation or Excessive Activity of Phosphatases
The ability of apoptotic targets to inhibit ERK1/2- and Akt-dependent signaling events, either basal activity or following induction by EGF or insulin (Figs. 1 and 4), implies that inhibition of these signaling pathways is generalized and not specific to the stimulatory factor or cytokine. To address this issue more specifically, we determined the effect of apoptotic, necrotic, and latex bead targets on surface expression of EGFR by BU.MPT responders. BU.MPT cells were incubated with targets for 30 min, as in our signaling studies, and then washed. EGFR expression was unaltered immediately following exposure of BU.MPT cells to targets (Fig. 9A) and remained unaltered for at least 18 h (Fig. 9B).
FIGURE 9.

Flow cytometric analysis of the effect of apoptotic, necrotic, and latex bead targets on surface expression of the EGF receptor by epithelial cells. Serum-starved BU.MPT cells were unstimulated (Vehicle) or stimulated with apoptotic cells (Apo), necrotic cells (Nec), or cell-sized latex beads for 30 min. Relative expression of the EGFR was determined cytofluorometrically either immediately (A) or 18 h (B) after stimulation. Surface staining by FITC-conjugated anti-EGFR (clone ICR10) or irrelevant control (rat IgG2a) antibody are depicted by open or shaded histograms, respectively.
We next explored the role of phosphatases in mediating inhibition by apoptotic targets. Enhanced phosphatase activity induced by apoptotic targets could contribute to modulation of the phosphorylation of ERK1/2- and Akt-dependent signaling in at least two fundamental ways. Apoptotic target-induced phosphatases may inhibit the normal activation of ERK1/2 and/or Akt by interfering with the activity of essential upstream signaling elements. Alternatively, activation of ERK1/2 and Akt may occur normally, and phosphatases may prematurely abort their activity by removing the phosphate groups responsible for activity. As shown in Fig. 10, none of a broad panel of phosphatase inhibitors prevented inhibition of phosphorylation of Akt in response to apoptotic targets. Chemical inhibitors used in these studies (and the major phosphatases they inhibit) included calyculin A (PP1 and PP2A (protein phosphatases 1 and 2A)); PTPI IV (SHP-2 (Src homology 2 domain-containing protein phosphatase-1), PTP-1B (protein-tyrosine phosphatase 1B), PTP-β, PTP-ϵ, PTP-μ, PTP-σ, and PTP-Meg-2); phenylarsine oxide (protein-tyrosine phosphatases); potassium bisperoxo(1,10-phenanthroline)oxovanadate and dipotassium bisperoxo(picolinato)oxovanadate V (most protein-tyrosine phosphatases plus PTEN (phosphatase and tensin homolog), an inhibitor of Akt-dependent signaling); and PTPI I (SHP-1 and PTP-1B). Phosphatase inhibitors similarly failed to prevent inhibition of phosphorylation of p90RSK (a downstream target of ERK1/2) in response to apoptotic targets (data not shown). Together, these data suggest that, at least with respect to the panel of phosphatase inhibitors used, modulation of Akt activity by apoptotic targets is not mediated by enhanced phosphatase activity.
FIGURE 10.

ERK1/2 and Akt responses following pharmacologic inhibition of phosphatases. Serum-starved BU.MPT cells were stimulated with apoptotic cells (Apo) for 30 min following a 2-h incubation in the presence or absence of each of the following phosphatase inhibitors at two different concentrations (C1 and C2): calyculin A (5 and 10 nm), PTPI IV (50 and 100 μm), phenylarsine oxide (PAO; 20 and 100 μm), potassium bisperoxo(1,10-phenanthroline)oxovanadate (bpV(phen); 1 and 5 μm), dipotassium bisperoxo(picolinato)oxovanadate V (bpV(pic); 1 and 5 μm), and PTPI I (10 and 20 μm). In all cases, non-adherent target cells were removed by washing, and BU.MPT cell lysates were probed for active Akt as shown. Equal loading was confirmed by probing for total Akt (supplemental Fig. 8).
DISCUSSION
Some Responses Elicited by Apoptotic Target Cells are Conserved and Ubiquitous
As cells undergo apoptosis, they acquire new cell-associated activities that modulate the function of neighboring viable cells (6, 11, 13–15). The best characterized of these is anti-inflammatory, but these new activities are not limited to inflammatory suppression. Apoptotic targets can also modulate the proliferation and survival of viable responder cells through their effects on proximal signaling pathways, especially those involving MAPK modules and Akt-dependent signaling (7, 12, 15). Because these effects have been characterized predominantly in primary mϕ cultures, it was unclear whether, like inflammatory suppression, they represented a ubiquitous response to apoptotic targets. To address this question, we studied the proximal signaling events induced following recognition of apoptotic target cells in kidney and other epithelial cells. Epithelial cells are of particular interest in this regard, because, in contrast to mϕ, they are essentially non-phagocytic and non-migratory.
A number of signaling responses, especially those involving inflammatory suppression, appear to be conserved across diverse cell lineages (7, 12–16, 20, 21). Our results suggest that the ability of apoptotic targets to modulate signaling events within MAPK-dependent pathways may also be widespread (Fig. 11). The distinct effects of apoptotic and necrotic target cells reflect the ability of mϕ (6) and epithelial cells (Fig. 3) to discriminate targets undergoing these two forms of death via distinct and non-competing receptors.
FIGURE 11.

Convergent and divergent signaling events elicited within viable cells in response to the recognition or engulfment of dead cells. The recognition and subsequent engulfment of dead cells by both professional and non-professional phagocytes is coupled with a number of early signaling events. These signaling events can be separated into two categories: those that appear to be conserved among all cell types (A) and those that vary depending upon the cell type (B). A, receptor-mediated discrimination of apoptotic and necrotic corpses elicits directionally opposite responses in proinflammatory transcription (denoted here as NFκB-dependent transcriptional activity) and the activity of the signaling kinases ERK1/2, JNK1/2, p38, and p90RSK. These signaling events are identical between mϕ and kidney proximal tubular epithelial cells (i.e. BU.MPT cells). B, mϕ and kidney proximal tubular epithelial cells differ with respect to the effect of dead cells on the activity of the kinase Akt. For mϕ, Akt activity is linked to phagocytosis, so that engulfment of dead targets, whether apoptotic or necrotic, leads to activation of Akt and subsequent phosphorylation of its downstream targets (including GSK3β and Bad). Phagocytosis of latex beads, which occurs in a receptor-independent manner, similarly leads to activation of Akt. In marked contrast, for kidney proximal tubular epithelial cells, Akt activity is linked to the recognition of dead targets. Receptor-mediated discrimination of apoptotic versus necrotic corpses elicits directionally opposite responses in Akt activity. Although apoptotic corpses inhibit Akt activity, necrotic corpses stimulate Akt activity. Latex particles, which are taken up in a receptor-independent manner, have no effect on Akt activity.
Akt-dependent Signaling Responses Elicited by Apoptotic Target Cells Vary with Cell Lineage and Tissue Origin
Although the ability of apoptotic targets to modulate signaling events within Akt-dependent pathways may also be widespread, lineage-specific variations exist in the nature of the response. As contrasted to mϕ (7, 12, 15), this variation manifests in several ways (Fig. 11). First, and most strikingly, although apoptotic targets activate Akt in mϕ (12, 15), they inhibit Akt in BU.MPT cells (Fig. 4). This is true for both basal and EGF- and insulin-induced Akt activity. Second, unlike mϕ, for which activation of Akt is linked to phagocytosis (12, 15), inhibition of Akt in BU.MPT cells is independent of phagocytosis and triggered exclusively by receptor-mediated recognition (Figs. 6 and 7). Finally, as opposed to mϕ (12, 15), the responses of BU.MPT cells to necrotic and apoptotic targets are oppositely directed, with necrotic targets leading to activation of Akt. The divergent Akt-dependent responses by BU.MPT cells to apoptotic and necrotic targets is consistent with the existence of distinct receptors for these two types of dead targets.
Apoptotic Cell Death Serves as a Sentinel of Environmental Change
Together with our previous work, these data suggest that the network of signaling responses to apoptotic targets, as observed in viable cells of multiple lineages, represents a ubiquitous dimension of immune and tissue homeostasis (6, 7, 11–16, 20, 21). Importantly, for both mϕ (7, 12) and BU.MPT cells,3 modulation of ERK1/2 and Akt in response to apoptotic or necrotic targets correlates with changes in cell proliferation and cell survival, respectively. We hypothesize that local alterations in the nature and extent of cell death, triggered by diverse events, such as ischemia, infection, aging, and acute injury, alert surrounding viable cells to environmental changes and stresses. In most cases, signaling events induced in responding cells by apoptotic and necrotic targets are diametrically opposed. Such a differential probably provides a “gauge” to neighboring viable cells by which to assess the overall severity of the environmental stress: sudden and catastrophic in the case of necrotic targets versus more gradual and potentially adaptable in the case of apoptotic targets.
This hypothesis may be used to understand the very different responses of mϕ and BU.MPT cells with respect to Akt signaling. We speculate that the critical difference lies in the ability of mϕ to migrate throughout the body, such that there is no one environment to which mϕ must adapt. Following their recruitment to areas of stress and injury, mϕ must begin the task of clearing dead cells and debris. Interaction with dead targets, whether apoptotic or necrotic, activates Akt in mϕ and thereby selectively enhances their viability. The resulting increase in longevity is in accord with their capacity as professional phagocytes.
In contrast, epithelial cells, such as BU.MPT cells, are predominantly non-phagocytic and fixed within tissues. An increased number of dead cells in their vicinity signals that there are changes in the environment. In this regard, the mode of cell death can provide a clue to the nature of that change and help to guide the response of neighboring viable cells. For example, in the setting of progressive atherosclerosis, chronic ischemia may result in a diminished delivery of oxygen and nutrients. The resulting increase in apoptotic cell death will partially inhibit Akt in nearby viable cells and lead to a reduced cell population. Once supply and demand are again matched, apoptotic cell death returns to base-line levels, and a new steady state is achieved. An increase of necrotic cell death, on the other hand, carries different implications. In this case, the environmental change is likely to be acute and drastic. Increased Akt activity may constitute the cellular equivalent of a “fight and fright” response. For example, vascular occlusion and infarction may produce a stress of such severity that no compensatory long term adaptation is possible, and any cell survival is of potential benefit.
We also observed variation in the Akt response by epithelial cells from different organs. Although our survey is too limited to permit definitive conclusions, such variation may reflect differences in the function and architectural organization of the organs. The kidney, for example, is composed of functional units known as nephrons. The critical question in terms of the direction of Akt modulation may relate to the relative benefit versus harm of retaining a damaged and only partially functional unit. In the case of the kidney, because a nephron normally reabsorbs close to 99% of its filtered sodium, even a mild loss of tubular epithelium could lead to a life-threatening urinary loss of plasma sodium (23). Shutdown of the nephron would therefore seem advantageous. An increase in apoptotic death within a nephron could lead to its shutdown via a positive feedback loop, with apoptotic death begetting further death through apoptotic target-mediated inhibition of Akt. These putative mechanisms require experimental confirmation in intact animals.
In summary, our data indicate the ubiquity of the cellular response to dead targets. We hypothesize that the complex response of viable cells to nearby apoptotic and necrotic targets plays a crucial role in homeostasis not only in the immune system but also in tissues and organs throughout the body. The signaling response to nearby dead target cells permits all cells to continuously monitor their environment for stress or change. Lineage-specific differences in the response of viable cells to adjacent dead targets probably reflect lineage-specific differences in the function and properties of responding cells.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants DK071678 (to V. A. P.), AG024234 (to D. S. U.), and DK059529 (to J. H. S.). This work was also supported by a GRIP Renal Innovations Program Award from Genzyme, Inc. (to J. S. L.), a Veterans Affairs Merit Award (to W. L.), a Pennsylvania State System of Higher Education Faculty Professional Development Grant (to A. L.), and Canadian Institutes of Health Research Grants MOP-67101 (to J. R.) and MOP-42391 (to J. R.) and Institute of Musculoskeletal Health and Arthritis Priority Announcement MUS-67101 (to J. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–9.
V. A. Patel, D. J. Lee, L. Feng, A. Antoni, W. Lieberthal, J. H. Schwartz, J. Rauch, D. S. Ucker, and J. S. Levine, manuscript in preparation.
- mϕ
- macrophage(s)
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated kinase
- JNK
- c-Jun N-terminal kinase
- FBS
- fetal bovine serum
- PI
- propidium iodide
- PBS
- phosphate-buffered saline
- EGF
- epidermal growth factor
- CFDA
- 5,6-carboxyfluorescein diacetate succinimidyl ester
- CMTMR
- 5,6-(((4-chloromethyl)-benzoyl)-amino)-tetramethylrhodamine
- FITC
- fluorescein isothiocyanate
- EGFR
- epidermal growth factor receptor
- PTPI
- protein-tyrosine phosphatase inhibitor.
REFERENCES
- 1.Voll R. E., Herrmann M., Roth E. A., Stach C., Kalden J. R., Girkontaite I. (1997) Nature 390, 350–351 [DOI] [PubMed] [Google Scholar]
- 2.Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M. (1998) J. Clin. Invest. 101, 890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonald P. P., Fadok V. A., Bratton D., Henson P. M. (1999) J. Immunol. 163, 6164–6172 [PubMed] [Google Scholar]
- 4.Huynh M. L., Fadok V. A., Henson P. M. (2002) J. Clin. Invest. 109, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freire-de-Lima C. G., Xiao Y. Q., Gardai S. J., Bratton D. L., Schiemann W. P., Henson P. M. (2006) J. Biol. Chem. 281, 38376–38384 [DOI] [PubMed] [Google Scholar]
- 6.Cocco R. E., Ucker D. S. (2001) Molec. Biol. Cell 12, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel V. A., Longacre A., Hsiao K., Fan H., Meng F., Mitchell J. E., Rauch J., Ucker D. S., Levine J. S. (2006) J. Biol. Chem. 281, 4663–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klune J. R., Dhupar R., Cardinal J., Billiar T. R., Tsung A. (2008) Mol. Med. 14, 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen G. Y., Tang J., Zheng P., Liu Y. (2009) Science 323, 1722–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savill J., Dransfield I., Gregory C., Haslett C. (2002) Nat. Rev. Immunol. 2, 965–975 [DOI] [PubMed] [Google Scholar]
- 11.Cvetanovic M., Ucker D. S. (2004) J. Immunol. 172, 880–889 [DOI] [PubMed] [Google Scholar]
- 12.Reddy S. M., Hsiao K. H., Abernethy V. E., Fan H., Longacre A., Lieberthal W., Rauch J., Koh J. S., Levine J. S. (2002) J. Immunol. 169, 702–713 [DOI] [PubMed] [Google Scholar]
- 13.Mitchell J. E., Cvetanovic M., Tibrewal N., Patel V., Colamonici O. R., Li M. O., Flavell R. A., Levine J. S., Birge R. B., Ucker D. S. (2006) J. Biol. Chem. 281, 5718–5725 [DOI] [PubMed] [Google Scholar]
- 14.Cvetanovic M., Mitchell J. E., Patel V., Avner B. S., Su Y., van der Saag P. T., Witte P. L., Fiore S., Levine J. S., Ucker D. S. (2006) J. Biol. Chem. 281, 20055–20067 [DOI] [PubMed] [Google Scholar]
- 15.Patel V. A., Longacre-Antoni A., Cvetanovic M., Lee D. J., Feng L., Fan H., Rauch J., Ucker D. S., Levine J. S. (2007) Autoimmunity 40, 274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monks J., Rosner D., Geske F. J., Lehman L., Hanson L., Neville M. C., Fadok V. A. (2005) Cell Death Differ. 12, 107–114 [DOI] [PubMed] [Google Scholar]
- 17.Sinha D., Wang Z., Price V. R., Schwartz J. H., Lieberthal W. (2003) Am. J. Physiol. Renal Physiol. 284, F488–F497 [DOI] [PubMed] [Google Scholar]
- 18.Sinha D., Wang Z., Ruchalski K. L., Levine J. S., Krishnan S., Lieberthal W., Schwartz J. H., Borkan S. C. (2005) Am. J. Physiol. Renal Physiol. 288, F703–F713 [DOI] [PubMed] [Google Scholar]
- 19.Jat P. S., Noble M. D., Ataliotis P., Tanaka Y., Yannoutsos N., Larsen L., Kioussis D. (1991) (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 5096–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parnaik R., Raff M. C., Scholes J. (2000) Curr. Biol. 10, 857–860 [DOI] [PubMed] [Google Scholar]
- 21.Wood W., Turmaine M., Weber R., Camp V., Maki R. A., McKercher S. R., Martin P. (2000) Development 127, 5245–5252 [DOI] [PubMed] [Google Scholar]
- 22.El-Achkar T. M., Dagher P. C. (2006) Nat. Clin. Pract. Nephrol. 2, 568–581 [DOI] [PubMed] [Google Scholar]
- 23.Thurau K., Boylan J. W. (1976) Am. J. Med. 61, 308–315 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.