

Abstract
The serotonin transporter (SERT) regulates extracellular levels of the neurotransmitter serotonin (5-hydroxytryptamine) in the brain by facilitating uptake of released 5-hydroxytryptamine into neuronal cells. SERT is the target for widely used antidepressant drugs, including imipramine, fluoxetine, and (S)-citalopram, which are competitive inhibitors of the transport function. Knowledge of the molecular details of the antidepressant binding sites in SERT has been limited due to lack of structural data on SERT. Here, we present a characterization of the (S)-citalopram binding pocket in human SERT (hSERT) using mutational and computational approaches. Comparative modeling and ligand docking reveal that (S)-citalopram fits into the hSERT substrate binding pocket, where (S)-citalopram can adopt a number of different binding orientations. We find, however, that only one of these binding modes is functionally relevant from studying the effects of 64 point mutations around the putative substrate binding site. The mutational mapping also identify novel hSERT residues that are crucial for (S)-citalopram binding. The model defines the molecular determinants for (S)-citalopram binding to hSERT and demonstrates that the antidepressant binding site overlaps with the substrate binding site.
Keywords: Diseases/Neurodegeneration, Methods/Computer Modeling, Methods/Site-directed Mutagenesis, Neurobiology/Neuroscience, Protein/Ligand Binding, Protein/Structure, Transport/Neurotransmitter
Introduction
The serotonin transporter (SERT)2 is an integral membrane protein that facilitates transport of serotonin (5-hydroxytryptamine, 5HT) across cellular membranes (1). In addition to peripheral endocrine functions, 5HT is a neurotransmitter in the brain; it is involved in control of several important physiological functions such as mood, appetite, and sexual behavior. Expressed mainly in the membrane of serotonergic neurons, SERT utilizes energetically favorable cotransport of Na+ to remove released 5HT from the extracellular space. Human SERT (hSERT) belongs to the solute carrier 6 (SLC6) transporter family along with highly homologous transporters for the neurotransmitters γ-aminobutyric acid, glycine, dopamine, and norepinephrine (2–4). These transporters are important drug targets for treatment of a wide range of neurological diseases. In particular, hSERT is the molecular target for widely used drugs for treatment of depression and anxiety. Also, psychostimulants such as amphetamine and 3,4-methylenedioxy-N-methylamphetamine (“ecstasy”) have hSERT as the molecular target (5–7).
The selective serotonin re-uptake inhibitors (SSRIs) are a class of antidepressant and anti-anxiety drugs that function as highly selective competitive inhibitors of hSERT (8). Although SSRIs are highly important for treatment of affective disorders (6), the molecular basis for their function, including location and structure of drug binding pockets, is largely unknown and a matter of debate (9, 10). Such information is important for understanding essential aspects of drug action, ranging from selectivity profile to therapeutic efficacy. Moreover, such information is indispensable for the development of new and improved drugs targeting hSERT. The primary impediment for elucidation of the structural mechanisms of hSERT inhibition is the lack of a three-dimensional structure of the protein. Still, several residues in SERT have been identified mainly by mutagenesis studies that modulate antidepressants potency (11–17). The use of comparative molecular modeling to create structural models of ligand-hSERT interactions has previously been hampered by the low phylogenetic and functional similarity between hSERT and available template proteins (18–21). However, high resolution crystal structures of a bacterial homolog to mammalian SLC6 transporters, LeuT (22, 23), have proven excellent templates for constructing experimentally validated models of substrate and drug binding pockets in human SLC6 transporters, including the human transporters for dopamine and γ-aminobutyric acid (24–32).
In this study, we provide an experimentally validated three-dimensional model of the binding site in hSERT for the SSRI (S)-citalopram (Lexapro®) using mutational analysis of hSERT paired with structure-activity data for (S)-citalopram analogs. LeuT structures are used to create homology models of hSERT, followed by docking of (S)-citalopram. Validation of binding models was performed based on the mutational dataset from 64 hSERT point mutants describing the contribution of residues in the putative 5HT binding pocket for (S)-citalopram inhibition. Characterization of (S)-citalopram analogs at selected point mutants provided evidence for the pairing of specific hSERT residues with the functional groups of (S)-citalopram.
EXPERIMENTAL PROCEDURES
Materials
Dulbecco's modified Eagle's medium, fetal bovine serum, trypsin, and penicillin-streptomycin were purchased from Invitrogen (Carlsbad, CA). Cell culture dishes and 96-well plates were from Nunc (Roskilde, Denmark). [3H]5HT (20.3 Ci/mmol) and MicroScint-20 scintillation mixture were obtained from PerkinElmer Life Sciences (Waltham, MA). (S)-Citalopram and all analogs were prepared by H. Lundbeck A/S, Copenhagen, Denmark.
Site-directed Mutagenesis
The mammalian expression plasmid pcDNA3.1 containing hSERT cDNA has been described previously (14). Generation of point mutations in pcDNA3.1-hSERT was performed by site-directed mutagenesis using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's recommendations, followed by sequencing of the entire hSERT coding sequence of each mutant construct (MWG Biotech, Martinsried, Germany).
Cell Culturing and Expression
COS7 cells (American Type Culture Collection, Rockville, MD) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified 5% CO2 environment. COS7 cells were transfected using TransIT DNA transfection reagent following the protocol supplied by the manufacturer (Mirus, Madison, WI). Prior to transfection confluent cells growing in monolayer were re-suspended in Dulbecco's modified Eagle's medium at a concentration of 1.3 × 106 cells/ml. Per 96-well plate, 6 μg of DNA and 18 μl of transfection reagent were mixed in 0.6 ml of Dulbecco's modified Eagle's medium and incubated at 20 °C for 20 min. Subsequently this mixture was added to the cell suspension, and immediately afterward the cells were dispensed into white 96-well plates at 50% confluence.
[3H]5HT Transport Measurements
Uptake assays were performed 40 h after transfection. Wells were washed twice with phosphate-buffered saline (in mm: NaCl, 137; KCl, 2.7; Na2HPO4, 4.3; and KH2PO4, 1.4, pH 7.3) containing 0.5 mm CaCl2 and 0.5 mm MgCl2 (PBSCM) prior to uptake experiments. In inhibition studies, triplicate wells were incubated with increasing concentration of inhibitors (IC50 determinations) or a fixed concentration of inhibitor (% inhibition) in PBSCM and 50 nm or 150 nm [3H]5HT at 20 °C for 30 min. Uptake was terminated by washing three times with PBSCM. For determination of Km values, cells were incubated with increasing concentration of unlabeled 5HT in PBSCM and a fixed concentration of 50 or 150 nm [3H]5HT at 20 °C for 30 min. Uptake was terminated by washing three times with PBSCM. The amount of accumulated radioligand was determined by solubilizing cells in scintillant (MicroScint20) with counting of plates in a Packard TopCounter (Packard Inc., Prospect, CT). Nonspecific uptake was determined in parallel by measuring uptake in non-transfected cells.
Data Calculations and Statistical Analysis
All data analysis was performed using Prism 4.0 software (GraphPad Inc., San Diego, CA). For determination of IC50 values, dose-response data from [3H]5HT uptake assays were fitted by Equation 1,
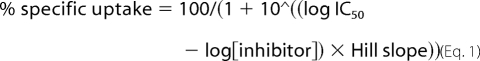 |
where IC50 is the concentration of inhibitor that produces a half-maximal inhibition of uptake. For determination of the Michaelis-Menten constant, data from Km assays were fitted by Equation 2.
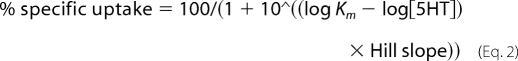 |
IC50 values were converted to Ki values using the Cheng-Prusoff equation (Equation 3) (33),
where [L] is the concentration of [3H]5HT. Wild type was always assayed in parallel with point mutants. Hence, mutant and wild-type Ki values were compared using a repeated measures analysis of variance followed by Dunnett's post hoc test unless otherwise indicated.
Homology Modeling
Starting from the crystal structures of LeuT in the occluded substrate-bound conformation (PDB code 2A65) (22) and the outward facing tryptophan-bound conformation (PDB code 3F3A) (23), an occluded and an outward facing homology model of hSERT was constructed using the MODELLER comparative modeling software package (34) based on a previously published alignment of SERT and LeuT (35). From each starting template, five models were produced, and for each of them four loop models were built. For each of the templates, the models with the lowest penalty were retained for analysis. Equivalent to the LeuT structures (22, 23), two Na+ ions were placed in the hSERT models using the corresponding LeuT crystal coordinates, and the Asp-98 side chain was constrained to point inward the cavity to assure optimal contact to the Na+ ions. According to analysis by the PROCHECK program (36), 90 and 91% of residues in the occluded and the outward-facing models, respectively, were in the most favorable regions of the Ramachandran plot. Residues in the occluded (five residues) and outward-facing (three residues) models that were found in disallowed regions of the Ramachandran plots were exclusively located in extracellular and intracellular loops.
Ligand Docking
Ligand docking into the hSERT models was performed using the Glide protocol (version 5.0) or the induced-fit docking (IFD) protocol that uses a combination of Glide and the Prime protocol (version 1.7) in the Schrödinger software package (Schrödinger, LLC, New York, NY) with default settings applied unless otherwise stated. Before docking, the Protein Preparation Wizard script within the Maestro interface (version 8.5) of Schrödinger was used to prepare the structures in both models. Default settings were used except that, for the occluded model, only the positions of the hydrogen atoms were minimized. For both docking approaches, standard precision scoring functions were used to score the resulting ligand-protein complexes. In general, for each docking experiment, the models containing the highest scoring binding mode were retained for further analysis (see supplemental Fig. S3).
For the initial IFD procedures, (S)-citalopram was docked into the substrate binding cavity of the occluded and the outward-facing hSERT models using Asp-98 as center for the docking. For the occluded model of hSERT, we found that the orientation of the side chains of Ile-172 and Phe-341 were major determinants for the contour of the binding site. Therefore, in separate calculations Ile-172 and Phe-341 were mutated to Ala during the initial stage of IFD of (S)-citalopram into the occluded hSERT model and added again later in the model refinement. During the experiment, the protein van der Waals scaling was furthermore set to 0.5 to maximize the search space. To search for further binding modes of (S)-citalopram in hSERT, we used a Glide docking procedure with default settings applied for docking of the inhibitor into the protein structure obtained from IFD of (S)-citalopram into the occluded hSERT model.
RESULTS
Generation of hSERT Mutant Library
SLC6 transporters are believed to share a similar topology of 12 transmembrane (TM) segments that are connected by short loop segments with intracellular N and C termini (Fig. 1). Structural alignment of SLC6 transporters and LeuT suggest that most family members adopt a similar structure in which the TM1 to TM10 segments form a cylindrical bundle that spans the cell membrane with the substrate binding pocket located in the center of the cylinder (35) (Fig. 1). Experimental data for hSERT and other mammalian SERTs indicate that (S)-citalopram bind within or overlapping with the substrate binding pocket (8, 12, 13, 17). Therefore, we initially focused on examining the role of residues in this region of hSERT for (S)-citalopram inhibition. Our aim was 2-fold: (i) identify residues that are key determinants for recognition of (S)-citalopram and (ii) probe the effect of varying amino acid side chains at these positions. Using a homology model of hSERT as an initial guide (17), we focused on residues located within 6 Å of the putative substrate binding site. This resulted in selection of 22 residues, which are located in TM1, TM3, TM6, or TM8 for mutational analysis (Fig. 1).
FIGURE 1.

A, schematic representation of the topology of hSERT. The putative substrate binding site is formed by residues located in TM1 (dark blue), TM3 (light blue), TM6 (aqua), and TM8 (cyan). White circles indicate the amino acid positions selected for mutagenesis. In B: Left, main-chain trace representation of a structural model of hSERT in complex with the substrate 5HT (17) with α-helices shown as helical ribbons. TMs 1, 3, 6, and 8 are highlighted using color coding as in A. The semi-transparent sphere outlines the location of the putative substrate binding site. Right, close-up view of the putative 5HT binding site in hSERT. Only TMs 1, 3, 6, and 8 forming the binding site are shown, whereas the remaining TMs have been omitted for clarity. 5HT and residues surrounding the substrate binding site are shown as stick representations, and the two sodium ions are shown as purple spheres. Carbons are colored according to the color code of the parent TM segment except for 5HT, which is yellow. Nitrogens and oxygens are dark blue and red, respectively. C, structure of (S)-citalopram.
Initially, we substituted each position with one or more residues having different physiochemical properties from the wild-type residue. For residues identified as key determinants for (S)-citalopram inhibition, additional substitutions were typically introduced. This resulted in a total of 64 point mutants (Fig. 2 and Table 1). The effect of mutations on hSERT function was assessed by determination of the Km for transport for each mutant using a [3H]5HT uptake assay (“Experimental Procedures”) (Table 1). For 11 mutants, specific [3H]5HT uptake could not be measured (Table 1), and none of these mutants could bind 2β-carbomethoxy-3β-(4-[125I]iodophenyl)tropane in intact cells (data not shown). Thus, these mutations either inhibit protein folding and surface expression or severely decrease the ability for hSERT to transport 5HT and bind 2β-carbomethoxy-3β-(4-[125I]iodophenyl)tropane. These mutants were not studied further.
FIGURE 2.

Effect on (S)-citalopram potency of mutations in the substrate binding pocket. Graphic summary of the fold change in Ki (shown on the x-axis) for (S)-citalopram induced by introduction of different point mutations at various positions (shown on the y-axis) in hSERT. The -fold change is calculated as Ki(wild-type)/Ki(mutant) or Ki(mutant)/Ki(wild-type) for mutations decreasing or increasing (S)-citalopram potency, respectively. Black data points located at the x = 0 position indicate the identity of the wild-type residue (one-letter amino acid coding). Gray shading of data points specifies that the mutation did not significantly affect Ki compared with wild type, whereas open circles specify a significant change (p < 0.05; repeated measures analysis of variance followed by Dunnett's post hoc test). Mutations producing >10-fold change in Ki and their corresponding positions are highlighted in blue. Error bars are ±S.E. and shown when larger than symbols. Ki values and statistics are shown in Table 1. Non-functional mutants are indicated on the right.
TABLE 1.
Impact of mutations on [3H]5HT uptake kinetics and inhibitory potency of (S)-citalopram
Transport activities, substrate Km values and (S)-citalopram Ki values were determined in a [3H]5HT uptake assay as described under “Experimental Procedures.” Results are presented as mean ± S.E. from at least three independent experiments each performed in triplicate. The fold changes in Ki values were calculated from paired experiments as Ki(WT)/Ki(mutant) or -Ki(mutant)/Ki(WT) for mutants increasing or decreasing, respectively, (S)-citalopram potency. Asterisks denote significantly different Ki value compared to wild type (p < 0.05; repeated-measures ANOVA followed by Dunnett's post hoc test). N.F. = non-functional.

The majority of functional mutants display Km values for 5HT transport comparable to wild type (0.98 μm) (Table 1). Tyr-95, Asn-177, and Thr-439 have previously been shown to contribute to 5HT binding (24, 37, 38). Accordingly, we found that mutation of these residues in addition to Ala-169, Ile-172, and Gly-442 led to increases in substrate Km (3–10 μm) (Table 1). For the majority of the 53 functional mutants, transport activity was between 15 and 60% compared with wild-type hSERT, whereas six mutants had severely impaired, but detectable, transport activity (5–15% relative to wild type) (Table 1). Further experiments are required to determine whether changes in transport activity are caused by altered protein expression or function.
Identification of Residues Critical for (S)-Citalopram Inhibition in hSERT
The Ki values for (S)-citalopram were determined at each functional point mutant from concentration-inhibition curves constructed from measurements of [3H]5HT uptake at varying concentrations of (S)-citalopram (“Experimental Procedures”) (Fig. 3 and Table 1). At 16 of the 22 selected positions, mutations induce a significant change in Ki. The large fraction of residues where mutation affects inhibition strongly corroborates the proposal that this region in hSERT contains the (S)-citalopram binding site. Mutations in six positions (Tyr-95, Asp-98, Ile-172, Asn-177, Phe-341, and Ser-438) induced from a 10-fold to more than a 400-fold shift in Ki and stood out as key determinants for (S)-citalopram inhibition (Figs. 2 and 3 and Table 1). Among these, Tyr-95, Asp-98, Ile-172, and Ser-438 have previously been reported as key residues for (S)-citalopram inhibition (11–13, 17), whereas the importance of Asn-177 and Phe-341 has not been recognized before.
FIGURE 3.

Analysis of (S)-citalopram potency at wild-type and mutant hSERT. Dose-response curves from representative experiments of inhibition by (S)-citalopram of [3H]5HT uptake in COS7 cells transfected with hSERT cDNA carrying point mutations at the critical amino acid positions Tyr-95, Asp-98, Ile-172, Asn-177, Phe-341, and Ser-438 as described under “Experimental Procedures.” Data points represent the mean from triplicate determinations of accumulated radioactivity in cells incubated with [3H]5HT in the presence of increasing concentrations of inhibitor. Uptake has been normalized to percent uptake of cells incubated in absence of inhibitor. Error bars are ±S.E. and shown when larger than symbols. The normalized data were plotted versus log of the molar concentration of competitor and fit to a nonlinear one-site competition curve as described under “Experimental Procedures.”
Contribution of TM1 and TM3 Residues to (S)-Citalopram Inhibition
Homology models of hSERT show that TM1 and TM3 contribute to forming the substrate binding site in hSERT (24, 35) (Fig. 1). Both TM segments have been subject to detailed mutagenesis studies, and several mutations in TM1 and TM3 can influence the binding of substrates and inhibitors (11–13, 39–41). Here, three TM1 residues (Tyr-95, Asp-98, and Gly-100) were subjected to mutagenesis and in agreement with others (12, 13, 39), we find that substitutions at Tyr-95 and Asp-98 strongly influences (S)-citalopram potency. Y95F induces only a modest loss of potency (4-fold), but removal of the aromatic moiety by introduction of valine (Y95V), glutamine (Y95Q), or alanine (Y95A) has pronounced negative effect on Ki (up to 436-fold increase) (Fig. 2 and Table 1). These results suggest that the aromatic moiety of Tyr-95, rather than the hydroxyl group, is important for (S)-citalopram inhibition. The closely located Asp-98 has previously been shown to be a key residue for both substrate and inhibitor binding. It has been suggested that Asp-98 forms a direct interaction with amino groups found in 5HT and all SERT inhibitors (11, 17, 24, 42). In concurrence with others (11), we find that extension of the acidic side chain by one methylene (D98E) caused a 15-fold loss of potency of (S)-citalopram.
Our analysis included 21 mutations across 10 positions in TM3. Mutation at six positions significantly affected (S)-citalopram inhibition (Fig. 2 and Table 1). In particular, we found Ile-172 highly sensitive to mutation, which is in accordance with earlier findings (13, 39). Specifically, Henry et al. (13) identified that the I172M mutation in hSERT decreased Ki of (S)-citalopram by almost three orders of magnitude. To further study the role of Ile-172, we introduced a range of additional substitutions at this position. Introduction of an aromatic ring (I172F) or a negative charge (I172E) increased Ki by 89- and 21-fold, respectively, whereas a relatively more polar glutamine (I172Q) induced a modest 4-fold increase. Interestingly, decreasing side-chain bulk size (I172A) increased potency of (S)-citalopram 4-fold. Combined with previous results, these data suggest that the hydrophobic side chain of Ile-172 mediates a direct hydrophobic interaction between hSERT and (S)-citalopram and/or has a key role in shaping the binding pocket. Asn-177 is putatively located one helical turn above Ile-172 on TM3 and has not been examined before. Interestingly, substitution of the side-chain amide group with a negative carboxylate group (N177E) slightly increased the potency of (S)-citalopram. In contrast, N177A and N177S display loss-of-potency (up to 10-fold), indicating that the amide side chain of Asn-177 is important for (S)-citalopram inhibition.
Contribution of TM6 and TM8 Residues to (S)-Citalopram Inhibition
TM6 and TM8 have generally received little attention in SERT mutagenesis studies, and these regions were prior to the availability of the LeuT structures not known to contribute to substrate or inhibitor binding. However, the LeuT structures have suggested that the position and orientation of TM6 and TM8 in SERT relates to TM1 and TM3, respectively, by a 2-fold symmetry axis. Indeed, we found that mutations in eight of the nine positions in TM6 and TM8 included in our study, induced significant changes in Ki values of (S)-citalopram (Fig. 2 and Table 1). Two positions, Phe-341 and Ser-438 in TM6 and TM8, respectively, are particularly sensitive to mutations. The role of Phe-341 in hSERT inhibition has not previously been studied. Mutation to alanine (F341A) decreases Ki modestly (3-fold), whereas introduction of longer aliphatic side chains (F341I and F341L) increases Ki by 2- and 6-fold, respectively. Interestingly, addition of a hydroxyl group to the aromatic ring of Phe-341 (F341Y) induced a dramatic 56-fold increase in Ki. We have previously reported that the subtle S438T mutation dramatically reduces the potency of (S)-citalopram (369-fold) (17). Combined with characterization of derivatives of (S)-citalopram, Ser-438 was established as being in close proximity of the dimethyl aminopropyl chain of (S)-citalopram (17). In concurrence with this, S438A only had a modest effect on Ki (3-fold loss of potency), indicating that the hydroxyl group of Ser-438 did not form a direct contact with (S)-citalopram. Removal of the β-hydroxy group of the neighboring Thr-439 (T439A) or replacement with a methyl group (T439V) significantly increased the potency of (S)-citalopram (6- and 2-fold, respectively), whereas removal of the γ-methyl group (T439S) had no significant effect. These results suggest that interaction of (S)-citalopram prefers a non-polar side chain in this part of the pocket.
Inhibition of [3H]5HT Uptake by Citalopram Analogs at Selected SERT Point Mutants
From the initial characterization of the mutant library, Tyr-95, Asp-98, Ile-172, Asn-177, Phe-341, and Ser-438 were identified as key determinants for (S)-citalopram inhibition (Fig. 2 and Table 1). We next sought to identify which part of the (S)-citalopram molecule that might be responsible for interaction with these residues. (S)-Citalopram can be divided into three regions: a cyano-substituted phtalane skeleton, a fluorophenyl group, and a dimethyl aminopropyl chain (Fig. 1). Both Asp-98 and Ser-438 have previously been suggested to be in close proximity to the dimethyl aminopropyl chain (11, 17), thus serving as important anchor points for the positioning of (S)-citalopram in hSERT. To identify specific interaction partners for the two remaining moieties in (S)-citalopram, the cyanophtalane and the fluorophenyl groups, the inhibitory activity of a set of citalopram derivatives with changes in these parts of citalopram were determined at mutants of the critical positions Tyr-95, Ile-172, Asn-177, and Phe-341 (Fig. 4). Also included in these experiments were mutants of Ala-173 and Val-343, because the side chains of these residues have been suggested to be orientated directly toward the center of the substrate binding pocket (43). For these experiments, we used racemic compounds because the pure (S)-enantiomers of the derivatives were not available to us. However, (R)-citalopram has more than 100-fold lower inhibitory potency at hSERT compared with (S)-citalopram (44, 45), thus we assume that the (R)-enantiomers of the derivatives do not contribute significantly to hSERT inhibition. The inhibitory activity of each citalopram derivative across wild type and mutants was assessed by measurement of percent inhibition produced at a single concentration (IC50 at wild-type hSERT for each compound; see supplemental Fig. S1).
FIGURE 4.

Effect of mutations on inhibition of hSERT by analogs of citalopram. A, view of the substrate binding pocket with the pocket surface shown as contour. Key residues Tyr-95, Asp-98, Ile-172, Ala-173, Asn-177, Phe-341, and Val-343 are shown as stick representations color coded as in Fig. 1. B, three-dimensional stick presentations of the structures of citalopram and analogs with molecule contours shown as mesh. Carbons are in yellow, nitrogens in dark blue, oxygens in red, fluorine in light blue, chlorine in green, and bromine in dark red. C, heat map representation of mutation-induced change in uptake inhibition by citalopram analogs carrying modifications around the cyanophtalane moiety (left) or fluorophenyl moiety (right). Values represent differences between the percent inhibition produced by a single inhibitor concentration of [3H]5HT uptake in COS7 cells expressing wild-type or mutant hSERT. For each analog, the test concentration was chosen to be the IC50 concentration for wild-type hSERT (in nm): citalopram, 59; chlorocitalopram, 49; bromocitalopram, 121; desfluoro-citalopram, 232; descyanocitalopram, 154; and 5-methylcitalopram, 70 (see supplemental Fig. S1). Data represent the mean difference from at least three independent experiments, where inhibition at wild-type and mutant hSERT was determined in triplicate.
For each compound, the difference in percent inhibition observed between wild type and mutant was plotted as a heat map (Fig. 4). First, we focused on the cyanophtalane moiety employing two derivatives in which the polar cyano group is substituted with a hydrogen atom (descyanocitalopram) or a non-polar methyl group (5-methylcitalopram). Compared with citalopram, both derivatives displayed similar level and type of sensitivity toward the vast majority of mutations. However, a different pattern was observed at V343N, where inhibition for all three compounds increased, but to a much larger extent for the descyano and 5-methyl derivatives (40 and 28%, respectively) than for citalopram (10%). These results suggest that Val343 is located vicinal to the cyanophtalane moiety of citalopram.
Next, we focused on the fluorophenyl group and employed three citalopram derivatives where the fluoro-atom was substituted with hydrogen (desfluorocitalopram), bromine (bromocitalopram), or chlorine (chlorocitalopram) (Fig. 4). In general, the majority of mutations produced similar effects on these derivatives as for citalopram. However, two interesting observations for mutations at the closely positioned residues Ile-172, Ala-173, and Asn-177 were made. The first observation pertains to Ile-172, where the mutations I172A and I172V, which change the size of the hydrophobic side chain, display differential effects on citalopram and desfluoro-citalopram compared with bromo- and chlorocitalopram (Fig. 4). When the phenyl ring contains a fluorine or a hydrogen in the para position, I172A leads to gain in inhibition (35 and 37%, respectively), whereas the presence of the larger bromo- and chloro-atoms leads to significant loss of inhibition (by 9 and 10%, respectively). A similar trend, although to a lesser extent, was observed at I172V: citalopram and desfluoro-citalopram tolerated this mutation (same degree of inhibition as compared with wild type), whereas the bromo- and chloro-derivatives lost inhibition (28 and 25%, respectively). These results suggest that Ile-172 is in proximity of the fluorophenyl group of citalopram in the binding pocket.
The second notable observation is made at the closely positioned residues Ala-173 and Asn-177. Here, we find that the A173S and N177E mutations increase inhibition for all tested compounds to a similar extent (11% to 17% for A173S; 24% to 28% for N177E) except for desfluorocitalopram, which experienced a markedly larger increase at A173S and N177E (38 and 43%, respectively). To further investigate this pattern, we generated full dose-response curves for citalopram and desfluoro-citalopram at the A173S and N177E mutants (Fig. 5). This confirmed that both mutations indeed improve the relative potency of desfluoro-citalopram significantly more than for citalopram. Notably, at both A173S and N177E mutations, desfluoro-citalopram and citalopram were equipotent, in contrast to wild-type hSERT where removal of the fluorine on the phenyl ring of citalopram led to a 5-fold loss of potency (Fig. 5 and supplemental Fig. S1). Importantly, Ala-173 and Asn-177 contribute to formation of the same surface region in the substrate binding pocket (Fig. 4A) (24). The finding, that increasing polarity in this part of the pocket had a selective effect on inhibitor potency when electron density was decreased at the para position in the phenyl ring, suggests that the fluorophenyl group is located in this region of the binding pocket.
FIGURE 5.

Effect of A173S and N177E on citalopram and des-fluorocitalopram potency. A, representative dose-response curves for inhibition by citalopram (•) and desfluorocitalopram (○) of wild type (left), A173S (middle), and N177E (right). Data points represent the mean from triplicate determinations. Error bars are ±S.E. and shown when larger than symbols. B, graphic summary of mutational effect on citalopram and desfluorocitalopram on inhibitory potency. Error bars are ±S.E. from at least three different experiments.
In summary, analyzing the effect of 23 mutations at six key positions on five (S)-citalopram analogs identified four mutations where the mutational effect was distinctly different among the tested analogs. This allowed us to establish Ile-172/Ala-173/Asn-177 as the anchor point for the fluorophenyl group on citalopram and the cyanophtalane group to potentially be oriented toward Val-343.
Molecular Modeling of the (S)-Citalopram Binding Site in hSERT
The results from the hSERT mutational analysis and the structure-function relationship study with citalopram analogs, combined with previous findings, was used to guide construction of molecular models of the (S)-citalopram binding site in hSERT. First, we generated three-dimensional models of hSERT using the structures of LeuT in the occluded substrate-bound state (22) and the more outward-facing tryptophan-bound state (23) as templates. Models were constructed using comparative modeling (34) based on the structure-based alignment of SERT and LeuT by Beuming et al. (35), resulting in two models for hSERT in an occluded and an outward-facing conformation, respectively (“Experimental Procedures”).
Next, (S)-citalopram was docked into the substrate binding pocket in both hSERT models using an IFD approach (see “Experimental Procedures”). Because both models are based on LeuT structures in which the substrate binding pocket contains relatively small ligands, leucine and tryptophan, the volume of the equivalent pockets in both of the hSERT homology models is too small to allow direct docking of larger ligands, including (S)-citalopram (24, 43, 46). The induced-fit approach overcomes this hindrance by allowing the side chains to move during the docking. The best scoring binding poses from these docking experiments were chosen for further analysis, resulting in two models, A and B (Fig. 6 and supplemental Figs. S2 and S3), based on the occluded and the outward-facing hSERT homology models, respectively.
FIGURE 6.

Orientation of (S)-citalopram in different models obtained from docking of the inhibitor into the substrate binding pocket of hSERT. A–D, models were generated by docking of (S)-citalopram into homology models of hSERT generated on the basis of two conformational different crystal structures of the bacterial leucine transporter LeuT as described under “Experimental Procedures.” Shown are cross-sectional illustrations of Models A–D. TMs 1, 3, 6, and 8 are shown as main-chain trace representation using the same coloring scheme as in Fig. 1, whereas the rest of the protein is omitted for clarity. (S)-Citalopram and key residues are shown as stick representations with the coloring scheme as in Fig. 1. A, IFD of (S)-citalopram into the occluded hSERT homology model (Model A). B, IFD of (S)-citalopram into the outward-facing hSERT homology model (Model B). C, glide docking of (S)-citalopram into the occluded hSERT homology model (Model C). D, IFD of (S)-citalopram into the occluded hSERT homology model (Model D).
Inspection of these models shows that the binding mode of (S)-citalopram is substantially different in the two models, because the two aromatic regions of (S)-citalopram are oriented in almost opposite directions in the binding pocket. In Model A, the dimethyl amino group of (S)-citalopram is in close proximity of the carboxylate group of Asp-98 (3 Å), the cyanophtalane group is positioned near a region formed by Ala-173, Asn-177, Ala-441, Leu-443, and Cys-473, while the fluorophenyl group is inserted in between Ile-172, Tyr-176, and Phe-335 (Fig. 6A and supplemental Fig. S3). In Model B, (S)-citalopram adopts a binding mode in which part of the cyanophtalane group is positioned above Tyr-176 and Phe-335 and the cyano group protrudes the aromatic lid formed by these two residues, pointing toward the extracellular vestibule (Fig. 6B and supplemental Fig. S3). Similar to Model A, the dimethyl amino group is located close to Asp-98, whereas the fluorophenyl group is located between Ile-172, Tyr-176, and Ser-438 (Fig. 6B and supplemental Fig. S3).
We found that, for both models, several features of (S)-citalopram binding were in conflict with key experimental observations of the mutational mapping; indicating that the models do not fully explain the bioactive orientation of (S)-citalopram in hSERT. Specifically, in Model B (S)-citalopram is located distantly from the critical residues Asn-177 and Phe-341 (>7 Å and >5 Å, respectively). In contrast, Model A shows that (S)-citalopram binds within 5 Å of all of 6 key residues, but (S)-citalopram assumes an orientation in which the positions of the fluorophenyl- and the cyanophtalane groups are inconsistent with observations from the structure-function relationships of the citalopram analogs. In Model A, the fluorophenyl and the cyanophtalane groups are essentially positioned opposite to what was expected from the structure-function analysis of citalopram derivatives (Fig. 6A and supplemental Fig. S3).
To further explore binding modes of (S)-citalopram, we used a standard Glide docking approach with the protein structure from Model A as starting point (“Experimental Procedures”). The primary difference to IFD is that in the Glide approach only the ligand is treated as a flexible molecule, and, by an extensive conformational search, probable conformations are generated, while the transporter structure remains fixed. The highest scoring binding mode was similar to Model A, but a distinct binding mode was also obtained and designated Model C (Fig. 6C and supplemental Figs. S2 and S3). In Model C, the fluorophenyl group is located in close proximity to the Ile-172/Ala-173/Asn-177 region, which is in agreement with the experimental observations. However, the cyanophtalane group is positioned between Gly-100, Tyr-176, and Phe-335 (Fig. 6C and supplemental Fig. S3), which is inconsistent with the experimental data. Furthermore, in Model C, Ser-438, which we have previously shown to be located close to the dimethyl amino group of (S)-citalopram (17), is located more than 5 Å from this moiety. Thus, Model C is also considered a poor candidate for deducing a bioactive conformation of (S)-citalopram in hSERT.
The inability of these docking approaches to produce appropriately verified models for (S)-citalopram binding in hSERT led us to examine the hSERT homology models used as the starting point in more detail. Specifically, the occluded hSERT model is based on a crystal structure of LeuT in complex with the relatively small ligand leucine (22). Therefore, the volume of the cavity in the hSERT model equivalent to the leucine binding pocket in LeuT may not allow the docking procedures to explore all possible (S)-citalopram conformations. We therefore inspected the occluded hSERT homology model, and, in agreement with others (46), we found that particularly the orientation of Phe-341 has great influence on the spaciousness of the binding pocket in hSERT. Ile-172 is positioned close to Phe-341, and both residues are highly critical for (S)-citalopram binding (13) (Fig. 2 and Table 1). Therefore, Ile-172 and Phe-341 were both mutated to Ala during IFD of (S)-citalopram into the occluded hSERT model and subsequently replaced in the refinement of the model (“Experimental Procedures”). The best scoring pose from this approach, Model D, shows a different orientation of (S)-citalopram compared with Models A, B, and C (Fig. 6 and supplemental Fig. S3). Specifically, the ligand adopts a binding mode in which the fluorophenyl and the cyanophtalane groups are positioned in the Ile-172/Ala-173/Asn-177 region and the Val-343 region, respectively, while the dimethylamino group is positioned close to Asp-98 and Ser-438. Importantly, Phe-341 adopts a significantly different conformation in Model D, thereby making room for the cyanophtalane group to accommodate the space between TM3 and TM6. Overall, the orientation of (S)-citalopram in Model D fulfills the two major requirements derived from the experimental data: (i) (S)-citalopram is located within 5 Å of all six critical residues Tyr-95, Asp-98, Ile-172, Asn-177, Phe-341, and Ser-438 and (ii) the orientation of the cyanophtalane-, fluorophenyl-, and dimethylaminopropyl groups of (S)-citalopram is consistent with the experimental data. In summary, docking of (S)-citalopram into homology models of hSERT provided a number of potential binding modes, of which Model D provides a reliable bioactive binding mode.
Specific Interactions of (S)-Citalopram and hSERT in the Binding Pocket
The model of (S)-citalopram binding to hSERT (Model D) demonstrates that the inhibitor is positioned such that the cyanophtalane-, fluorophenyl-, and dimethylaminopropyl moieties occupy three distinct sub-sites within the substrate binding pocket (Fig. 7). The alkyl chain containing the dimethyl amino group is accommodated by a sub-pocket formed mainly by the unwound segments in the helical breaks of TM1 and TM6 (Fig. 7, A and B, site A). Here, the backbone carbonyl groups of Tyr-95, Ala-96, Val-97, Phe-335, Ser-336, and Leu-337 along with the side-chain carboxylate group on Asp-98 provide an electronegative environment that surrounds the tertiary amino group of the ligand. A comparison of the orientation of the dimethylaminopropyl chain of (S)-citalopram in our model with the ethylamine chain of 5HT in a model of hSERT (24) shows that these similar ligand moieties assume almost identical orientation and contacts. Many monoamine transporter inhibitors contain a charged amino group, which is essential for high affinity binding and are sensitive to mutations at Asp-98 (11).
FIGURE 7.

(S)-Citalopram binding in the hSERT substrate binding pocket (Model D). A, the (S)-citalopram binding pocket viewed from the extracellular side with the pocket surface shown as transparent contour. The three functional groups on the ligand occupy three distinct sub-pockets within the pocket: the dimethylaminopropyl chain in site A, the fluorophenyl group in site B, and the cyanophtalane group in site C. (S)-Citalopram is displayed as sticks in yellow. B, close-up view of the dimethylaminopropyl chain in site A. Protein side chains and backbone groups forming possible direct contacts to the aminopropyl moiety are shown as stick representations. Carbons are shaded according to the color code of the parent TM segment (same coloring scheme as in Fig. 1). The surface pocket contour is shaded according to electronegativity with increasing intensity of red signifying increasing electronegativity. C, a hydrophobic bulge between site B and site C is formed by the side chains of Ile-172 and Phe-341.
As we have previously reported (17), the S438T mutation induces a decrease of >2000-fold in affinity of (S)-citalopram. The effect of introducing a methyl group at this position in hSERT could be compensated for by reciprocal removal of a methyl group from the inhibitor, indicating that S438T introduce a steric clash between the dimethylamino group and the γ-methyl group of Ser-438. The present model supports this putative role of the S438T mutation by showing the Ser-438 side chain is orientated parallel to the dimethylaminopropyl chain in a distance of <3 Å (Fig. 7B).
Focusing on the accommodation of the cyanophtalane and the fluorophenyl groups of (S)-citalopram, the phtalane ring system is orientated perpendicularly to the bottom part of the pocket, pointing toward a crevice formed by TM6 and TM10 (Fig. 7, A and C, site C), whereas the fluorophenyl group protrudes into a sub-pocket between TM3 and TM8 (Fig. 7, A and C, site B). The side chains of the critical residues Ile-172 and Phe-341 connect to form a hydrophobic bulge located between the fluorophenyl and the cyanophtalane sub-pockets (Fig. 7C, sites B and C). The hydrophobic side chain of Ile-172 is aligned with the fluorophenyl ring of (S)-citalopram and connects with the phenyl group of Phe-341, the latter forming a hydrophobic surface where the cyanophtalane moiety of (S)-citalopram is binding (Fig. 7C). The role of Phe-341 for (S)-citalopram inhibition have not previously been examined, whereas Ile-172 is well characterized as a major determinant for binding of several inhibitors (13, 39). We propose that mutations such as I172M and F341Y cause dramatic changes in (S)-citalopram affinity by reshaping the hydrophobic bulge formed by these residues such that the ability of the fluorophenyl and cyanophtalane groups to bind in their respective sub-pockets is decreased. Thus, Ile-172 and Phe-341 are probably not direct contact points for (S)-citalopram, but are key determinants for controlling alignment of the inhibitor. This is supported by the characterization of (S)-citalopram analogs at I172M and F341Y. Here, we observed that the mutational effects on inhibitor potency were independent of inhibitor structure (Fig. 4). This role of Ile-172 is consistent with a recent study by Walline et al. (39) of the effect of I172M on the inhibitory potency of amphetamine analogs in hSERT. Here, it was suggested that the effects of I172M on potency of amphetamine analogs is probably not caused by disruption of direct inhibitor-protein interactions, but rather may result from a change in the tertiary structure of the inhibitor binding pocket (39).
DISCUSSION
We have performed a systematic mutational analysis of residues forming the putative substrate binding pocket in hSERT to delineate their role as structural determinants for high affinity binding of the antidepressant drug (S)-citalopram. Our site-directed mutagenesis experiments identified two new hSERT residues, Asn-177 and Phe-341, which are highly critical for (S)-citalopram potency in addition to four previously described residues Tyr-95, Asp-98, Ile-172, and Ser-438. Multiple side-chain substitutions at these six residues, as well as other positions were performed, and the mutants were examined for differential effect on the potency of citalopram and analogs of citalopram. Hereby, we obtained a comprehensive mutational mapping of these residues and their importance for the interaction between (S)-citalopram and hSERT. We used the resulting experimental dataset for construction of three-dimensional models of the (S)-citalopram binding pocket in hSERT. These models were obtained by docking (S)-citalopram into models of hSERT, based on LeuT structures. Careful evaluation of the models reveals one model (Model D; Figs. 6 and 7) where the binding mode of (S)-citalopram is consistent with the constraints derived from the experimental data, particularly regarding the pairing of specific functional groups of (S)-citalopram with specific hSERT residues.
A number of important features regarding the molecular mechanism of (S)-citalopram inhibition of hSERT emerges from this study: First, the mutational analysis clearly identifies residues within the substrate binding pocket as critical determinants for high affinity inhibition. Second, as evidenced by docking, (S)-citalopram fits well into the substrate binding pocket of hSERT modeled on the basis of LeuT in the substrate-bound occluded conformation. Together, these findings support the proposal that this site in hSERT (referred to as S1) is the location for the (S)-citalopram high affinity binding site, as also proposed previously (10, 13, 17, 47). Comparison of our (S)-citalopram model with models for 5HT (24, 42) show that the binding sites for 5HT and (S)-citalopram overlaps and confirms that (S)-citalopram is a competitive antagonist of 5HT binding as previously demonstrated by several pharmacological studies (8, 48–50).
Recent studies have suggested that antidepressants, including SSRIs, can bind to a second site (denoted S2) in the extracellular vestibule of hSERT (51–53). In LeuT, and in our homology models, the S2 site is located above the substrate binding pocket separated mainly by a hydrophobic lid formed by a conserved pair of tyrosine (Tyr-176 in hSERT) and phenylalanine (Phe-335 in hSERT) residues in TM3 and TM6, respectively. In the present study, we did not consider the S2 pocket as a potential site for (S)-citalopram binding, because we previously found that non-conservative mutations of residues around the S2 vestibule produced only modest changes (<2-fold change) on (S)-citalopram potency (17). The lack of effect on (S)-citalopram inhibitory potency of mutations within the proposed S2 pocket is in agreement with the findings in the present study and previous studies, which consistently demonstrate that (S)-citalopram potency is highly sensitive to mutations in the S1 substrate binding pocket (13, 17). Therefore, it appears unlikely that the proposed S2 pocket is relevant for high affinity binding of (S)-citalopram in hSERT.
Several LeuT-based models of inhibitor binding to mammalian SLC6 transporters have been reported, including a few addressing antidepressant binding in hSERT (30, 31, 43, 46, 54). A model for (S)-citalopram, based on the substrate-bound LeuT structure, was recently presented (43). This study did not include experiments to probe the role of specific hSERT residues for (S)-citalopram binding to validate the binding mode of the ligand in the resulting model, which is most similar to Model A in the present study. As discussed above we found that conclusions from the mutational analysis did not support this model.
Inhibitors of monoamine transporters have found unparalleled use in the treatment of depression with more than 30 drugs currently approved (6). Among the pharmacological characteristics that determine the clinical properties of these drugs, the selectivity among the monoamine transporters in general, and the selectivity ratio between hSERT and the human norepinephrine transporter (hNET) in particular, is regarded a key determinant for clinical efficacy in depressive disorders. Development of newer generations of antidepressants have focused on a balanced activity across hSERT and hNET; this is exemplified by the hSERT-selective inhibitors such as (S)-citalopram and fluoxetine (Prozac®), the hNET-selective inhibitors such as reboxetine (Edronax®) and atomoxetine (Strattera®), or the dual inhibitors of hSERT and hNET such as duloxetine (Cymbalta®) and venlafaxine (Effexor®). Although the genes for hSERT and hNET have been known for more than two decades, progress in understanding the molecular basis for the hSERT/hNET selectively profile of these classes of antidepressants, including (S)-citalopram, has been sparse. The present model and the mutational dataset could constitute a starting point for identification of specific residues in hSERT and hNET that are responsible for (S)-citalopram selectivity. Notably, among the residues in our model that form the (S)-citalopram binding pocket several are not conserved in hNET, including two of the six critical residues, Tyr-95 and Ile-172. Indeed, mutation of Tyr-95 to Phe, as found in hNET, decreased the potency of (S)-citalopram, whereas mutating Ile-172 to Val, as found in hNET, had no significant effect on (S)-citalopram potency (Fig. 2 and Table 1). However, further studies are needed to fully elucidate the molecular details for hSERT selectivity of (S)-citalopram.
In conclusion, we have generated a model of the (S)-citalopram binding site in hSERT by combining mutational analysis and comparative modeling. The model allows for a rational explanation of previous and current mutations and provides novel insight into the structural details for interaction of an antidepressant drug with hSERT and can guide future studies aiming at identifying molecular determinants for hSERT/hNET selectivity of antidepressants.
Supplementary Material
Acknowledgments
We thank Annette Bjørn for excellent technical assistance and Dr. Anders A. Jensen for fruitful discussions and helpful guidance of the experimental work and the preparation of the manuscript. We thank Dr. Eric Gouaux for providing coordinates of the tryptophan-bound LeuT structure before release date.
This work was supported by the Drug Research Academy (Ph.D. stipend to J. A.), H. Lundbeck A/S, the Danish Ministry of Science, Technology and Innovation (Industrial Ph.D. stipend to A. M. J.), the Danish Council for Strategic Research NABIIT programme (to O. T.), and The Danish Council for Independent Research, Medical Sciences (to L. O.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- SERT
- serotonin transporter
- hSERT
- human serotonin transporter
- 5HT
- 5-hydroxytryptamine (serotonin)
- SSRI
- selective serotonin reuptake inhibitor
- IFD
- induced-fit docking
- hNET
- human norepinephrine transporter
- TM
- transmembrane helice
- SLC6
- solute carrier 6
- PBSCM
- phosphate-buffered saline containing CaCl2 and MgCl2.
REFERENCES
- 1.Masson J., Sagné C., Hamon M., El Mestikawy S. (1999) Pharmacol. Rev. 51, 439–464 [PubMed] [Google Scholar]
- 2.Gether U., Andersen P. H., Larsson O. M., Schousboe A. (2006) Trends Pharmacol. Sci. 27, 375–383 [DOI] [PubMed] [Google Scholar]
- 3.Rudnick G. (2006) J. Membr. Biol. 213, 101–110 [DOI] [PubMed] [Google Scholar]
- 4.Chen N. H., Reith M. E., Quick M. W. (2004) Pflugers Arch. 447, 519–531 [DOI] [PubMed] [Google Scholar]
- 5.Moltzen E. K., Bang-Andersen B. (2006) Curr. Top. Med. Chem. 6, 1801–1823 [DOI] [PubMed] [Google Scholar]
- 6.Andersen J., Kristensen A. S., Bang-Andersen B., Strømgaard K. (2009) Chem. Commun. 3677–3692 [DOI] [PubMed] [Google Scholar]
- 7.Torres G. E., Gainetdinov R. R., Caron M. G. (2003) Nat. Rev. Neurosci. 4, 13–25 [DOI] [PubMed] [Google Scholar]
- 8.Apparsundaram S., Stockdale D. J., Henningsen R. A., Milla M. E., Martin R. S. (2008) J. Pharmacol. Exp. Ther. 327, 982–990 [DOI] [PubMed] [Google Scholar]
- 9.Rudnick G. (2007) ACS Chem. Biol. 2, 606–609 [DOI] [PubMed] [Google Scholar]
- 10.Henry L. K., Meiler J., Blakely R. D. (2007) Mol. Interv. 7, 306–309 [DOI] [PubMed] [Google Scholar]
- 11.Barker E. L., Moore K. R., Rakhshan F., Blakely R. D. (1999) J. Neurosci. 19, 4705–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barker E. L., Perlman M. A., Adkins E. M., Houlihan W. J., Pristupa Z. B., Niznik H. B., Blakely R. D. (1998) J. Biol. Chem. 273, 19459–19468 [DOI] [PubMed] [Google Scholar]
- 13.Henry L. K., Field J. R., Adkins E. M., Parnas M. L., Vaughan R. A., Zou M. F., Newman A. H., Blakely R. D. (2006) J. Biol. Chem. 281, 2012–2023 [DOI] [PubMed] [Google Scholar]
- 14.Kristensen A. S., Larsen M. B., Johnsen L. B., Wiborg O. (2004) Eur. J. Neurosci. 19, 1513–1523 [DOI] [PubMed] [Google Scholar]
- 15.Larsen M. B., Elfving B., Wiborg O. (2004) J. Biol. Chem. 279, 42147–42156 [DOI] [PubMed] [Google Scholar]
- 16.Mortensen O. V., Kristensen A. S., Wiborg O. (2001) J. Neurochem. 79, 237–247 [DOI] [PubMed] [Google Scholar]
- 17.Andersen J., Taboureau O., Hansen K. B., Olsen L., Egebjerg J., Strømgaard K., Kristensen A. S. (2009) J. Biol. Chem. 284, 10276–10284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravna A. W. (2006) World J. Biol. Psychiatry 7, 99–109 [DOI] [PubMed] [Google Scholar]
- 19.Ravna A. W., Edvardsen O. (2001) J. Mol. Graphics Model. 20, 133–144 [DOI] [PubMed] [Google Scholar]
- 20.Ravna A. W., Sylte I., Dahl S. G. (2003) J. Pharmacol. Exp. Ther. 307, 34–41 [DOI] [PubMed] [Google Scholar]
- 21.Ravna A. W., Sylte I., Kristiansen K., Dahl S. G. (2006) Bioorg. Med. Chem. 14, 666–675 [DOI] [PubMed] [Google Scholar]
- 22.Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 23.Singh S. K., Piscitelli C. L., Yamashita A., Gouaux E. (2008) Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Celik L., Sinning S., Severinsen K., Hansen C. G., Møller M. S., Bols M., Wiborg O., Schiøtt B. (2008) J. Am. Chem. Soc. 130, 3853–3865 [DOI] [PubMed] [Google Scholar]
- 25.Forrest L. R., Tavoulari S., Zhang Y. W., Rudnick G., Honig B. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 12761–12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forrest L. R., Zhang Y. W., Jacobs M. T., Gesmonde J., Xie L., Honig B. H., Rudnick G. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10338–10343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y. W., Rudnick G. (2006) J. Biol. Chem. 281, 36213–36220 [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y., Zomot E., Kanner B. I. (2006) J. Biol. Chem. 281, 22092–22099 [DOI] [PubMed] [Google Scholar]
- 29.Zomot E., Bendahan A., Quick M., Zhao Y., Javitch J. A., Kanner B. I. (2007) Nature 449, 726–730 [DOI] [PubMed] [Google Scholar]
- 30.Beuming T., Kniazeff J., Bergmann M. L., Shi L., Gracia L., Raniszewska K., Newman A. H., Javitch J. A., Weinstein H., Gether U., Loland C. J. (2008) Nat. Neurosci. 11, 780–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paczkowski F. A., Sharpe I. A., Dutertre S., Lewis R. J. (2007) J. Biol. Chem. 282, 17837–17844 [DOI] [PubMed] [Google Scholar]
- 32.Wein T., Wanner K. T. (2010) J. Mol. Model. 16, 155–161 [DOI] [PubMed] [Google Scholar]
- 33.Cheng Y., Prusoff W. H. (1973) Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 34.Sali A., Blundell T. L. (1993) J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 35.Beuming T., Shi L., Javitch J. A., Weinstein H. (2006) Mol. Pharmacol. 70, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 36.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 37.Adkins E. M., Barker E. L., Blakely R. D. (2001) Mol. Pharmacol. 59, 514–523 [DOI] [PubMed] [Google Scholar]
- 38.Lin F., Lester H. A., Mager S. (1996) Biophys. J. 71, 3126–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walline C. C., Nichols D. E., Carroll F. I., Barker E. L. (2008) J. Pharmacol. Exp. Ther. 325, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J. G., Sachpatzidis A., Rudnick G. (1997) J. Biol. Chem. 272, 28321–28327 [DOI] [PubMed] [Google Scholar]
- 41.Chen J. G., Rudnick G. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 1044–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufmann K. W., Dawson E. S., Henry L. K., Field J. R., Blakely R. D., Meiler J. (2009) Proteins 74, 630–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jørgensen A. M., Tagmose L., Jørgensen A. M., Topiol S., Sabio M., Gundertofte K., Bøgesø K. P., Peters G. H. (2007) ChemMedChem 2, 815–826 [DOI] [PubMed] [Google Scholar]
- 44.Hyttel J., Bøgesø K. P., Perregaard J., Sánchez C. (1992) J. Neural Transm. Gen. Sect. 88, 157–160 [DOI] [PubMed] [Google Scholar]
- 45.Sánchez C., Bergqvist P. B., Brennum L. T., Gupta S., Hogg S., Larsen A., Wiborg O. (2003) Psychopharmacology 167, 353–362 [DOI] [PubMed] [Google Scholar]
- 46.Tavoulari S., Forrest L. R., Rudnick G. (2009) J. Neurosci. 29, 9635–9643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henry L. K., Defelice L. J., Blakely R. D. (2006) Neuron 49, 791–796 [DOI] [PubMed] [Google Scholar]
- 48.Talvenheimo J., Nelson P. J., Rudnick G. (1979) J. Biol. Chem. 254, 4631–4635 [PubMed] [Google Scholar]
- 49.Humphreys C. J., Levin J., Rudnick G. (1988) Mol. Pharmacol. 33, 657–663 [PubMed] [Google Scholar]
- 50.Graham D., Esnaud H., Habert E., Langer S. Z. (1989) Biochem. Pharmacol. 38, 3819–3826 [DOI] [PubMed] [Google Scholar]
- 51.Zhou Z., Zhen J., Karpowich N. K., Law C. J., Reith M. E., Wang D. N. (2009) Nat. Struct. Mol. Biol. 16, 652–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Z., Zhen J., Karpowich N. K., Goetz R. M., Law C. J., Reith M. E., Wang D. N. (2007) Science 317, 1390–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh S. K., Yamashita A., Gouaux E. (2007) Nature 448, 952–956 [DOI] [PubMed] [Google Scholar]
- 54.Jørgensen A. M., Tagmose L., Jørgensen A. M., Bøgesø K. P., Peters G. H. (2007) ChemMedChem 2, 827–840 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.