

Abstract
The light-activated enzyme protochlorophyllide oxidoreductase (POR) catalyzes an essential step in the synthesis of the most abundant pigment on Earth, chlorophyll. This unique reaction involves the sequential addition of a hydride and proton across the C17=C18 double bond of protochlorophyllide (Pchlide) by dynamically coupled quantum tunneling and is an important model system for studying the mechanism of hydrogen transfer reactions. In the present work, we have combined site-directed mutagenesis studies with a variety of sensitive spectroscopic and kinetic measurements to provide new insights into the mechanistic role of three universally conserved Cys residues in POR. We show that mutation of Cys-226 dramatically alters the catalytic mechanism of the enzyme. In contrast to wild-type POR, the characteristic charge-transfer intermediate, formed upon hydride transfer from NADPH to the C17 position of Pchlide, is absent in C226S variant enzymes. This suggests a concerted hydrogen transfer mechanism where proton transfer only is rate-limiting. Moreover, Pchlide reduction does not require the network of solvent-coupled conformational changes that play a key role in the proton transfer step of wild-type POR. We conclude that this globally important enzyme is finely tuned to facilitate efficient photochemistry, and the removal of a key interaction with Pchlide in the C226S variants significantly affects the local active site structure in POR, resulting in a shorter donor-acceptor distance for proton transfer.
Introduction
The light-activated enzyme protochlorophyllide oxidoreductase (POR)3 catalyzes the reduction of protochlorophyllide (Pchlide) (Fig. 1A), a key reaction in the chlorophyll biosynthetic pathway that triggers a profound transformation in plant development (1–3). Although the reaction can also be catalyzed by a light-independent Pchlide reductase in non-flowering land plants, algae, and cyanobacteria, higher plants only contain POR and are therefore completely reliant on light for the greening process (1, 2). The light dependence of POR presents a unique opportunity to study catalysis at low temperatures and on ultrafast timescales, both of which are usually inaccessible for the majority of enzymes (1). Recent advances in our understanding of the catalytic mechanism of POR illustrate why it is an important generic model for studying enzyme catalysis and reaction dynamics (4–12).
FIGURE 1.

The light-driven reduction of Pchlide. A, trans addition of hydrogen across the C17=C18 double bond of Pchlide to form Chlide in the chlorophyll biosynthesis pathway is catalyzed by the light-driven enzyme, protochlorophyllide oxidoreductase (POR). B, three-dimensional view of the active site of POR based on the structural homology model (17), showing the proposed position of the conserved Cys residues and the putative Tyr proton donor (numbering in T. elongatus POR). The protein backbone is indicated in gray (as a ribbon), the NADPH is shown in yellow, Pchlide is in green, the Tyr is in cyan, and the Cys residues are in red.
Low temperature spectroscopy indicates that the catalytic cycle of the enzyme comprises an initial light-driven reaction that involves hydride transfer from the pro-S face of NADPH to the C17 of Pchlide to form a charge-transfer complex (4, 5). This facilitates the subsequent protonation of the C18 position of the Pchlide molecule from a conserved Tyr residue during the first of the dark reactions (5–7). By using laser photoexcitation studies, we have shown that these two sequential enzymatic H-transfer reactions occur by quantum mechanical tunneling (8). Proton tunneling is coupled to promoting motions in the enzyme that facilitate the reaction by compression of the proton tunneling barrier. At physiological temperatures, hydride transfer also depends on compressive motions localized within the enzyme active site. At cryogenic temperatures, these motions are frozen with concomitant effects on the rate and energetics of the hydride transfer reaction (8). Studies of bulk solvent effects have revealed that solvent-slaved motions control proton, but not hydride, tunneling. This indicates that proton transfer is influenced by a long-range dynamic effect extending from solvent to the enzyme active site (9, 10). The catalytic cycle is completed by a series of ordered product release and cofactor binding steps, which are also linked to conformational changes in the enzyme (11, 12). Ultrafast measurements have also revealed that spectral changes on the picosecond time scale are consistent with the need for conformational change before the hydride transfer step (13–16). Of particular note is that prior excitation with a laser pulse leads to a more efficient conformation of the active site and an enhancement in the catalytic efficiency of the enzyme (16).
Three cysteine residues, namely Cys-37, Cys-199, and Cys-226 (numbering is for POR from Thermosynechococcus elongatus) are absolutely conserved throughout all POR enzymes and are thought to be located in, or close to, the enzyme active site (Ref. 17 and Fig. 1B). The involvement of Cys residues in substrate binding and catalysis has been demonstrated by the use of thiol modification reagents (18, 19). An additional photo-protective role, which involves pigment coordination by Cys residues between POR A and POR B isoforms, has recently been proposed for the barley enzyme (20). Cys-199 and Cys-226 variants were unable to produce Chlide and accumulated singlet oxygen, suggesting that energy transfer between adjacent PORs had been disrupted (20). However, a detailed understanding of the role of the conserved Cys residues in the catalytic mechanism of POR is currently lacking. We now present an in-depth kinetic and spectroscopic analysis of a number of single, double, and triple Cys variants of POR and show that mutations to the Cys-226 residue gives rise to a remarkable change in the catalytic mechanism of the enzyme.
EXPERIMENTAL PROCEDURES
Sample Preparation
All chemicals were obtained from Sigma-Aldrich, except NADPH (Melford Laboratories) and D2O (Goss Scientific). Recombinant POR from the thermophilic cyanobacteria T. elongatus BP-1 was overexpressed in Escherichia coli and purified as described previously (11). Site-directed mutagenesis of the por gene was performed using the QuikChange kit (Stratagene) to mutate residues Cys-37, Cys-199, and Cys-226 to a Ser residue. The following primers (Sigma-Aldrich) were used to generate all single, double, and triple mutated variants: C37S forward primer 5′- CAC GTT ATA ATG GCC TCC CGC AAT CTT GAA AAA GC-3′; C37S reverse primer 5′-GCT TTT TCA AGA TTG CGG GAG GCC ATT ATA ACG TG-3′; C199S forward primer 5′-CCT ACA AAG ACA GCA AGC TCT CCA ATA TGC TGA CGG CAC G-3′; C199S reverse primer 5′-CGT GCC GTC AGC ATA TTG GAG AGC TTG CTG TCT TTG TAG G-3′; C226S forward primer 5′-TCC CTT TAC CCC GGT AGT GTG GCC GAC ACA CCC-3′; C226S reverse primer 5′-GGG TGT GTC GGC CAC ACT ACC GGG GTA AAG GGA-3′.
The correct mutations were confirmed by DNA sequencing (Lark Technology), and variant protein was produced as described previously (11). The Pchlide pigment was produced and purified as described previously (11). Deuterated pro-S NADPH (NADP2H) was purified and analyzed for chemical and isotopic purity as described (21). For solvent isotope effect measurements, POR was deuterated by exchange into a deuterated buffer system containing 50 mm Tris, pH 7.5, 100 mm NaCl, and 1 mm dithiothreitol.
Multiple Turnover (Steady-state) Kinetics and Substrate Binding Measurements
Steady-state activity measurements were carried out as described previously (22) using a Cary 50 spectrophotometer (Varian Inc.). The binding of NADPH coenzyme was monitored using FRET (23) in a Cary Eclipse fluorimeter (Varian Inc.), and the binding of Pchlide was measured by following the red-shift in absorbance at 642 nm, essentially as described (4, 23).
Low Temperature Spectroscopy Studies
Low temperature fluorescence spectra of POR loaded with NADPH and Pchlide were measured in 44% glycerol and 20% sucrose containing 50 mm Tris-HCl, pH 7.5, 0.1% Genapol, 0.1% 2-mercaptoethanol using a Cary Eclipse fluorimeter (Varian Inc.). The excitation light was provided from a xenon arc light source at 450 nm. Excitation and emission slit widths were 3 nm. Low temperature absorbance spectra were measured in the same buffer using a Cary 50 spectrophotometer (Varian Inc.). For all measurements, 1-ml samples were maintained in an Opstistat DN liquid nitrogen cryostat (Oxford Instruments Inc.) at the desired temperature to initiate the reaction before cooling to 77 K at an approximate rate of 10 K per minute to record spectra.
Laser Photoexcitation Measurements
Absorption transients at 696 and 681 nm were measured using laser photoexcitation of dark assembled enzyme-NADPH-Pchlide ternary complexes. Samples were excited at 450 nm, using an OPO of a Q-switched Nd-YAG laser (Brilliant B, Quantel) in a cuvette of 1-cm pathlength as described previously (8). Rate constants were measured at the stated temperature from the average of at least five time dependent absorption measurements by fitting to a single exponential function. For studies of the temperature dependence of rate constants, data were fitted to the Eyring equation. For viscosity studies, glycerol solutions were prepared by weight and calculation of solution viscosity was as described (24). The effect of viscosity on the observed rate was fitted to Equation 1 (25, 26) to describe the contribution of the protein friction to the total friction of the system,
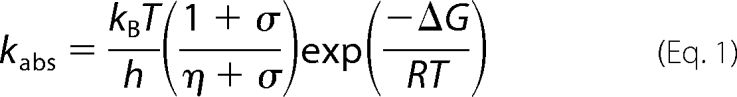 |
where σ, in units of viscosity, is the contribution of the protein friction and η is the absolute viscosity.
RESULTS
Initial Characterization of Cys Variants
The catalytic activity of wild-type POR and the Cys variants were measured initially under steady-state conditions (Table 1). Variants carrying the C226S mutation showed a significant reduction in activity compared with the wild-type enzyme (∼6–7% of wild-type activity), whereas variant enzymes with mutations at only the Cys-37 and Cys-199 positions retained wild-type levels of activity. The role of the Cys residues in substrate binding was determined by measuring the ability of each variant form to bind either NADPH or Pchlide. FRET measurements from Trp residue(s) in the protein to the bound NADPH coenzyme (23) were used to calculate the dissociation constant for NADPH. The apparent dissociation constant (Kd) for the C199S and C226S enzymes was similar to that of wild-type POR (0.021 ± 0.009 μm and Table 1), whereas all variants with the C37S mutation had a higher Kd for NADPH. Moreover, the affinity for NADPH is further reduced in the double and triple variants with the C37S mutation. The red-shift in the absorbance maximum of Pchlide upon binding to the POR-NADPH complex was used to measure the dissociation constant for Pchlide (4, 23). Pigment binding was not influenced by mutations to either Cys-37 or Cys-199, but showed a significant decrease in affinity for all variant enzymes with the C226S residue (Table 1), indicating the importance of this residue in Pchlide binding.
TABLE 1.
Steady-state kinetic and substrate binding parameters for wild-type and mutant forms of POR
All measurements were performed at 25 °C as described under “Experimental Procedures.”
| Enzyme | kcat | Relative activity | Kd for NADPH | Kd for Pchlide |
|---|---|---|---|---|
| s−1 | % | μm | μm | |
| Wild type | 0.165 ± 0.002 | 100.0 ± 1.2 | 0.021 ± 0.009 | 5.6 ± 0.6 |
| C37S | 0.164 ± 0.005 | 99.4 ± 3.0 | 0.055 ± 0.002 | 5.6 ± 0.5 |
| C199S | 0.162 ± 0.002 | 98.2 ± 1.2 | 0.028 ± 0.009 | 5.2 ± 0.3 |
| C226S | 0.0118 ± 0.0003 | 7.2 ± 0.2 | 0.019 ± 0.001 | 23.0 ± 1.3 |
| C37S/C199S | 0.163 ± 0.003 | 98.8 ± 1.8 | 0.20 ± 0.084 | 5.4 ± 0.5 |
| C37S/C226S | 0.0113 ± 0.0003 | 6.8 ± 0.2 | 0.21 ± 0.045 | 33.6 ± 4.0 |
| C199S/C226S | 0.0110 ± 0.0001 | 6.6 ± 0.1 | 0.041 ± 0.008 | 34.1 ± 4.7 |
| C37S/C199S/C226S | 0.0117 ± 0.0002 | 7.1 ± 0.1 | 0.26 ± 0.079 | 28.4 ± 2.2 |
Low Temperature Measurements Reveal a Catalytic Mechanism That Lacks the A696 Intermediate for the Cys-226 Variants
Previously, the catalytic intermediates in the POR reaction have been trapped by continuous illumination at low temperatures and analyzed by cryogenic absorbance and fluorescence spectroscopy, allowing the hydride and proton transfer steps to be identified (4–6). The initial hydride transfer reaction involves the formation of a non-fluorescent charge-transfer species with a broad absorbance band at 696 nm (4), which is converted to the POR-Chlide-NADP+ product complex, with a fluorescence band at 684 nm and an absorbance band at 681 nm after the subsequent proton transfer step (6). Consequently, we have used similar low temperature measurements to characterize the intermediates formed in the reaction catalyzed by the Cys variants. By taking into account the respective Kd values for Pchlide bound to the wild-type enzyme and Cys variants, we ensured that identical starting concentrations of ternary complex were used in these experiments. Comparable spectra were observed in the cryogenic fluorescence measurements for the wild type and Cys variants (Fig. 2A). In addition, the low temperature absorbance spectra showed that variant enzymes with mutations at only the Cys-37 and Cys-199 positions proceeded via a catalytic mechanism identical to that in wild-type POR (Fig. 2B). However, all enzymes with a mutation at the Cys-226 position lacked the charge-transfer intermediate with a broad absorbance band at 696 nm (Fig. 2C), which is formed by hydride transfer from NADPH below 180 K for wild-type POR. Moreover, formation of the subsequent proton transfer intermediate, with an absorbance band at 681 nm, occurs at much lower temperatures (below 180 K) for the C226S variant compared with wild-type POR (Fig. 2C). Significantly, no additional spectral bands were observed in other regions of the absorbance spectrum for the C226S variant (supplemental Fig. S1). It should also be noted that for all enzymes (i.e. wild type and variants), the proton transfer species is converted into the absorbance band of free Chlide at 670 nm above 240 K, confirming that these latter steps are unaffected in the variant enzymes (Ref. 7 and supplemental Fig. S2).
FIGURE 2.

Cryogenic spectroscopic analysis of the catalytic intermediates formed in the wild-type and variant POR enzymes. A, 77 K fluorescence emission spectra of 1.5 μm Pchlide and 250 μm NADPH in the presence of either 20 μm wild-type POR, C37S or C199S and 69.1 μm C226S, recorded after illumination at 230 K for 10 min. The inset shows the 77 K fluorescence emission spectra of wild-type POR and the C226S variant, recorded before (control) and after illumination at 180 K for 10 min. B and C, 77 K absorbance difference spectra of 10 μm Pchlide and 250 μm NADPH in the presence of either 20 μm wild-type POR (B) or 42.3 μm C226S variant enzyme (C), recorded after illumination for 10 min at different temperatures ranging from 77 to 280 K. The difference spectra were obtained by using a non-illuminated sample as a blank. The arrows indicate the formation and disappearance of the different absorbance bands at higher temperatures. D, typical kinetic traces of 1.5 μm Pchlide and 250 μm NADPH in the presence of either 20 μm wild-type POR, C37S or C199S and 69.1 μm C226S, showing the rate of increase in fluorescence at 684 nm upon illumination at 230 K.
The rate of formation of the POR-Chlide-NADP+ product complex was measured for all of the enzymes at 230 K by following the increase in fluorescence at 684 nm, using the fluorescence excitation source to simultaneously trigger catalysis and monitor fluorescence (Fig. 2D). The rate of formation of this species under continuous illumination conditions was significantly faster for all variant enzymes containing the C226S mutation compared with wild-type POR and the variant enzymes with no mutation at the Cys-226 position (supplemental Table S1).
Single Turnover Laser Excitation Measurements Confirm Reaction Mechanism
We have recently shown that the catalytic reaction of POR can be triggered by using a 6-ns laser pulse tuned to the Soret region of the Pchlide absorbance spectrum, allowing the rates of both the hydride and proton transfer steps to be determined (8). We have used similar laser photoexcitation measurements at 298 K to assess the effects on hydride and proton transfer (i.e. formation of the absorbance bands at 696 nm and 681 nm) for all of the variant enzymes (Fig. 3). To ensure that similar levels of ternary enzyme-substrate complex were used in the experiments it was necessary to use higher concentrations of enzyme for the mutant enzymes, based on the respective Kd values for Pchlide binding. The rates of formation and disappearance of the 696 nm absorbance band were found to be similar to wild-type POR for all variant enzymes that lacked the mutation of the Cys-226 residue (supplemental Table S1). However, there was clearly no increase in absorbance at 696 nm for any of the variant enzymes that contain the C226S residue (Fig. 3A). Instead, a monoexponential increase in absorbance at 681 nm was observed upon photoexcitation for the C226S variants enzymes with a rate constant of 5.34 ± 0.05 × 104 s−1 (Fig. 3B). This contrasts with wild-type POR (Fig. 3B) and the remaining variants (supplemental Fig. S3), where laser transients at 681 nm are biphasic and represent both the hydride transfer (from the broad absorbance band at 696 nm) and proton transfer steps (supplemental Table S1).
FIGURE 3.

Single turnover kinetics of Chlide formation in the wild-type and C226S enzymes measured by laser photoexcitation experiments. A, typical kinetic traces measured at 696 nm over 180 μs for the wild type (black) and C226S variant (red) following photoexcitation with a 6-ns laser pulse at 450 nm. The inset shows typical kinetic transients over 4.5 μs. B, typical kinetic traces measured at 681 nm over 180 μs for the wild type (black) and C226S variant (red) following photoexcitation with a 6-ns laser pulse at 450 nm. C, laser-induced difference spectra for the C226S variant, created by measuring the absorbance at the respective wavelength after 0, 2.5, and 150 μs. The inset shows the equivalent laser-induced difference spectra for the wild-type enzyme. All traces were collected at 25 °C as described under “Experimental Procedures” and were fitted to a single exponential to obtain rate constants. Samples contained 15 μm Pchlide and 250 μm NADPH in the presence of either 92.2 μm C226S or 50 μm wild-type POR.
Kinetic transients were subsequently measured between 660 and 710 nm to generate laser-induced difference spectra of the reaction intermediates for wild-type POR and the C226S variant (Fig. 3C). With wild-type enzyme, the broad absorbance band at ∼695–700 nm is formed after 2.5 μs and is converted into the POR-Chlide-NADP+ product complex, which has an absorbance peak at ∼680 nm (Fig. 3C, inset). In contrast, the ∼680-nm absorbing species is the only intermediate observed upon photoexcitation in the C226S variant (Fig. 3C), and this forms on a much faster timescale (i.e. starts to appear within 2.5 μs) than for wild-type POR.
Further Characterization of the Kinetics of Formation of the 681 nm Absorbance Band of C226S
We also performed laser photoexcitation studies with the C226S variant using both pro-S NADP2H and a deuterated buffer system to analyze the isotopic dependence associated with the formation of the POR-Chlide-NADP+ (supplemental Fig. S4 and Table 2). A solvent isotope effect (SIE) of ∼3.6 at 298 K was measured for C226S, which is significantly larger than for wild-type POR. In addition, a primary kinetic isotope effect (KIE) of close to unity with pro-S NADP2H suggests that proton transfer (and not hydride transfer) is rate limiting in the formation of Chlide catalyzed by the C226S variant.
TABLE 2.
The rates of product formation for the C226S variant in the presence of either pro-S NADPH or pro-S NADP2H and in either H2O or 2H2O buffers
All rates were measured at 25 °C as described under “Experimental Procedures.”
| Sample | Rate of product formation | Isotope effect |
|---|---|---|
| s−1 × 104 | ||
| NADPH in H2O | 5.34 ± 0.05 | 1 |
| NADP2H in H2O | 4.90 ± 0.08 | 1.08 ± 0.13 |
| NADPH in 2H 2O | 1.43 ± 0.04 | 3.60 ± 0.14 |
| NADP2H in 2H 2O | 1.36 ± 0.06 | 3.73 ± 0.09 |
We also measured the temperature dependence in both H2O and D2O (278–323 K) for Chlide formation in the C226S variant, which reports on the nature of any fast (sub ps) coupled promoting motions (8). The data, analyzed in the form of Eyring plots (Fig. 4A) to provide thermodynamic parameters for the reaction (supplemental Table S2) reveal that the rate of Chlide formation in the C226S variant is less dependent on temperature (36.6 kJ mol−1) compared with wild-type POR (53.7 kJ mol−1). In addition, the SIE is measurably independent of temperature and the prefactor ratio (A′H/A′D = 4.1) is similar in value to the observed SIE, suggesting that promoting motions are less dominant for proton transfer in the variant enzyme.
FIGURE 4.

Temperature and viscosity dependence of the rate constants for Chlide formation in the C226S variant. A, Eyring plot of ln (kobs/T) versus 1/T for the rate of formation of the absorbance band at 681 nm in the presence of either H2O or D2O for the C226S variant. Data are shown fitted to the Eyring equation and activation enthalpies, ΔH‡, and activation entropies, ΔS‡, are shown in supplemental Table S2. The inset shows the temperature dependence of the SIE. B, dependence of the rate constants for Chlide formation on solvent viscosity for wild-type and C226S POR. Data are shown fitted to Equation 1. In all cases, the error bars are calculated from the average of at least five kinetic traces.
We have shown previously that the rate of proton transfer in wild-type POR is strongly dependent on the solvent viscosity, suggesting that a network of long-range solvent-slaved protein motions are associated with the proton tunneling reaction (9). In contrast, the rate constant for Chlide formation in the C226S variant is less dependent on solvent viscosity (Fig. 4B). The data were fitted to Equation 1 to quantify the sensitivity of the reaction to solvent viscosity, in terms of the contribution of the protein friction (σ) required for Chlide formation. The much higher value obtained for the C226S variant (σ = 7.68 ± 0.66 cP; ΔG‡ = 46.0 ± 0.04 kJ) compared with wild-type POR (σ = 0.15 ± 0.05 cP; ΔG‡ = 47.8 ± 0.03 kJ) suggests a significant reduction in viscosity dependence.
DISCUSSION
The light-driven enzyme POR catalyzes a key reaction in chlorophyll biosynthesis and is an important model system for studying the mechanism of enzymatic hydride and proton transfer reactions (1). It has been proposed that one or more conserved Cys residues are important for activity (18–20), although a detailed demonstration of their exact role in catalysis is currently lacking. Consequently, in the present study we have analyzed a number of single, double, and triple site-directed variants of the three conserved Cys residues in a thermophilic POR, by using a variety of kinetic and spectroscopic methods to probe key aspects of the catalytic cycle. Two of the conserved Cys residues, Cys-37 and Cys-199, appear to have a relatively minor role in catalysis as variants retained wild-type levels of steady-state activity. However, binding of the NADPH coenzyme is weakened by mutations to the Cys-37 residue, which is in close proximity to the conserved nucleotide binding domain at the N-terminal region of the protein (17, 27). Hence, it is possible that Cys-37 may affect the proposed interaction of the neighboring Arg-38 residue with the 2′-ribose-phosphate group of the NADPH molecule. It is also interesting to note that the affinity for NADPH is much lower in the multiple C37S variants, suggesting that the additional mutations may have a cooperative effect on NADPH binding.
Conversely, the third conserved Cys residue, Cys-226, has a major role in the catalytic mechanism of POR. The importance of this residue is illustrated by the considerable reduction in steady-state activity and the compromised binding of the Pchlide substrate for all C226S variants. Cys-226 has a key role in forming an active ternary enzyme-substrate complex, suggesting that pigment binding could be mediated directly by Cys-226, or the effects might be longer range (e.g. through local folding changes on mutating Cys-226). However, more significantly, the C226S variants catalyze the photoreduction of Pchlide by using a different catalytic mechanism to wild-type POR. The charge-transfer intermediate, with a broad absorbance band at 696 nm, which is formed by hydride transfer from the NADPH molecule in wild-type POR (4), is completely absent in the C226S variant enzymes. Consequently, the only species that could be observed in the low temperature and laser photoexcitation measurements was the POR-Chlide-NADP+ product complex, with a characteristic absorbance peak at ∼680 nm (6).
Recent studies on wild-type POR, using deuterated cofactors and buffers, has revealed that the isotope effects associated with each step are specific probes for hydride and proton transfer and have provided conclusive proof of a sequential mechanism (8). For the C226S variants, the rate of formation of the ternary product complex has a SIE of ∼3.7 and a KIE of unity (within error) at 298 K, implying that proton transfer only is rate limiting in this step. Hence, the Pchlide molecule must accept hydrogen from both NADPH and the putative proton donor (Tyr-193) in a single observable kinetic step. This may involve an alternative hydrogen transfer mechanism in which an electron from NADPH is initially transferred upon photoexcitation, followed by the rate-limiting proton transfer (from Tyr-193 to substrate) before the remaining proton and electron are transferred from NADPH (Fig. 5). This final step should be relatively rapid, thereby explaining the lack of a KIE when measurements were repeated with the deuterated coenzyme.
FIGURE 5.

Proposed scheme of the hydrogen transfer mechanism in the C222S variant.
Previous studies on the role of the bulk solvent have shown that the POR-Chlide-NADP+ product complex can only form above the solvent glass transition temperature (10) and that the rate of formation is dependent on the viscosity of the solvent (9). Hence, for the wild-type enzyme, proton transfer is driven by a long-range network of coupled motions from the solvent to the enzyme active site (9, 10). However, in the C226S variant, formation of the ternary enzyme-product complex occurs below the solvent glass transition temperature and on a much faster timescale than in wild-type POR. Moreover, the rate of this step is less influenced by changes to the solvent viscosity, all of which points to a significant reduction in the level of long-range solvent-slaved protein motions that are required for the wild-type enzyme. In addition, according to the recognized model for environmentally coupled hydrogen tunneling (28, 29), the temperature dependence of the rate in protonated and deuterated buffers can provide important information on the nature of any fast (sub ps) promoting motions, which are coupled to the tunneling reaction. Consequently, the temperature-independent SIE associated with this step suggests that proton transfer proceeds in the absence of any dominant fast promoting motions in the C226S variant (30). The lack of any coupled protein dynamics contrasts with the situation in wild-type POR, where the SIE associated with the proton transfer reaction is strongly temperature dependent and implies a key role for coupled promoting motions (8). It is possible that the mutation of the Cys-226 residue alters the local active site structure in POR, resulting in a shorter donor-acceptor distance for proton transfer. The homology models that have been developed for POR suggest that Cys-226 interacts with ring A of the Pchlide molecule (17, 20), which may be important for ensuring that NADPH is in close proximity to the C17 position of Pchlide for efficient hydride transfer upon photoexcitation. Thus, removal of this interaction in the C226S variant could significantly affect the position of the Pchlide molecule in the enzyme active site, thereby increasing the distance between NADPH and the C17 of Pchlide, while simultaneously shortening the distance between the putative proton donor (Tyr-193) and the C18 position. Consequently, this would remove the need for any significant protein motions for the proton transfer step and helps to explain the change in catalytic mechanism in the C226S variant.
In summary, we have used a variety of detailed spectroscopic and kinetic techniques to analyze the role of the conserved Cys residues in the catalytic mechanism of the light-driven enzyme, POR. We find that Cys-37 and Cys-199 have a relatively minor role in catalysis, whereas a mutation to the Cys-226 residue causes a remarkable change in the mechanism of the hydrogen transfer reactions. In the absence of a crystal structure for POR, our results can be rationalized in terms of the reported homology models of the enzyme (17, 20), where Cys-226 interacts with the Pchlide molecule, thus maintaining an optimum alignment with the NADPH cofactor and the proton donor. Our studies provide new insight into the mechanistic role of the conserved Cys residues and suggest that the active site is finely tuned to facilitate efficient photochemistry.
Supplementary Material
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC), UK.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1–S4.
- POR
- NADPH:protochlorophyllide oxidoreductase
- Pchlide
- protochlorophyllide
- Chlide
- chlorophyllide
- KIE
- kinetic isotope effect
- SIE
- solvent isotope effect.
REFERENCES
- 1.Heyes D. J., Hunter C. N. (2005) Trends Biochem. Sci. 30, 642–649 [DOI] [PubMed] [Google Scholar]
- 2.Lebedev N., Timko M. P. (1998) Photosyn. Res. 58, 5–23 [Google Scholar]
- 3.Aronsson H., Sundqvist C., Dahlin C. (2003) Plant Mol. Biol. 51, 1–7 [DOI] [PubMed] [Google Scholar]
- 4.Heyes D. J., Ruban A. V., Wilks H. M., Hunter C. N. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11145–11150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heyes D. J., Heathcote P., Rigby S. E., Palacios M. A., van Grondelle R., Hunter C. N. (2006) J. Biol. Chem. 281, 26847–26853 [DOI] [PubMed] [Google Scholar]
- 6.Heyes D. J., Ruban A. V., Hunter C. N. (2003) Biochemistry 42, 523–528 [DOI] [PubMed] [Google Scholar]
- 7.Menon B. R., Waltho J. P., Scrutton N. S., Heyes D. J. (2009) J. Biol. Chem. 284, 18160–18166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heyes D. J., Sakuma M., De Visser S. P., Scrutton N. S. (2009) J. Biol. Chem. 284, 3762–3767 [DOI] [PubMed] [Google Scholar]
- 9.Heyes D. J., Sakuma M., Scrutton N. S. (2009) Angew Chem. Int. Ed. Engl. 48, 3850–3853 [DOI] [PubMed] [Google Scholar]
- 10.Durin G., Delaunay A., Darnault C., Heyes D. J., Royant A., Vernede X., Hunter C. N., Weik M., Bourgeois D. (2009) Biophys. J. 96, 1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyes D. J., Hunter C. N. (2004) Biochemistry 43, 8265–8271 [DOI] [PubMed] [Google Scholar]
- 12.Heyes D. J., Sakuma M., Scrutton N. S. (2007) J. Biol. Chem. 282, 32015–32020 [DOI] [PubMed] [Google Scholar]
- 13.Heyes D. J., Hunter C. N., van Stokkum I. H., van Grondelle R., Groot M. L. (2003) Nature Struct. Biol. 10, 491–492 [DOI] [PubMed] [Google Scholar]
- 14.Dietzek B., Kiefer W., Hermann G., Popp J., Schmitt M. (2006) J. Phys. Chem. B 110, 4399–4406 [DOI] [PubMed] [Google Scholar]
- 15.Zhao G. J., Han K. L. (2008) Biophys. J. 94, 38–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sytina O. A., Heyes D. J., Hunter C. N., Alexandre M. T., van Stokkum I. H., van Grondelle R., Groot M. L. (2008) Nature 456, 1001–1004 [DOI] [PubMed] [Google Scholar]
- 17.Townley H. E., Sessions R. B., Clarke A. R., Dafforn T. R., Griffiths W. T. (2001) Proteins 44, 329–335 [DOI] [PubMed] [Google Scholar]
- 18.Oliver R. P., Griffiths W. T. (1981) Biochem. J. 195, 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heyes D. J., Martin G. E., Reid R. J., Hunter C. N., Wilks H. M. (2000) FEBS Lett. 483, 47–51 [DOI] [PubMed] [Google Scholar]
- 20.Buhr F., El Bakkouri M., Valdez O., Pollmann S., Lebedev N., Reinbothe S., Reinbothe C. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 12629–12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pudney C. R., Hay S., Sutcliffe M. J., Scrutton N. S. (2006) J. Am. Chem. Soc. 128, 14053–14058 [DOI] [PubMed] [Google Scholar]
- 22.McFarlane M. J., Hunter C. N., Heyes D. J. (2005) Photochem. Photobiol. Sci. 4, 1055–1059 [DOI] [PubMed] [Google Scholar]
- 23.Heyes D. J., Menon B. R., Sakuma M., Scrutton N. S. (2008) Biochemistry 47, 10991–10998 [DOI] [PubMed] [Google Scholar]
- 24.Hay S., Pudney C. R., Sutcliffe M. J., Scrutton N. S. (2008) Angew Chem. Int. Ed. Engl. 47, 537–540 [DOI] [PubMed] [Google Scholar]
- 25.Ansari A., Jones C. M., Henry E. R., Hofrichter J., Eaton W. A. (1992) Science 256, 1796–1798 [DOI] [PubMed] [Google Scholar]
- 26.Ivković-Jensen M. M., Kostić N. M. (1997) Biochemistry 36, 8135–8144 [DOI] [PubMed] [Google Scholar]
- 27.Birve S. J., Selstam E., Johansson L. B. (1996) Biochem. J. 317, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuznetsov A. M., Ulstrup J. (1999) Can. J. Chem. 77, 1085–1096 [Google Scholar]
- 29.Knapp M. J., Rickert K., Klinman J. P. (2002) J. Am. Chem. Soc. 124, 3865–3874 [DOI] [PubMed] [Google Scholar]
- 30.Masgrau L., Roujeinikova A., Johannissen L. O., Hothi P., Basran J., Ranaghan K. E., Mulholland A. J., Sutcliffe M. J., Scrutton N. S., Leys D. (2006) Science 312, 237–241 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.