

Abstract
FGFRL1 (fibroblast growth factor receptor like 1) is the fifth and most recently discovered member of the fibroblast growth factor receptor (FGFR) family. With up to 50% amino acid similarity, its extracellular domain closely resembles that of the four conventional FGFRs. Its intracellular domain, however, lacks the split tyrosine kinase domain needed for FGF-mediated signal transduction. During embryogenesis of the mouse, FGFRL1 is essential for the development of parts of the skeleton, the diaphragm muscle, the heart, and the metanephric kidney. Since its discovery, it has been hypothesized that FGFRL1 might act as a decoy receptor for FGF ligands. Here we present several lines of evidence that support this notion. We demonstrate that the FGFRL1 ectodomain is shed from the cell membrane of differentiating C2C12 myoblasts and from HEK293 cells by an as yet unidentified protease, which cuts the receptor in the membrane-proximal region. As determined by ligand dot blot analysis, cell-based binding assays, and surface plasmon resonance analysis, the soluble FGFRL1 ectodomain as well as the membrane-bound receptor are capable of binding to some FGF ligands with high affinity, including FGF2, FGF3, FGF4, FGF8, FGF10, and FGF22. We furthermore show that ectopic expression of FGFRL1 in Xenopus embryos antagonizes FGFR signaling during early development. Taken together, our data provide strong evidence that FGFRL1 is indeed a decoy receptor for FGFs.
Keywords: Cell/Differentiation, Development Differentiation, Genetics/Human, Membrane/Proteins, Organisms/Xenopus, Protease, Receptors, Fibroblast Growth Factors
Introduction
The fibroblast growth factors (FGFs)2 constitute a family of heparin-binding polypeptides that are involved in a multitude of biological processes such as cellular growth, differentiation, and organogenesis (1, 2). In humans and in mice, the FGF family comprises 22 members (3), of which FGF1 (acidic FGF) and FGF2 (basic FGF) are the most thoroughly studied. Most of the cellular effects of the FGF ligands are mediated by four structurally related tyrosine kinase receptors, designated FGFR1–4. The binding of an FGF ligand to its receptor induces the dimerization of the receptor, followed by the autophosphorylation of tyrosine residues in the cytoplasmic domain and downstream signal transduction (1).
FGFRL1, located on the short arm of chromosome 4 in humans, is the fifth and most recently discovered member of the FGFR family (4, 5). With an amino acid sequence similarity of up to 50%, its ectodomain closely resembles that of the four conventional FGFRs. Its intracellular domain, however, comprises only 100 amino acid residues and completely lacks the tyrosine kinase domain needed for FGF-mediated transphosphorylation and signal transduction. During embryonic development of the mouse, FGFRL1 is expressed in cartilaginous bone precursors, the diaphragm and tongue muscles, the heart and the aorta, the lung, the pancreas, and the kidney. Mice with a targeted disruption of the FGFRL1 gene die immediately after birth because of a hypoplastic diaphragm, which is unable to inflate the lungs (6). In addition to the lethal diaphragm defect the mutant mice display skeletal alterations, craniofacial dysplasia, heart valve defects, and embryonic anemia (7). Perhaps most strikingly, the homozygous null embryos also fail to develop functional metanephric kidneys (8). This defect in kidney development is characterized by an absence of nephrogenesis and by reduced and erratic ureteric branching morphogenesis, ultimately leading to severely dysplastic, rudimentary kidneys without any filtration capacity. Moreover, two recent studies (7, 9) suggested that FGFRL1 could be involved in the etiology of human Wolf-Hirschhorn syndrome, which is a congenital malformation caused by the loss of the end of the short arm of chromosome 4 (10), a loss that sometimes includes the FGFRL1 gene.
Because of the close homology to the classical FGFRs and its structural features, FGFRL1 has been suggested to interfere with or modulate FGF signaling. Indeed, we have shown that overexpressed FGFRL1 can inhibit the expression of an FGF-inducible reporter gene construct in cultured cells (11). Based on expression pattern comparisons, it was proposed that the Xenopus FGFRL1 is part of a synexpression group with FGF8 and likely modulates FGF8-mediated activation of one or several FGFRs (12). The structure of FGFRL1 suggests that it could function as a negative regulator of FGFR signaling. Its ectodomain harbors the structural requirements for FGF binding, and it was shown to actually bind FGF2 in cell culture experiments (5, 13). It could therefore act as a dominant-negative FGFR by ligand-mediated dimerization with a conventional FGFR monomer, thereby preventing transphosphorylation and subsequent signal transduction. Alternatively, it could function as a decoy receptor that competes for FGF ligand binding with other FGFRs. In this context, FGFRL1 could exert its decoy receptor function either in its membrane-bound state or as a soluble form in the extracellular space. In both settings, it is a critical prerequisite that FGFRL1 binds FGF ligands with sufficient affinity to efficiently sequester ligands from other FGFRs. The complex phenotype of the FGFRL1 mutant mice (6–8) suggests that it is likely to interact with more FGFs than only FGF2, because FGF2-deficient mice display only subtle alterations (14).
In the present study we present several lines of evidence supporting the hypothesis that FGFRL1 is a negative regulator of FGF signaling. We demonstrate that the FGFRL1 ectodomain is shed from the cell membranes of HEK293 and differentiating C2C12 myoblasts, generating soluble receptors potentially capable of ligand scavenging. As determined by ligand dot blot analysis, cell-based binding assays, and surface plasmon resonance analysis, the soluble FGFRL1 ectodomain and the membrane-bound receptor are able to bind several FGF ligands with high affinity. Furthermore, we show that overexpression of FGFRL1 in Xenopus embryos antagonizes FGFR signaling in early development of the larvae.
EXPERIMENTAL PROCEDURES
Cell Culture
Human embryonic kidney (HEK293) and C2C12 cells were obtained from the ATCC and cultivated in DMEM with 10% FBS under standard conditions. The differentiation of C2C12 cells and primary myoblasts was induced by switching the medium to DMEM containing 1% FBS. The mouse primary myoblasts were isolated from the tongue of C57BL/6 embryos at embryonic day 18.5. The tongues were minced with a surgical blade in ice-cold PBS, and the fragments were incubated in 0.15% (w/v) collagenase (Roche Applied Science) in serum-free DMEM at 37 °C under constant shaking for 1 h, followed by the same period of incubation in 0.5% (w/v) trypsin/EDTA (Invitrogen) at 37 °C. The cell solution was then washed in DMEM with 10% FBS and passed through a 100-μm cell strainer. The cells were then cultured in DMEM containing 20% FBS and penicillin/streptomycin (Invitrogen) until differentiation was induced with DMEM containing 1% FBS and penicillin/streptomycin.
FGFRL1 Shedding
C2C12 cells were grown to confluence in DMEM containing 10% FBS, and differentiation was induced with DMEM containing 1% FBS. Every 24 h, the medium was completely replaced, and the conditioned supernatants were collected. FGFRL1 in the supernatants was purified with heparin-Sepharose, eluted with SDS sample buffer, and analyzed by Western blotting. Supernatants of the FGFRL1 expressing HEK293 cells were also conditioned for 24 h and subjected to the same treatment. For the shedding inhibition experiments, FGFRL1 overexpressing HEK293 cells were grown to confluence and rinsed with PBS, and the growth medium was exchanged with serum-free medium containing one of the following inhibitors: pepstatin A (Fluka; 5 μg/ml), leupeptin (Sigma; 5 μg/ml), GM6001 (Chemikon; 10 μm), furin inhibitor Dec-RVKR-CMK (Calbiochem; 30 μm), Bace Inhibitor II (Z-VLL-CBO; Merck; 2.5 μm), phorbol 12-myristate 13-acetate (Sigma; 10 ng/ml). The cells were incubated with the inhibitors for 24 h, and FGFRL1 was heparin-purified and subjected to Western blot analysis.
FGFRL1 Expression
The reading frames for human (AJ277437) and mouse (AJ293947) FGFRL1 were inserted into the BamHI/XbaI site of the expression vector pcDNA3.1 carrying a puromycin resistance cassette. (Invitrogen). The truncated versions for the human and the mouse protein were derived from the full-length constructs by PCR. These covered the nucleotide sequences for amino acids 1–416 of the human and 1–408 of the murine receptor (FGFRL1ΔC). For the stable expression in HEK293 cells, the cells were transfected with the FGFRL1 constructs in the pcDNA3.1 vector, and stably expressing cells were selected with 2 μg/ml puromycin over 14 days. All of the stable HEK293 cell lines used in this study were pooled transfectants, not clonal cell lines.
Genomic Sequencing
The coding polymorphism rs4647930 (P362Q) was verified in 96 normal British caucasians. The DNA samples for this purpose (human random control DNA panel HRC-1) were obtained from the European Collection of Cell Cultures (Salisbury, UK). The last exon of the FGFRL1 gene was amplified by PCR with the primer pair TACAGCTTCCGCAGCGCCTTCCTCAC/CTGCCTTCGTCTGCAGCTCCGTCCTC, and the resulting product of 806 bp was sequenced by cycle sequencing, followed by capillary electrophoresis.
Recombinant FGFRL1 Protein
The soluble, c-Myc-tagged FGFRL1 ectodomain was expressed in mammalian cells as described previously (15).
Ligand Dot Blot
All of the ligands were purchased from Peprotech, except FGF2 (Invitrogen), FGF3, FGF12, and FGF22 (R & D Systems) and FGF23 (Prospec). The ligands were reconstituted at 100 ng/μl in the buffers recommended by the manufacturers. All of the ligands (200 ng each) were spotted onto a nitrocellulose membrane, which was then blocked in 3% BSA in PBS. c-Myc-tagged purified FGFRL1 ectodomain at ∼50 μg/ml in PBS containing 0.5% BSA and 0.05% Triton X-100 was incubated with the membrane for several hours, followed by brief washing and detection with a mouse anti c-Myc antibody (Zymed Laboratories Inc., 1:500 in PBS, 1% BSA) and an alkaline phosphatase-coupled secondary antibody (Sigma; 1:30000 in PBS, 1% BSA) for 20 min each. The secondary antibody was visualized with precipitating alkaline phosphatase color substrate.
Northern Blotting
Total RNA was isolated from C2C12 cells with the proprietary TRIzol reagent (Sigma). The RNA was separated on a 1% agarose gel and processed for Northern blotting as previously described (13).
Western Blotting
C2C12 cells, primary cells and HEK293 cells were lysed in SDS containing sample buffer, and the protein mixture was directly loaded onto 10% SDS-polyacrylamide gels. After heparin purification of cell culture supernatants, the beads were heated in SDS sample buffer to elute all bound protein. The eluate was directly subjected to polyacrylamide gel separation on 10% gels. Semi-dry blotting was utilized to transfer proteins to nitrocellulose, followed by blocking with BSA and antibody detection with a goat polyclonal anti-human FGFRL1 antibody (R & D Systems) and alkaline phosphatase-coupled secondary antibody (Sigma). As a loading control, the blots were reprobed with a mouse monoclonal glyceraldehyde-3-phosphate dehydrogenase antibody (Anawa).
Cell-based Ligand Binding Assay
FGF1, FGF2, FGF3, and FGF12 were labeled with DyLight 547-NHS ester (Pierce) as recommended by the manufacturer and stabilized with 1 mg/ml bovine serum albumin. Living cells that had been stably transfected with a human FGFRL1ΔC construct were chilled on ice to prevent receptor internalization and incubated for 30 min with the labeled FGFs (1 μg/ml) and heparin (7.5 μg/ml) in serum-free DMEM. The cells were then thoroughly rinsed with cold PBS, fixed with 4% w/v paraformaldehyde, counterstained with 4′,6′-diamino-2-phenylindole, and inspected by epifluorescence microscopy. HEK293 cells that did not express FGFRL1 were used as a control.
Surface Plasmon Resonance Analysis
The interaction of FGF ligands with FGFRL1 was analyzed using a Biacore X instrument (Biacore International AB). Because our initial studies indicated that FGFRL1 interacts directly with the carboxymethylated dextran of regular CM5 chips, probably through its heparin-binding site (15), C1 chips lacking carboxymethylated dextran were utilized. FGF3 was immobilized on the chip according to the instructions of the supplier. Recombinant FGFRL1 protein was prepared as described (15). Increasing concentrations of recombinant protein were injected over the sensor chip at a flow rate of 10 μl/min. After 180 s, HBS-EP buffer was passed over the sensor surface to monitor the dissociation phase. At the end of each measurement, the sensor surface was regenerated by injection of 2 m NaCl in 100 mm sodium acetate, pH 4.5.
Immunofluorescence
The immunofluorescent stainings were prepared as described previously (11).
Mass Spectrometry
The shed FGFRL1 receptor was purified from HEK293 cell culture supernatant by heparin-Sepharose purification as described above. The FGFRL1 bands were excised from Coomassie-stained SDS-PAGE gels and subjected to tryptic and chymotryptic digestion followed by peptide separation by HPLC (Waters Alliance HT2795) and analysis on a Bruker Esquire3000plus Ion Trap Mass spectrometer.
Xenopus Experiments
In vitro fertilization, embryo culture, staging, and microinjection were performed as described (16, 17). The following constructs were used for in vitro RNA synthesis: human FGFRL1 (AJ277437), mouse FGFRL1 (AJ293947), Xenopus FGFR1 (18), and dominant-negative XFD (18). RNA synthesis was performed as described (17) except that the purification was done by phenol-chloroform extraction. RNA injections were then performed into both blastomeres of two-cell stage embryos using 300 pg of XFD, human or mouse FGFRL1 RNA/blastomere. RNA encoding the lineage tracer nuclear β-galactosidase was coinjected at 250 pg/blastomere. For rescue experiments, FGFR1 RNA was coinjected at 1 ng together with XFD or human or mouse FGFRL1. For control embryos injected with XFD or human or mouse FGFRL1 RNA alone, the amount of lineage tracer RNA was raised to 1.25 ng to adjust for the total amount of RNA injected. The embryos were raised to stage 35/36, stained for β-galactosidase activity, and fixed. Subsequently, the embryos were analyzed by visual inspection, and pictures were taken using a Stemi-2000 stereoscopic microscope (Zeiss).
RESULTS
FGFRL1 Is Shed from the Cell Membrane
During mouse embryogenesis, FGFRL1 is strongly expressed in the developing muscles of the diaphragm and the tongue (19). We also knew from previous studies that murine C2C12 myoblasts, a widely used cell culture model for myogenesis, strongly express FGFRL1 during their differentiation into multinucleated myotubes (6). We therefore set out to study FGFRL1 in this cell culture model of myogenesis. When we tried to analyze FGFRL1 expression in differentiating C2C12 cells, we could not detect any receptor protein in the cell lysates, although the cells began to express FGFRL1 mRNA when they initiated differentiation (Western and Northern blot in Fig. 1A). This caused us to speculate that FGFRL1 could be shed from the cell surface into the cell culture medium. Because FGFRL1 binds to heparin, we subjected the cell culture supernatant to heparin purification and analyzed the eluate for the presence of FGFRL1 protein. Indeed, we obtained a robust Western blot signal that corresponded well to the mRNA levels detected in the Northern blot experiment (Fig. 1A). This finding indicates that most or even all FGFRL1 protein is released from the cells in a soluble form.
FIGURE 1.

FGFRL1 is shed from the surface of C2C12 and HEK293 cells. A, C2C12 myogenic stem cells were induced to differentiate into myotubes by serum withdrawal over 7 days of culture. Northern blot analysis of FGFRL1 mRNA (bottom panel) and Western blot detection of FGFRL1 protein in lysates of differentiating C2C12 myoblasts (top panel) and in the corresponding supernatants (middle panel). B, primary myoblasts obtained from tongue muscle are shown before and after 3 days of in vitro differentiation (top panels; bar, 100 μm). The Western blots (bottom panel) show that undifferentiated, proliferating myoblasts (left top panel) secrete very little FGFRL1, whereas differentiating myoblasts (right top panel) released a larger amount of FGFRL1 into the cell culture medium over 3 days of culture. No FGFRL1 could be detected in the cell lysates of myoblasts and myotubes. C, Western blot analysis of FGFRL1 in the cell culture medium (sup) and in the cell lysate (lys) of HEK293 wild type (WT) cells and HEK293 cells overexpressing FGFRL1 (HEK RL1). Half of purified supernatant and of the lysed cell layers was loaded onto the gel. D, stable overexpression of full-length (RL1 full) and C-terminally truncated FGFRL1 (RL1 ΔC) in HEK293 cells. The immunofluorescence analysis (upper panels) shows that the full-length receptor resides mostly in intracellular vesicles, whereas RL1ΔC accumulates at the cell membrane. Bar, 20 μm. Western blot (bottom panel) detection of FGFRL1 in cell lysates (left top panel) and in the supernatants (right top panel). Note that both proteins were shed in equal amounts. E, protease inhibitor treatment of stably transfected HEK293 cells shedding FGFRL1. The cells were incubated in serum-free medium containing the indicated inhibitors for 24 h. The supernatants were then heparin-purified, and the eluates were subjected to Western blot detection of FGFRL1. None of the inhibitors had any effect on the shedding of FGFRL1.
In line with the results from the C2C12 cells, differentiating primary myoblasts obtained from mouse tongue muscle also released increasing amounts of FGFRL1 protein into the cell culture supernatant during their differentiation into myotubes (Fig. 1B). Again, we could not detect any FGFRL1 protein in the cell lysates of the primary myoblasts and myotubes, indicating that the receptor is efficiently shed from these cells (Fig. 1B).
To test whether FGFRL1 is also shed from the membrane of other cells, we overexpressed human FGFRL1 in human embryonic kidney (HEK293) cells. Western blot analysis of the cell culture supernatant revealed that a significant amount of FGFRL1 protein was shed from the HEK293 cells (Fig. 1C). As expected, the protein isolated from the supernatant was significantly smaller (Mr = 52 kDa) than the membrane-bound receptor (Mr = 67 kDa) from the cell lysate. The shed receptor from the HEK293 cells was of identical size as the endogenous protein released from C2C12 cells, suggesting that the site of cleavage is either identical or at least in the same region (data not shown).
Characteristics of the Protease
We next asked whether the subcellular distribution of FGFRL1 affects its shedding from the membrane of HEK293 cells. Utilizing pulse-chase experiments on living cells with a monoclonal antibody against FGFRL1, we have previously demonstrated that the newly synthesized full-length receptor is first transported to the plasma membrane followed by the internalization into intracellular vesicles. Deletion of the intracellular domain disrupts the internalization, resulting in the accumulation of FGFRL1 in the cell membrane (Ref. 11; for a schematic drawing of the FGFRL1 domain structure please see Fig. 3, top panel). The immunofluorescent staining in Fig. 1D shows the subcellular distribution of the full-length (FGFRL1 full) and the C-terminally truncated FGFRL1 (FGFRL1ΔC) in HEK293 cells. Although there was a clear difference in the distribution of the receptor, we could not detect any difference in the amount of the shed 52-kDa FGFRL1 ectodomain in the supernatants of the two cell lines (Western blot in Fig. 1D). Because the absence of cleaved receptor in the cell lysates of FGFRL1 overexpressing HEK293 cells (Fig. 1, C and D) excludes intracellular processing, we assume that a transient residence of the full-length receptor in the plasma membrane is sufficient for its cleavage. Unlike the shedding from the C2C12 and the primary myoblasts, proteolytic processing at the surface of the HEK293 cells is incomplete and affects only a fraction of the overexpressed FGFRL1. From the amounts detected in cell lysates and supernatants of the HEK cells, we estimated that ∼10% of the overexpressed protein is shed from the cells.
FIGURE 3.

Mass spectrometric analysis of the cleavage sites. A schematic representation of the FGFRL1 receptor is shown. The black bars beneath the scheme indicate the sequence coverage of the FGFRL1 ectodomain, which was 94% of the nonglycosylated peptides. The gaps in coverage most likely represent sites of N-glycosylation, indicated by CHO. Shown below the receptor scheme are the most C-terminal peptides that were identified for the human RL1–362Q and RL1–362P receptors and the mouse FGFRL1. Arrows indicate the proteolytic cleavage sites. Note that the exchange of proline 362 by glutamine results in a shift of the cleavage site to the polymorphic glutamine residue. The original cleavage site is still present in the RL1–362Q protein and is indicated by an arrow with a question mark.
We also attempted to identify the protease that sheds FGFRL1 from the membrane. Because most cell surface shedding is mediated by the ADAM (a disintegrin and metalloprotease)-type of metalloproteases (20), we tried to inhibit the shedding of FGFRL1 with the broad spectrum MMP Inhibitor GM6001 or with 10 mm of EDTA under serum-free conditions. We also tested an inhibitor of the furin proprotein convertase (Dec-RVKR-CMK) and the broad spectrum protease inhibitors leupeptin and pepstatin, which inhibit serine/cysteine and aspartic proteases, respectively, for their inhibitory effect. None of the tested inhibitors had any effect on the extent of receptor shedding (Fig. 1E). Likewise, the addition of the β-secretase inhibitor Z-VLL-CBO or the phorbolester phorbol 12-myristate 13-acetate or the presence of 10% FBS in the culture medium did not affect the protease (data not shown). For the moment, the identity of the FGFRL1 shedding protease remains elusive.
A Polymorphism Enhances FGFRL1 Shedding
If FGFRL1 functions as a soluble decoy receptor, its shedding must not destroy the putative ligand-binding site, which is located between Ig-like domains II and III (13). We set out to determine the exact site of cleavage to exclude the possibility that shedding affected the ligand-binding domains. Judging from the size of the soluble FGFRL1 ectodomain, we estimated that the site of cleavage should be in the membrane-proximal region. Upon screening of the SNP data bank of the NCBI, we discovered that there is an amino acid exchanging SNP in this region of the human FGFRL1 gene, resulting in the exchange of proline at position 362 in the membrane-proximal region to a glutamine residue (RL1–362Q). All other mammals sequenced so far carry a proline at this position. Further sequencing of 96 healthy human donors in our laboratory revealed that 41% of British caucasian individuals were heterozygous for this SNP, whereas 53% of the donors were homozygous for the allele with a proline at position 362 (RL1–362P). Only six individuals (6%) were homozygous for the RL1–362Q allele (Fig. 2A). Because this polymorphism was located near the putative shedding site, we included it in our analysis of the FGFRL1 cleavage site.
FIGURE 2.

A polymorphism in the membrane-proximal region affects the shedding of FGFRL1 from HEK293 cells. A, genomic sequencing of exon 6 of the FGFRL1 gene from 96 healthy British donors. A single nucleotide polymorphism results in the exchange of proline 362 in the membrane-proximal region to glutamine. The percentages of individuals carrying the respective alleles are shown. B, stable, cytomegalovirus promoter-driven overexpression of C-terminally deleted FGFRL1 constructs corresponding to the RL1–362P (left panel) and the RL1–362Q (middle panel) allele as well as the murine protein (right panel). No obvious differences in expression levels or subcellular distribution were detected. Bar, 20 μm. C, Western blot analysis of the three proteins in cell layers and in cell culture supernatants. No difference in overall expression in the cell layer was detected (bottom panel). Significantly more FGFRL1 was purified from supernatants of the RL1–362Q expressing cells, indicating that the shedding of this FGFRL1 variant is enhanced (middle panels).
We investigated first whether the polymorphism had any effect on the shedding of FGFRL1. To this end we generated C-terminally truncated FGFRL1 variants corresponding to both human alleles and to the murine FGFRL1 protein and stably overexpressed the three constructs in HEK293 cells. The protein distribution in the cells was analyzed by immunofluorescent staining (Fig. 2B), and no obvious difference in subcellular distribution could be detected between the constructs. As evidenced by the immunofluorescent staining and by the Western blot in Fig. 2C, overall expression levels in the three cell lines were comparable. However, the amount of FGFRL1 found in the supernatant was strongly increased with the glutamine bearing (RL1–362Q) construct, indicating an enhanced efficiency of shedding. Because only a fraction of the overexpressed FGFRL1 was shed from the membrane (see above), the enhanced shedding did not result in detectable depletion of the cellular pool of the RL1–362Q protein.
Analysis of the Cleavage Sites
The enhanced shedding of the RL1–362Q protein could either be due to an alternative site of cleavage or due to a change in the conformation of the juxtamembrane region leading to enhanced accessibility of the same site. We isolated the shed receptor proteins to address this question and to identify the site of cleavage. Shed FGFRL1 proteins were purified by heparin-Sepharose chromatography followed by SDS-PAGE. The Coomassie-stained gel bands were then subjected to tryptic digestions for liquid chromatography-mass spectrometry-based analysis. We achieved 94% sequence coverage of the nonglycosylated peptides of the ectodomain and 80% coverage of all peptides N-terminal from the putative cleavage sites. For all three constructs a C-terminal peptide was detected that ended with an amino acid that is not a site of tryptic digestion, indicating that the receptor is cleaved by the shedding protease at that position. As shown in Fig. 3, the proline to glutamine exchange at position 362 of the human receptor indeed resulted in an alternative cleavage site. Both the mouse wild type as well as the human RL1–362P protein were cleaved between serine 369 and serine 370 (serine 365 and serine 366 for the mouse), whereas the glutamine-bearing receptor was cleaved eight amino acids further N-terminally between proline 361 and glutamine 362. A chymotryptic digest of the RL1–362Q receptor confirmed that the most C-terminal peptide ended with proline 361. The P362Q exchange in the membrane-proximal region thus led to the enhanced shedding of the human FGFRL1 receptor from HEK293 cells because the glutamine residue at position 362 constituted an alternative, more effective cleavage site for the unidentified protease. It should be mentioned that the original cleavage site is still present in the RL1–362Q receptor (indicated by the arrow with the question mark in Fig. 3) and could still be subject to cleavage. Because we purified only the heparin-binding ectodomain of FGFRL1, the resulting soluble octapeptide QGPPVASS would have escaped our mass spectrometric detection.
On a side note, three of four putative N-linked glycosylation sites in the FGFRL1 ectodomain were indirectly confirmed by our mass spectrometric analysis (Fig. 3), because those peptides were not detected despite virtually full coverage of the nonglycosylated peptides. In addition, the signal peptide was found to be shorter than predicted in the Swiss Prot data base (amino acids 1–24), because a peptide from the shed receptor corresponding to the amino acids 18–26 was detected.
FGFRL1 Binds FGF Ligands
Although we did not identify the protease that mediates the shedding from the C2C12 cells and the HEK293 cells, we succeeded in mapping the site of cleavage to the membrane-proximal region. This leaves all three extracellular Ig-like domains intact, including Ig domains II and III, which are the ligand-binding domains of the four classical FGFRs. The ability of the shed ectodomain to bind FGF ligands would be a prerequirement, if FGFRL1 indeed functioned as a soluble decoy receptor. To investigate this, we employed a ligand dot blot assay with the soluble FGFRL1 and tested all commercially available FGFs for their ability to bind FGFRL1. For this purpose, a soluble, c-Myc-tagged FGFRL1 ectodomain was overexpressed in HEK293 cells. Sufficient amounts of highly purified, glycosylated FGFRL1 ectodomain were obtained after heparin purification from serum-free supernatants. All of the FGFs (epidermal growth factor served as a negative control) were spotted onto a nitrocellulose membrane and incubated with soluble FGFRL1. After washing, bound receptor was detected with an anti-Myc antibody. This ligand dot blot analysis revealed that FGFRL1 bound to a number of ligands (Fig. 4). We consistently observed the strongest signals for FGF3, FGF4, FGF8, FGF10, and FGF22, whereas less but still considerable binding was detected for FGF2, FGF5, FGF17, and FGF 23. Very little to no binding was observed for FGF1, FGF6, FGF7, FGF9, FGF12, FGF16, FGF19, FGF20, FGF21, and epidermal growth factor.
FIGURE 4.
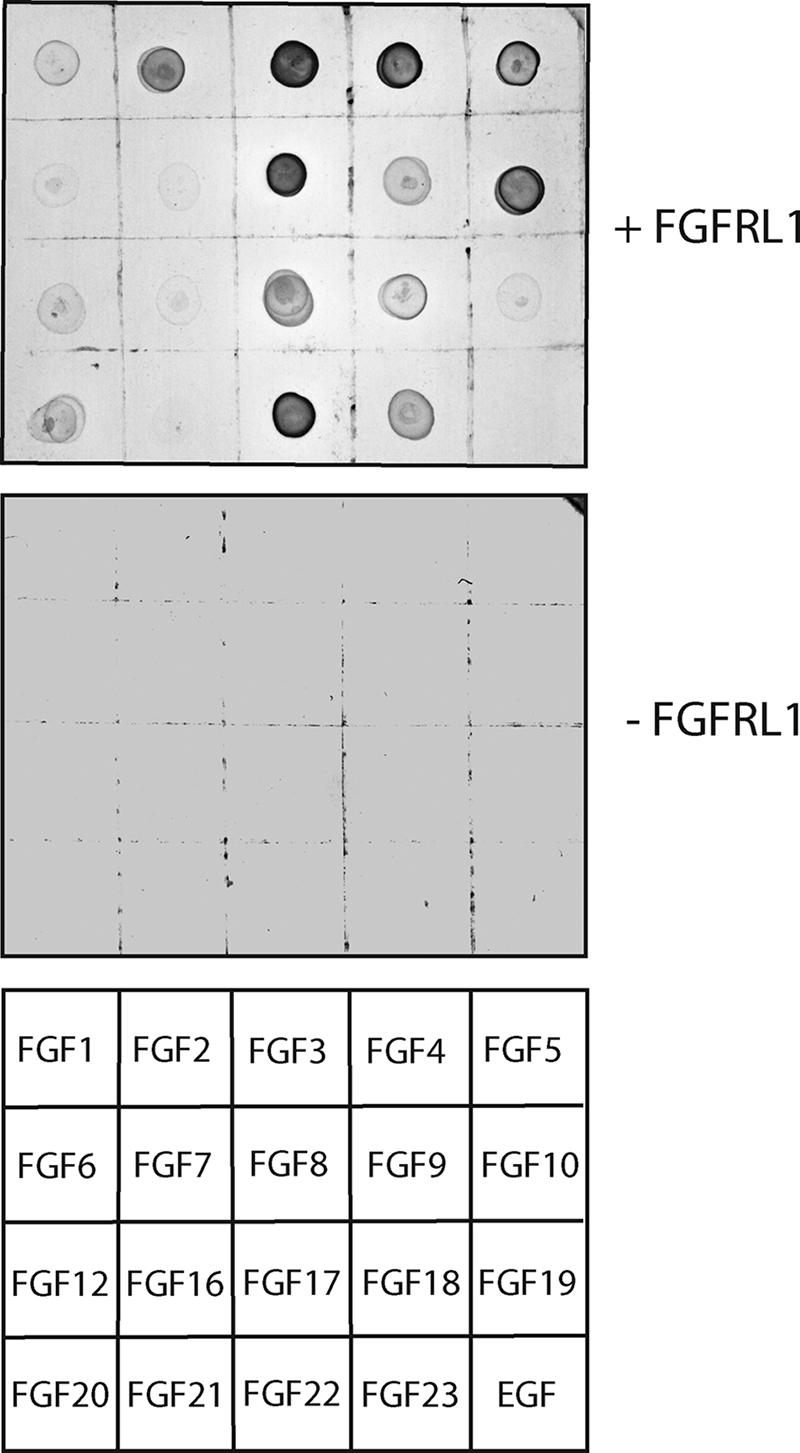
Ligand dot blot analysis reveals differential FGF binding preferences of human FGFRL1. Soluble, Myc-tagged FGFRL1 was used to probe a blot of spotted recombinant FGFs (200 ng each). In the control blot, Myc-tagged FGFRL1 was omitted. The bottom panel shows the positions of the spotted FGFs and the control epidermal growth factor on the blot.
Because it is unusual that FGF2 binds to an FGF receptor but FGF1 does not (21), we wanted to confirm some of the dot blot results with an alternative experimental approach. FGF1, FGF2, FGF3, and FGF12 were labeled by the covalent attachment of the red fluorescent dye DyLight 547. Living HEK293 cells that stably overexpress the C-terminally deleted FGFRL1 on the cell surface (Fig. 5, green) were cooled on ice to prevent receptor internalization and incubated with the labeled ligands. After washing, the cells were fixed, and bound FGFs were visualized by fluorescence microscopy. As shown in Fig. 5, labeled FGF2 and FGF3 clearly bound to the FGFRL1 expressing cells. In contrast, FGF1 and FGF12 did not bind, which is in line with the dot blot results. None of the labeled ligands bound to HEK293 cells that did not overexpress FGFRL1, which excludes unspecific binding or binding to endogenous receptors. The cell-based assay also demonstrated that not only the soluble protein but also the native, membrane-bound receptor is able to bind FGF ligands.
FIGURE 5.

A fluorescent binding analysis on living cells reveals differential FGF binding preferences of human FGFRL1. HEK293 cells expressing FGFRL1 were incubated with selected fluorescently labeled FGFs (DyLight 547, red channel). The subcellular localization of FGFRL1 (Cy-2, green channel) is shown in the top panel. The right panels show the cell nuclei after staining with 4′,6′-diamino-2-phenylindole. Note that FGF2 and FGF3 bind to the surface of the FGFRL1 expressing cells, whereas FGF1 and FGF12 do not bind. Bar, 20 μm.
With regard to the decoy receptor hypothesis, the observed binding should be of sufficient affinity to compete for ligand binding with the other FGFRs. We already knew that the binding must be of rather high affinity because the ligand-bound receptor did not dissociate in the dot blot experiments even after repeated and prolonged washing. Because we consistently observed the strongest signal with FGF3 in the dot blot assay, we decided to determine the affinity of FGF3 to FGFRL1 utilizing a surface plasmon resonance assay. To this end, recombinant FGF3 was coupled to a Biacore sensor chip, and increasing concentrations of soluble, recombinant FGFRL1 were injected over the chip. As shown in Fig. 6, the binding of FGFRL1 to FGF3 showed typical binding and dissociation curves for different concentrations of FGFRL1, resulting in a very low Kd of 4.16 × 10−9 m.
FIGURE 6.

Surface plasmon resonance analysis of the binding of FGFRL1 to FGF3. FGF3 was coupled to a Biacore sensor chip and increasing concentrations (3.13, 6.25, 12.5, 25, and 50 nm) of FGFRL1 ectodomain were injected over the sensor chip. The binding and dissociation kinetics shown in the diagram were integrated by the Biacore analysis software and resulted in a dissociation constant (Kd) of 4.16 × 10−9 m.
Ectopic Expression of FGFRL1 Antagonizes FGF Signaling during Xenopus Development
Our data demonstrate that FGFRL1 is able to bind FGFs both in its soluble and in its membrane-bound state. The high affinity binding observed in the dot blot experiments and measured by plasmon resonance analysis supports the notion that FGFRL1 could function as a negative regulator of FGF signaling. We wanted to test this notion in vivo using a developmental model organism. It was previously demonstrated that overexpression of XFD, a truncated FGFR1, disrupts the development of Xenopus embryos by interfering with FGF signaling in a dominant-negative fashion (22). The resulting embryos presented with gastrulation defects that subsequently affected trunk and tail formation and impaired notochord and muscle development (22). If FGFRL1 interfered with FGF signaling in vivo, injection of FGFRL1 mRNA into Xenopos embryos should produce a similar phenotype.
We therefore microinjected human and mouse FGFRL1 mRNA into Xenopus embryos at the two-cell stage and compared the effects on embryonic development with those of embryos injected with XFD mRNA. As expected, injection of XFD mRNA resulted in the typical posteriorventral truncation phenotype in 93% (n = 57) of the injected embryos (Fig. 7). In line with our hypothesis, injection of mouse and human FGFRL1 reproduced the XFD phenotype in 79% (n = 43) and 65% (n = 40) of the injected embryos, respectively. The dominant-negative effects of XFD expression are reversed by the coinjection of FGFR1 mRNA, which competitively restores FGF-mediated signaling (18). For the XFD construct, coinjection of FGFR1 mRNA reduced the percentage of affected larvae from 100 (n = 10) to 40% (n = 13). For mouse and human FGFRL1, we observed a decrease from 71 (n = 14) to 29% (n = 14) and from 50 (n = 12) to 11% (n = 9) of affected embryos, respectively. The rescue of the FGFRL1-induced mutant phenotype by the coinjection of FGFR1 clearly demonstrates that FGFRL1 interferes with FGF signaling in vivo.
FIGURE 7.

FGFRL1 antagonizes FGF signaling in Xenopus development. Both blastomeres of two-cell stage embryos were coinjected with XFD, mouse FGFRL1 (mFGRL1), or human FGFRL1 (hFGFRL1) mRNA (300 pg/blastomere) and 250 pg of mRNA for the lineage tracer nuclear β-galactosidase. Control embryos injected with lineage tracer only were used to determine stage 35/36 as the end point of development, where all embryos were fixed and processed for β-galactosidase activity. All of the injected embryos are shown beside selected examples presented in close-up views. A, left and right sides of injected control embryos develop normally (top panel). Embryos injected with XFD, mouse FGFRL1, or human FGFRL1 display similar phenotypes (bottom panel). B, coinjection of FGFR1 mRNA together with XFD, mFGFRL1, or hFGFRL1 mRNA rescues the XFD phenotype.
DISCUSSION
Since its discovery about a decade ago, it has been hypothesized that FGFRL1 might act as a decoy receptor that negatively regulates FGF signaling. The concept of such a molecule is certainly not unprecedented, because there are numerous membrane-bound or secreted receptors that specifically bind to ligands but are incapable of signal transduction and thus inhibit the action of their respective ligand(s). One thoroughly studied example is the soluble tumor necrosis factor receptor family member osteoprotegerin. Secreted by osteoblasts, osteoprotegerin acts as a decoy receptor for RANKL (receptor activator for nuclear factor κB ligand) that inhibits RANKL-mediated osteoclast differentiation (23). Also well characterized is the membrane-bound receptor IL1 receptor 2, which binds IL1 but lacks the cytoplasmic Toll-IL1 receptor motif needed for signal transduction. It thereby sequesters ligands from the active IL1R1 and attenuates IL1-mediated inflammatory signals (24). Particularly in the immune system, decoy receptors are widely used to “fine-tune” cellular responses to cytokines (25). In the realm of growth factors, the kinase-deficient ErbB3 functions as a decoy receptor by scavenging neuregulin ligands away from other ErbB receptors (26).
Here we presented several lines of evidence that support the concept of FGFRL1 being a “decoy,” a receptor that interferes with FGFR activation by competitive ligand binding. The receptor is proteolytically released from cell membranes of differentiating myoblasts at a time when the cells need to exit the cell cycle and when FGF-mediated proliferative signals need to be shut down or modulated. Furthermore we showed that it binds to several FGFs with high affinity, which is a prerequisite for a putative ligand scavenging receptor function. Finally, we demonstrated that the effect of FGFRL1 expression in developing Xenopus embryos closely resembles that of a known dominant-negative FGFR and that it can be reversed by coexpression of FGFR1.
Shedding of FGFRL1
The severe diaphragm phenotype of the FGFRL1-deficient mice (6, 7) clearly shows that FGFRL1 is critical for the development of this muscle, yet a simple defect in myoblast differentiation can be excluded as the underlying cause. Although there are about 40% fewer myofibers in the mutant diaphragms, the remaining myotubes are normally differentiated and express the full range of differentiation markers (6). Moreover, a successful, stable RNA interference knockdown of FGFRL1 in C2C12 myoblasts did not significantly alter their differentiation into myotubes in vitro.3 In addition, we did not observe any overt differentiation defects in primary, FGFRL1-deficient myoblasts isolated from tongues and diaphragm muscles of our mice.4 The biological effect of FGFRL1 during muscle development is most likely of a more subtle or complex nature. During mouse embryogenesis, FGFR4, FGF8, and Sprouty1, a negative regulator of FGF signaling, constitute a signaling axis that governs the balance between myogenic stem cell renewal and differentiation (27–30). In theory, FGFRL1 could be another player in this finely tuned system, perhaps by interfering with the amount of available FGF8. The loss of FGFRL1 function in our mice could disrupt the balance of myogenic stem cell proliferation and differentiation, leading to a lower number of available myoblasts that eventually form the diaphragm myofibers. Here we showed that FGFRL1 is released as a soluble receptor from myogenic stem cells in a cell culture system.
Because we could not detect any membrane-associated receptor in the cell layer, we assume that most or all FGFRL1 is shed from the cells before or during their differentiation. If this occurred in vivo, it would result in an accumulation of soluble FGFRL1 in the extracellular space, where the receptor could bind to FGF ligands such as FGF8 and prevent the ongoing activation of cellular FGFRs. From previous fluorescence resonance energy transfer and coprecipitation studies, we know that FGFRL1 forms constitutive dimers (15). Because the four conventional FGFRs dimerize upon binding of FGF ligands and heparan sulfate chains (1), it seems plausible that the constitutive FGFRL1 dimers would be capable of ligand binding after the shedding from the membrane.
Analysis of the Cleavage Sites
Our analysis of the cleavage site revealed that the site of proteolysis is in close proximity to an amino acid exchanging polymorphism in the human FGFRL1 gene. Quite surprisingly, the exchange of proline at position 362 of the human protein to a glutamine residue altered the shedding efficiency from HEK293 cells. This was due to a shift in the site of cleavage from serine 372 to the polymorphic glutamine residue at position 362. Because we consistently detected larger amounts of shed receptor in the supernatants of cells expressing the glutamine bearing receptor, we believe that Gln362 constitutes an alternative, more efficient site of cleavage. At this point we are not convinced that there is any biological significance in this finding, because six of the sequenced individuals were homozygous for the polymorphism but were listed as healthy donors. Moreover, the data are based on the overexpression of the receptor in heterologous cells, which might affect the kinetics or the stoichiometry of the protease-FGFRL1 interaction. However, because of experiments with several cell lines that endogenously express high levels of FGFRL1 mRNA, we do know that the shedding of FGFRL1 is not a ubiquitous process that takes place in every cell. None of the cells tested (A204 rhabdomyosarcoma, SW1353 chondrosarcoma, MG63 osteosarcoma) secreted any soluble FGFRL1 into the culture medium, indicating that they are devoid of FGFRL1 sheddase activity.4 This suggests that the protease responsible for the cleavage of FGFRL1 is subject to regulated expression in only a subset of cells or tissues. Because none of the conventional protease inhibitors tested had any effect on receptor shedding, the identity of the protease remains elusive.
FGF Ligand Binding
The notion of FGFRL1 being a decoy receptor for FGF ligands depends on the FGF binding capability of the receptor. Therefore, we used several experimental approaches to determine whether FGFRL1 binds FGFs and to investigate the ligand binding preferences. The dot blot and cell-based FGF binding assays clearly demonstrated that FGFRL1 binds to several FGFs with a high apparent affinity. Moreover, both the membrane-bound and the soluble receptors bind to FGFs. The Kd values of 4 × 10−9 m (4 nm) for FGFRL1 and FGF3 obtained by surface plasmon resonance analysis indicate that the interaction has an affinity that is about 1 order of magnitude higher than the affinity of FGF3 to its cognate receptor FGFR2b (31). The same applies to the affinity of FGFR2 toward its physiologic ligands, which are in the 10–100 nm range (31). Ibrahimi et al. (31) report comparably low Kd values only for mutant FGFR2 carrying single amino acid substitutions in the ligand-binding site. Hence, the measured affinity of FGFRL1 to FGF3 supports the notion that FGFRL1 acts as a decoy receptor.
With regard to the ligand binding preferences, it is interesting that FGFRL1 displayed strong binding to several members of the FGF7 family (FGF3, FGF10, and FGF22) but did not bind FGF7 itself. An explanation could lie in a generally lower affinity of FGF7 to FGFRs, because the Mohammadi group reported significantly higher Kd values for FGFR2b and FGF7 (1 × 10−5 m) than for FGFR2b and FGF3 (3.6 × 10−7 m) (32). It is also unusual that FGFRL1 binds FGF2 but shows no binding to FGF1 (21, 32). However, we confirmed this finding with the cell-based binding assay. Again, we detected robust binding of FGF2 but none of FGF1. Taken together, we can conclude that the ligand specificity of FGFRL1 does not conform to the rule that a certain receptor binds all ligands of an FGF family (21). This unusual ligand binding profile and the high affinity binding to some ligands suggest that the mechanism of binding could be different from the other FGFRs. On the other hand, single amino acid mutations in FGFR2 have been shown to result in promiscuous ligand binding (31), indicating that ligand receptor specificities are somewhat flexible and subject to small alterations in amino acid structure. A crystal structure of FGFRL1, ideally in a complex with a bound ligand, would likely reveal differences in the binding site and explain the discrepancies to the other FGFRs.
Concerning the biological significance, FGFRL1 also bound FGF4 and FGF8, which, together with FGF3, FGF10, and FGF22, consistently displayed the most prominent binding in the dot blot assay. FGF4 has been shown to be important for the development of the heart valves (33), a process that is disturbed in the FGFRL1-deficient mice (7). As already mentioned above, FGF8-mediated FGFR4 activation governs the balance between myoblast renewal and differentiation in developing skeletal muscle, a balance that could be disrupted in the hypoplastic diaphragms of the FGFRL1-deficient mice. Intriguingly, a recent study has shown that FGFRL1 can directly interact with FGFR4, lending more credibility to a modulation of FGFR4 and possibly FGF8 activity by FGFRL1 (34). Furthermore, FGF8 is of critical importance for the initiation and maintenance of nephrogenesis in the metanephric kidney, and its conditional inactivation in the metanephros results in virtually the same kidney phenotype as observed in the FGFRL1 knock-out mice (8, 35, 36). For Wnt-mediated nephrogenic signals, it has been shown that the β-catenin response is only transient and has to be shut down for nephron development to proceed (37). Hence it is conceivable that a similar nephrogenic FGF signal has to be tightly controlled by FGFRL1 to exert the full effect. Given the broad ligand binding preferences and the high binding affinities, FGFRL1 could serve as an efficient scavenging receptor, preventing the activation of conventional FGFRs in various tissues, thereby fine-tuning the tissue response to these ligands.
Xenopus Experiments
Because all of the data discussed above provided only indirect evidence that FGFRL1 functions as a decoy receptor, we finally set out to demonstrate that FGFRL1 can actually antagonize FGF signaling in vivo. We chose the Xenopus model system because it is relatively simple to introduce a new gene by microinjection of mRNA. In addition, the effect of a known dominant-negative regulator of FGF signaling, a truncated FGFR1 (XFD), had already been thoroughly described (18, 38, 39), enabling us to compare its effects to those of FGFRL1 expression. In fact, ectopic FGFRL1 expression almost exactly reproduced the XFD phenotype, albeit with a slightly lower penetrance. As with XFD, the FGFRL1 induced phenotype could be reversed by the coinjection of FGFR1 mRNA, indicating that it is indeed FGF signaling that is perturbed by FGFRL1. At present, we do not know the precise identity of the Xenopus FGF ligands that FGFRL1 scavenges when it is expressed in the developing embryos. It is likely that FGFRL1 binds several of the endogenous FGFs that are expressed in Xenopus embryos. Because the effects of FGFRL1 injection resemble those of XFD and could be reversed by FGFR1 injection, it is likely that FGFRL1 has overlapping ligand specificities with FGFR1 in the Xenopus embryos.
Conclusion
Taken together, our data suggest that FGFRL1 is a negative regulator of FGFR signaling. Whether it exerts its function as a soluble decoy receptor or in its membrane-bound state cannot be derived from our data. Because both forms are capable of ligand binding, it could very well be a combination of both. Judging from the developmental defects in the FGFRL1-deficient mice, it is likely needed for the fine-tuning of FGFR activity in tissues such as cartilage, the early kidney rudiment, bone, and the diaphragm muscle. Because the lethal phenotype of the FGFRL1-deficient mice severely hampers further research into the effects of FGFRL1, it would certainly be beneficial to generate conditional transgenic mice either overexpressing FGFRL1 or harboring tissue-specific deletions of this interesting receptor molecule.
This work was supported by Swiss National Science Foundation Grants 31003A0-113806, 31003A-127046, and 3100A0-101964; European Community Grant LSHG-CT-2004-005085; and funds from the Swiss Foundation for Research on Muscular Diseases.
T. Rieckmann and B. Trueb, unpublished data.
F. Steinberg and B. Trueb, unpublished observation.
- FGF
- fibroblast growth factor
- FGFR
- fibroblast growth factor receptor
- XFD
- truncated Xenopus FGFR1
- IL
- interleukin
- FBS
- fetal bovine serum
- BSA
- bovine serum albumin
- DMEM
- Dulbecco's modified Eagle's medium
- PBS
- phosphate-buffered saline.
REFERENCES
- 1.Beenken A., Mohammadi M. (2009) Nat. Rev. Drug Discov. 8, 235–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itoh N., Ornitz D. M. (2004) Trends Genet. 20, 563–569 [DOI] [PubMed] [Google Scholar]
- 3.Itoh N., Ornitz D. M. (2008) Dev. Dyn 237, 18–27 [DOI] [PubMed] [Google Scholar]
- 4.Wiedemann M., Trueb B. (2000) Genomics 69, 275–279 [DOI] [PubMed] [Google Scholar]
- 5.Sleeman M., Fraser J., McDonald M., Yuan S., White D., Grandison P., Kumble K., Watson J. D., Murison J. G. (2001) Gene 271, 171–182 [DOI] [PubMed] [Google Scholar]
- 6.Baertschi S., Zhuang L., Trueb B. (2007) FEBS J. 274, 6241–6253 [DOI] [PubMed] [Google Scholar]
- 7.Catela C., Bilbao-Cortes D., Slonimsky E., Kratsios P., Rosenthal N., Te Welscher P. (2009) Dis. Model. Mech 2, 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerber S. D., Steinberg F., Beyeler M., Villiger P., Trueb B. (2009) Dev. Biol. (in Press) [DOI] [PubMed] [Google Scholar]
- 9.Engbers H., van der Smagt J. J., van't Slot R., Vermeesch J. R., Hochstenbach R., Poot M. (2009) Eur. J. Hum. Genet. 17(1), 129–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergemann A. D., Cole F., Hirschhorn K. (2005) Trends Genet. 21, 188–195 [DOI] [PubMed] [Google Scholar]
- 11.Rieckmann T., Zhuang L., Flück C. E., Trueb B. (2009) Biochim. Biophys. Acta 1792, 112–121 [DOI] [PubMed] [Google Scholar]
- 12.Hayashi S., Itoh M., Taira S., Agata K., Taira M. (2004) Dev. Dyn 230, 700–707 [DOI] [PubMed] [Google Scholar]
- 13.Trueb B., Zhuang L., Taeschler S., Wiedemann M. (2003) J. Biol. Chem. 278, 33857–33865 [DOI] [PubMed] [Google Scholar]
- 14.Miller D. L., Ortega S., Bashayan O., Basch R., Basilico C. (2000) Mol. Cell. Biol. 20, 2260–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rieckmann T., Kotevic I., Trueb B. (2008) Exp. Cell Res. 314, 1071–1081 [DOI] [PubMed] [Google Scholar]
- 16.Brändli A. W., Kirschner M. W. (1995) Dev. Dyn 203, 119–140 [DOI] [PubMed] [Google Scholar]
- 17.Helbling P. M., Saulnier D. M., Robinson V., Christiansen J. H., Wilkinson D. G., Brändli A. W. (1999) Dev. Dyn. 216, 361–373 [DOI] [PubMed] [Google Scholar]
- 18.Amaya E., Musci T. J., Kirschner M. W. (1991) Cell 66, 257–270 [DOI] [PubMed] [Google Scholar]
- 19.Trueb B., Taeschler S. (2006) Int. J. Mol. Med. 17, 617–620 [PubMed] [Google Scholar]
- 20.Huovila A. P., Turner A. J., Pelto-Huikko M., Kärkkäinen I., Ortiz RM. (2005) Trends Biochem. Sci. 30, 413–422 [DOI] [PubMed] [Google Scholar]
- 21.Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M., Ornitz D. M. (2006) J. Biol. Chem. 281, 15694–15700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amaya E., Stein P. A., Musci T. J., Kirschner M. W. (1993) Development 118, 477–487 [DOI] [PubMed] [Google Scholar]
- 23.Leibbrandt A., Penninger J. M. (2008) Ann. N. Y. Acad. Sci. 1143, 123–150 [DOI] [PubMed] [Google Scholar]
- 24.Mantovani A., Locati M., Vecchi A., Sozzani S., Allavena P. (2001) Trend in Immunol. 22, 328–336 [DOI] [PubMed] [Google Scholar]
- 25.Mantovani A., Bonecchi R., Martinez F. O., Galliera E., Perrier P., Allavena P., Locati M. (2003) Int. Arch Allergy Immunol. 132, 109–115 [DOI] [PubMed] [Google Scholar]
- 26.Bogdan S., Klämbt C. (2001) Curr. Biol. 11, 292–295 [DOI] [PubMed] [Google Scholar]
- 27.Armand A. S., Laziz I., Chanoine C. (2006) Biochim. Biophys. Acta 1763, 773–778 [DOI] [PubMed] [Google Scholar]
- 28.Lagha M., Kormish J. D., Rocancourt D., Manceau M., Epstein J. A., Zaret K. S., Relaix F., Buckingham M. E. (2008) Genes Dev. 22, 1828–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Scheven G., Alvares L. E., Mootoosamy R. C., Dietrich S. (2006) Development 133, 2731–2745 [DOI] [PubMed] [Google Scholar]
- 30.Yu S., Zheng L., Trinh D. K., Asa S. L., Ezzat S. (2004) Lab. Invest. 84, 1571–1580 [DOI] [PubMed] [Google Scholar]
- 31.Ibrahimi O. A., Zhang F., Eliseenkova A. V., Itoh N., Linhardt R. J., Mohammadi M. (2004) Hum. Mol. Genet. 13, 2313–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohammadi M., Olsen S. K., Ibrahimi O. A. (2005) Cytokine Growth Factor Rev. 16, 107–137 [DOI] [PubMed] [Google Scholar]
- 33.Sugi Y., Ito N., Szebenyi G., Myers K., Fallon J. F., Mikawa T., Markwald R. R. (2003) Dev. Biol. 258, 252–263 [DOI] [PubMed] [Google Scholar]
- 34.Bushell K. M., Söllner C., Schuster-Boeckler B., Bateman A., Wright G. J. (2008) Genome Res. 18, 622–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grieshammer U., Cebrián C., Ilagan R., Meyers E., Herzlinger D., Martin G. R. (2005) Development 132, 3847–3857 [DOI] [PubMed] [Google Scholar]
- 36.Perantoni A. O., Timofeeva O., Naillat F., Richman C., Pajni-Underwood S., Wilson C., Vainio S., Dove L. F., Lewandoski M. (2005) Development 132, 3859–3871 [DOI] [PubMed] [Google Scholar]
- 37.Kuure S., Popsueva A., Jakobson M., Sainio K., Sariola H. J. (2007) Am. Soc. Nephrol. 18, 1130–1139 [DOI] [PubMed] [Google Scholar]
- 38.Hongo I., Kengaku M., Okamoto H. (1999) Dev. Biol. 216, 561–581 [DOI] [PubMed] [Google Scholar]
- 39.Kroll K. L., Amaya E. (1996) Development 122, 3173–3183 [DOI] [PubMed] [Google Scholar]