

Abstract
This work investigates the direct-type action of radiation (involving electron addition and electron abstraction) on DNA. Specifically, the effects of DNA hydration, conformation and packing on free radical yields are examined. The fact that these variables are interdependent complicates the analysis of how each variable affects free radical yields. The hydration dependence of free radical yields in films of both Li and Na DNA was examined. At low levels of DNA hydration (less than 25 waters per nucleotide), the relatively high free radical yields and the lack of water-derived radicals are evidence that damage transfer from the DNA solvation shell to the DNA molecule occurs. The scatter of measured free radical yields is significant (50–70%) in Li DNA, while in Na DNA it is much less (<25%). Our conclusions hinge upon two known differences between Li DNA and Na DNA: (1) At low DNA hydrations, the conformation of Na DNA undergoes several changes with increasing hydration, while the conformation of Li DNA is relatively constant over the same range. (2) Compared to Na DNA, Li DNA is more prone to self-associate, giving rise to macroscopic and microscopic crystalline domains in Li DNA films. The greater scatter of free radical yields in Li DNA films is therefore attributed to variability in packing. By virtue of the greater reproducibility of free radical yields in Na DNA films, the effects of DNA packing, conformation and hydration can be ascertained. In Na DNA, hydration-dependent changes in free radical yields are attributed primarily to changes in DNA packing.
INTRODUCTION
A track of ionizing radiation, intersecting hydrated DNA, deposits its energy in both DNA and its surrounding water. The resulting DNA damage is traditionally grouped into two categories: “direct-effect” damage, due to direct ionization of DNA, and “indirect-effect” damage, due to attack of DNA by radicals (mainly solvated electrons and hydroxyl radicals) that are produced from ionizations in the surrounding medium. DNA damage can also arise from the transfer of holes and dry (unsolvated) electrons from the DNA solvation shell to the DNA molecule. The products from these unsolvated electrons and holes are indistinguishable from direct-effect damage products. Hence damage from the direct ionization of the DNA and damage from electron and hole transfer processes are collectively referred to as “direct-type” damage (1). In other words, direct-type damage describes the attachment of unsolvated electrons and holes to the DNA target, regardless of the origin of these electrons and holes.
DNA hydration has been shown to influence the yields of direct-type damage generated in DNA exposed to ionizing radiation (2–11). The reported evidence supports the damage transfer hypothesis: Electrons and holes generated in the DNA solvation shell transfer to DNA, augmenting direct-type damage to DNA. As a result, DNA and its surrounding solvation waters [~22 waters per nucleotide (7, 12–14)] comprise a discrete target mass for the formation of direct-type damage. The target mass may be different for electrons and holes; i.e., the boundary beyond which damage transfer no longer occurs, and beyond which water radicals are generated, may be different for electrons and holes. In addition to its role in damage transfer, the hydration layer surrounding DNA influences the course of the reactions and stability of the radicals trapped on DNA. Water outside the solvation layer (bulk water) forms crystalline ice upon freezing. DNA hydration is often expressed in terms of the amount of water per nucleotide; this is abbreviated as Γ and has units of moles of water per mole of nucleotide.
The importance of hydration in the radiosensitivity of DNA has long been recognized (15–17). An understanding of the role of hydration in direct-type damage is necessary for an accurate quantification of the DNA target mass, which in turn is essential to predicting DNA damage in vivo. Determination of the DNA target mass is particularly relevant because the concentration of DNA in the cell nucleus is high (18). From studies on crystallized nucleosome core particles, we estimate the in vivo DNA hydration to be of the order of 60 waters per nucleotide (19, 20).
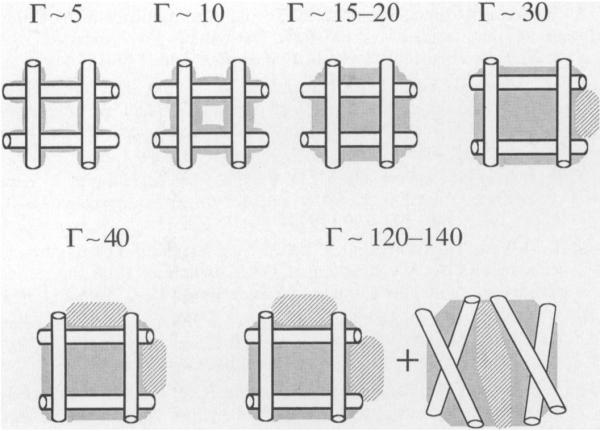
DNA packing can also influence the quantitative damage to irradiated DNA (1, 21, 22). Microdosimetry considerations are important when considering the relatively small volume occupied by the densely packed DNA in the nucleus of a cell. Local packing order, however, is equally important, and can readily be investigated with DNA model systems. We define DNA packing as the molecular arrangement of molecules within a DNA aggregate. DNA packing can influence the extent of combination reactions that precede electron and hole trapping and thus can influence the free radical yields. For example, a packing order that promotes the separation of radicals on separate DNA strands would hinder combination and thus enhance free radical yields. In contrast, the localization of multiple radicals to the same DNA strand would decrease free radical yields by virtue of the more extensive combination along the DNA helix. Figure 1 gives an illustration of this phenomenon; the probability that multiple radicals will localize to the same DNA duplex is greater for loosely aggregated DNA (as depicted in Fig. 1a) than for tightly packed DNA (as depicted in Fig. 1b).
FIG. 1.
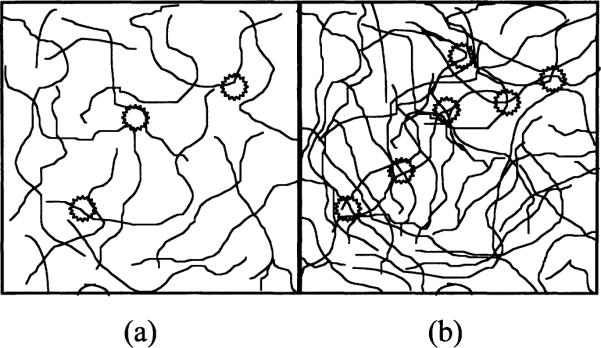
Loose compared to compact DNA packing. The thin lines represent double-stranded DNA; the stars represent clusters of damage, deposited by ionizing radiation. The number of damage clusters is dependent upon the amount of DNA present. The number of DNA radicals formed per cluster (not the number of clusters) determines the free radical yield. DNA acts as both an electron and a hole scavenger. The clusters of damage in the more loosely packed DNA system (panel a) are in the vicinity of fewer DNA molecules than that of the more densely packed DNA (panel b). Consequently, in the latter case, radicals will tend to localize to relatively more DNA molecules. The separation of radicals on different DNA double strands would tend to hinder combination reactions (and thus promote radical trapping) compared to the localization of radicals to the same DNA double strand.
DNA packing also influences free radical yields through its effect on DNA hydration; i.e., coordination of one DNA molecule with another can alter the pattern and structure (23) of solvation. Tight DNA packing can hinder the binding of some outer-shell solvation waters; alternatively, loosely packed DNA is more readily hydrated.
In vivo, DNA is normally tightly compacted into chromatin via its association with histone and nonhistone proteins (18). These proteins function, in part, to prevent DNA-DNA clustering (24); additionally, they confer radio-protection (25). Chromatin compaction is released in transcriptionally active DNA; additionally, the packing order of chromatin undergoes changes during cell replication and cell development. Previous work has shown that “open” chromatin structure is more radiosensitive than “condensed” chromatin (25–27). The relative radiosensitivity of polyamine-induced compacted DNA supports this hypothesis (28–30). In the condensed state, DNA maintains its closely associated waters (31), but the overall extent of waters surrounding DNA is reduced. As a result, indirect-type damage, arising from HO., H. and eaq− attack on DNA, is diminished (25). Direct-type damage transfer processes may be affected similarly.
The damage transfer mechanism in hydrated DNA can be examined by measuring free radical yields in irradiated DNA (4–7, 9). If complete transfer occurs from water to DNA, the free radical yield in aggregate DNA (DNA and its surrounding water) should be independent of Γ. In other words, an incremental increase in the aggregate DNA mass, due to added solvation water, should be associated with a proportional increase in the amount of direct-type damage to DNA. This assertion assumes that free radical combination reactions occur to the same extent in both the DNA and DNA solvation shell.
Because DNA scavenges the precursors of HO. and H. (holes and electrons, respectively), the damage transfer hypothesis predicts that HO. and H. (and eaq) will not be formed in the solvation shell (2, 4, 5, 7). The yield of these species is low in ice, since combination reactions predominate [attributable to greater electron and hole mobility in the ice phase (14, 32, 33)]. Hence the free radical yield in frozen aggregate DNA decreases as the hydration is extended beyond the boundaries for electron and hole transfer. Identification of these boundaries is difficult in samples where the distribution of hydration water lacks uniformity.
Sevilla and coworkers have shown that HO. is not detected in relatively dry DNA (Γ < 8), but is detected when Γ > 8, suggesting that all holes do not transfer to DNA in this regime of hydration (8 > Γ > 22) (8, 9). This inference is consistent with the observation that diamagnetic DNA base products, originating from HO. attack, are detected in DNA samples with Γ ≥ 13.2 (11).
Measurements of free radical yields in variably hydrated DNA have been shown to be consistent with the damage transfer hypothesis (4, 7). This agreement is particularly evident at higher hydrations (Γ > 22), where the free radical yield decreases with increasing hydration. However, the data at lower hydrations (Γ < 22) are inconclusive. Our group observed significant scatter, which we attributed to a variability in the conformation and packing of DNA (4). Sevilla and coworkers reported an increase in free radical yield with increasing water content (7); Hüttermann and coworkers observed a similar relationship (3). This dependence of free radical yield on hydration at low humidities may be attributable to hydration-dependent changes in DNA conformation. Indeed, Swarts et al. conclude that conformational changes play a significant role in the yields of various DNA base damage products (10, 11). Another possibility is that the DNA solvation shell waters stabilize free radical damage on the DNA molecule (6); this radical stabilization effect reflects the packing order of the DNA aggregate (22).
Our group has recently shown that, in variably hydrated RNA polymer powders, the free radical yields are less variable compared to those of calf thymus DNA powders (5). Conceivably, this effect arises from the increased uniformity of RNA polymer conformation (34) and packing. As expected from the damage transfer hypothesis, the free radical yield in the poly(A)·poly(U) duplex is independent of solvation shell hydration. In contrast, the free radical yield in poly(U) decreases as Γ increases; this dependence on hydration is attributed to a hydration-dependent variability in base stacking.
With samples of RNA polymer films, the scatter in free radical yield is reduced further and, in all three polymer samples studied [poly(A)·poly(U), poly(U), poly(A)], the free radical yield is relatively independent of solvation shell hydration (5). In RNA polymer films, where the translucent, glassy appearance suggests a homogeneous arrangement, it is believed that the packing is more uniform than that of opaque powders.
In the present study, DNA is examined in the form of films because, at least on a macroscopic scale, the films retain uniformity for 2 < Γ < 20. We provide evidence that films of Na DNA give relatively small scatter in free radical yields for Γ < 20. In sharp contrast, films of Li DNA give relatively large scatter in free radical yields. With changes in hydration, Li DNA maintains a relatively homogeneous conformation, whereas Na DNA undergoes conformational changes. Thus the free radical yields in frozen DNA are more sensitive to changes in packing than to changes in the relative mass of water or changes in DNA conformation.
MATERIALS AND METHODS
Film Preparation
Various methods of DNA film preparation have been reported; they include: “wet-spinning” DNA fibers, in a parallel arrangement, around a spindle (35); stroking DNA gels (36) or viscous DNA solutions (13, 37, 38) with a spatula; oven-drying a DNA gel (39); and allowing a sample of polymer powder, rolled onto a plate, to humidify (5). Our novel approach, involving the slow evaporation of a dilute DNA solution, offers several advantages: It is much easier to implement than wet spinning; it does not require a custom-designed wet-spinning apparatus; and, unlike earlier approaches, it is useful for growing large, visibly uniform films.
DNA films were prepared from calf thymus Na DNA (Sigma Chemical Co.). The DNA was dissolved in an XC1 solution, where X represents the Na or Li cation. For Li DNA, the solution was dialyzed against a LiCl solution.
The DNA was then ethanol-precipitated, redissolved in water [A260 ~ 12–15 OD units, corresponding to ~0.60–0.80 mg/ml (40, 41)] and filtered. For Li DNA films, the solution was subsequently sonicated to break the DNA into smaller fragments. The careful on–off sonication procedure breaks the lengthy DNA strands into shorter (200 to 5000 base pairs) double-stranded pieces while minimizing the amount of single-strandedness produced (42). The excess salt in solution (of the order of 0.05–0.1 M) also promoted double-strandedness (34). The UV absorbance of the DNA solution did not change with sonication, or by sonication at ~0°C, confirming that the DNA solution is primarily double-stranded. The DNA solution was then evaporated in a petri dish until a clear, uniform film of DNA was formed.
The film was twice dialyzed in an XC1, 80:20 ethanol:water solution to control the salt content (43). The film was cut into thin strips and put into thin-walled Suprasil quartz tubes. The DNA samples were placed in a chamber, equilibrated against a saturated NaOH solution (5% relative humidity) for 3 days, and then weighed. Three of these samples were subsequently vacuum-dried (<0.02 Torr) to determine the amount of removable water in the 5% relative humidity DNA films (4). Two methods were used to humidify the DNA samples: humidifying 5% relative humidity DNA, and dehumidifying highly hydrated DNA.
For the humidified DNA samples, the 5% relative humidity samples were placed into humidity chambers (with known relative humidities) for a duration greater than 4 weeks. During this time, the DNA films became hydrated to a steady state. Γ was calculated from the measured mass of these samples.
For the dehumidified DNA samples, water was added directly to the DNA films to raise Γ to ~120 to 150 waters per nucleotide. The samples were sealed and allowed to equilibrate for 3 days. They were then unsealed and placed into chambers, held at selected relative humidities, to remove water. The extent of dehydration was controlled by the relative humidity and the duration of drying. Samples were not allowed to equilibrate in the humidity chambers; rather they were removed when the desired hydration, calculated from the mass of the samples, was reached. The sample tubes were then sealed and allowed to equilibrate for >1 week.
A sample of the Li DNA film was redissolved in water (~0.13 mg/ml), and the UV absorbance as a function of temperature was measured. The hyperchromicity (35% increase in UV absorption, at 260 nm, upon melting) indicated that the film comprised primarily double-stranded DNA.
In the calculation of Γ, two assumptions were made: (1) 2.5 mol of water per nucleotide could not be removed by vacuum drying (10, 11), and (2) roughly 6% of the dry weight of DNA was attributable to excess salt (43).
Sample Cooling and Irradiation
Samples were cooled to low temperatures (ranging from 4 to 293 K) in a Janis cryostat (44). While in the cryostat, the sample was irradiated with X rays generated by a Varian/EimaOc EG-76H X-ray tube. The X-ray tube was run at a voltage of 70 keV and a current of 20 mA. The low-energy X rays were attenuated by beryllium windows in both the X-ray tube and Janis Dewar and by a 0.001” aluminum filter on the X-raytube.
Calculation of Dose Rate
The dose rate in the Janis Dewar was determined from free radical dose-response curves in L-alanine pellets as measured by EPR (45, 46). Cylindrical pellets were made from a 9:1 (mass:mass) mechanical mixture of alanine:paraffin. The dimensions of the pellets, diameter 1.0 mm and length 3 mm, allowed them to be placed in the same quartz tubes used for DNA samples and to be positioned at the same location normally occupied by the DNA samples. The concentration of radicals, relative to an EPR standard, is measured as a function of exposure time. This function is linear up to 160 kGy (45), well beyond the doses employed here. The free radical yield for the alanine pellets was calibrated using a 60Co-γ-ray source of known dose rate (4). Because there is a slight realignment of the beryllium windows as the temperature of the Janis Dewar is changed from room temperature to 77 K, dosimetry was also done at lower temperatures. Irradiation was carried out at 77 K in both the Janis Dewar and the 60Co source. It should be noted that the free radical yield of alanine at 77 K is approximately 2.1 times less than the free radical yield at 293 K and that dose saturation begins to occur at a dose of ~25 kGy.
The calculated dose rate of the X-ray tube, for a sample loaded into a Suprasil quartz tube and positioned in the Janis Dewar, is 24 ± 3 kGy/h.
EPR Measurements
The EPR measurements were taken with a Varian E-12 Q-band spectrometer. The waveguide and sample cavity were incorporated into a Janis cryostat with low-temperature capability (4 K and above). A ruby chip standard (from the National Institute of Standards and Technology), with a known number of unpaired electrons, was mounted along the side of the sample cavity. For each sample spectrum, a ruby spectrum was taken under the same conditions; these spectra do not overlap. The concentration of free radicals in the sample was determined by comparing the double integral of the EPR spectrum of the sample with that of the ruby. Baseline subtractions were performed prior to each integration.
The calibration of the ruby standard is checked periodically. In doing so, we have recognized that in some of our work reported earlier (1, 4, 5, 47) the effective number of spins (6 × 1014) was subject to a measurement error due to partial power saturation of the primary standard. Several other standards, including TEMPO derivatives, CuSO4 and irradiated alanine, were used to determine the effective number of spins in the ruby chip. These standards were calibrated against a National Institute of Standards and Technology ruby bar, and against each other, at room temperature using an X-band Bruker EPR spectrometer. The work reported here employs the corrected value of 3.1 × 1014 ± 25% effective spins at the center sample position. Our previous yields (1, 4, 5, 47) should therefore be multiplied by a correction factor of 0.5 to obtain the absolute yields. It should be stressed that the relative values of free radical yield are significantly more accurate than the absolute values, estimated at ±10–15% compared to ±25–50%, respectively.
The concentration of trapped free radicals increased linearly with absorbed radiation dose. The slope of this linear dependence is directly related to the number of trapped free radicals generated per unit of absorbed energy. The absorbing mass in the calculation of free radical yield was the aggregate of DNA and all surrounding water (hydration water and bulk water).
RESULTS AND DISCUSSION
Free Radical Yields in Li DNA
The hydration dependence of the free radical yields in Li DNA films is shown in Fig. 2. Although the experimental data are consistent with the damage transfer hypothesis, they exhibit significant scatter, of the order of 50 to 70%. Similar scatter was observed in powder samples of calf thymus Na DNA (4). In these samples, the scatter was ascribed to variability in DNA packing and conformation. Our experiments are particularly sensitive to sample variability since they are 5 to 10 times smaller than the samples used by others. By the reasoning outlined below, the observed scatter in the Li DNA data is considered as evidence that this variability is due mainly to changes in DNA packing rather than conformation.
FIG. 2.
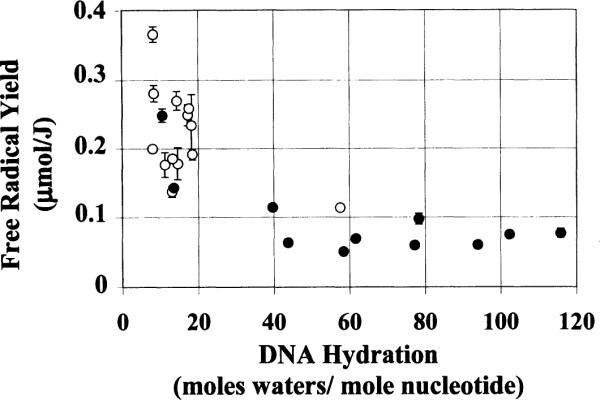
Yield of radicals in Li DNA as a function of DNA hydration. Each data point represents one dose-response curve determined from four to five data points. The error bars are ± 1 standard deviation (SD) of the slope of the dose-response curve. Open circles represent humidified samples and filled circles represent dehumidified samples.
DNA conformation is known to be influenced by a number of factors, including DNA hydration (48, 49), DNA packing interactions (50), salt concentration and the DNA counterion (e.g. Na+ compared to Li+). Na DNA, for example, undergoes several conformational changes as a function of relative humidity: denatured DNA (very low relative humidity) ↔ B-like form (below 45% relative humidity) ↔ A form (between 45% and 92% relative humidity) ↔ B form (above 92% relative humidity) (12). The A form differs significantly from the B form in structure (see Tables 1 and 2). Li DNA, on the other hand, undergoes a change in conformation (C ↔ B) at a Γ of ~10 to 12 and no changes with further increases in Γ. Important here is the fact that the B and C conformations are structurally similar (see Tables 1 and 2) (34). Hence the Li DNA structure remains relatively constant over the full range of hydration.
TABLE 1.
Structural Characteristics of Different DNA Conformation Forms
| Per nucleotide |
Groove width |
Groove depth |
||||||
|---|---|---|---|---|---|---|---|---|
| Conformation type | Pitch (Å) | Helical symmetry | Axial rise (Å) | Turn angle (degrees) | Minor (Å) | Major (Å) | Minor (Å) | Major (Å) |
| A | 28.2 | 11 | 2.56 | 32.7 | 11.0 | 2.7 | 2.8 | 13.5 |
| B | 33.8 | 10 | 3.38 | 36.0 | 5.7 | 11.7 | 7.5 | 8.5 |
| C | 31.0 | 9.33(3) | 3.32 | 38.6 | 4.8 | 10.5 | 7.9 | 7.5 |
Notes. The A conformation possesses structural characteristics that are significantly different than those of the B and C conformations. The A conformation also exhibits unique base-stacking characteristics. The bases are tilted (~20Å) with respect to the helical axis. Along with a smaller turn angle, the large base tilt causes interstrand base overlap. In contrast, the bases in the B and C conformations are roughly parallel (~ − 6° tilt) to the helical axis and thus undergo only intrastand stacking. The most striking difference between the A conformation and the B and C conformations is the sugar puckering mode (for A DNA it is C3′endo, while for B and C it is C3′−exo or a similar variant). Sugar puckering describes the conformation of the ribose sugar group and affects the macroscopic helical arrangement. The groove dimensions are affected primarily by the dislocation of the DNA base pairs with respect to the helical axis. Table and text are derived from ref. (34).
TABLE 2.
Hydration-Dependent Changes in Conformation: Na+ Compared to Li+ DNA
 |
Notes. Both Na DNA and Li DNA undergo conformational changes with hydration. However, the Na DNA changes are more significant due to the large differences between A and B conformations of DNA (see Table 1).
aRelative humidity.
The scatter in free radical yields from films of Li DNA therefore is not attributable to variability in DNA conformation. By deduction, DNA packing is the critical variable. Packing-mediated variability in yields of DNA damage products has also been reported in studies of strand breaks (30).
Li DNA characteristically organizes into crystalline arrangements. In wet-spun fibers, this crystalline order is highly uniform (39, 49). The technique of wet-spinning DNA promotes DNA crystallinity through the packing of long strands of DNA parallel to one another. In contrast, film formation by slow evaporation of high-molecular-weight DNA disrupts the uniform crystalline order (49). In our samples, sonication was used to break the DNA into short fragments (roughly in the range of 200 to 5000 base pairs in length). Short DNA fragments, by virtue of their intrinsic rigidity (24), readily form crystalline domains (42, 51–53). Hence it is likely that our Li DNA films are heterogeneously ordered, possessing both crystalline (39) and amorphous domains. We propose that packing heterogeneity is the main source of scatter in the Li DNA data.
Free Radical Yields in Na DNA
The hydration dependence of free radical yield in Na DNA films is depicted in Fig. 3. The reduction in scatter, relative to that in Li DNA, is readily apparent. This reduction in scatter can be explained by a more homogeneous DNA packing order in the Na DNA films compared to that in Li DNA films. Packing homogeneity in DNA films is expected from previous studies on films (5).
FIG. 3.
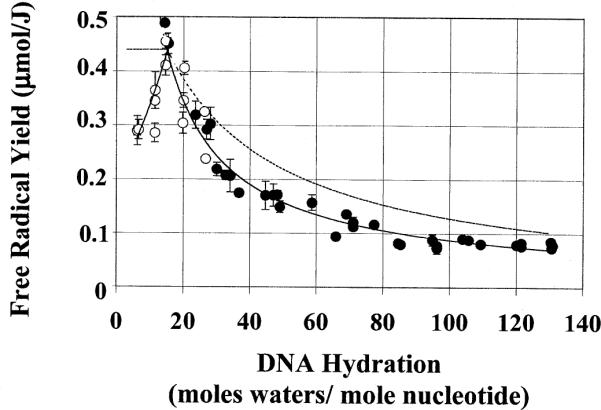
Yield of radicals in Na DNA as a function of DNA hydration. Each data point represents one dose-response curve determined from four to five data points. The error bars are ± 1 SD of the slope of the dose-response curve. The solid line represents a power regression of the experimental data performed independently for Γ ≤ 15 and for Γ ≥ 15. For a given hydration of DNA, the relative amounts of water and DNA can readily be calculated. In this work, a 6% dry mass is assigned to excess NaCl. Assuming a target mass free radical yield and target mass hydration, the theoretical hydration dependence of the free radical yields in DNA can be modeled. The dashed line represents the theoretical curve, assuming a target mass hydration of 15 and a target mass yield of 0.46 μmol/J. The solid lines represent power regression of the data performed independently for Γ ≤ 15 and Γ ≥ 15; their only purpose is to help delineate the trend in the data. Open and filled circles are as defined in the legend to Fig. 2.
Films prepared from wet-spun Na DNA fibers are less uniformly crystalline than similarly prepared Li DNA films. In addition, Na DNA films, initially prepared from wet-spun fibers and subsequently cycled through a dilute state, possess no preferred orientation (49). This indicates that Na DNA films, prepared by evaporation of a dilute DNA solution (as our films are prepared), are disordered. These films possess no macroscopic crystallinity. Microcrystalline domains may be present; however, these would constitute only a small fraction of the Na DNA sample (49).
The packing order in Na DNA film samples is therefore expected to be relatively homogeneous for a given DNA hydration. In contrast, the packing in Li DNA films varies between different samples (see above) even at the same Γ. We therefore propose that the differences in packing and packing heterogeneity between the Li DNA films and the Na DNA films account for differences in the reproducibility of the data for free radical yield.
Hydrogen Atom Yields
Dry electrons react with H2O and H30+, giving rise to hydrogen atoms (H.). The damage transfer hypothesis predicts that H. will not be formed in relatively dry DNA. Rather electrons will rapidly transfer to DNA. As the degree of hydration is increased, beyond the boundary for electron transfer, the signal of trapped H. is expected to grow in. The trapped H. yield is a measure of the amount of crystalline ice present in the sample, though frozen glassy regions may also yield H.. Trapped H. possesses a distinguishing EPR spectrum, with a large hyperfine splitting of 50.5 mT.
The hydration dependence of the yields in both Na and Li DNA is shown in Fig. 4. While the yields of H. arise from the same samples as the DNA radical yields, unlike the scatter in the DNA radical yields, the scatter in yields of H. does not differ significantly between the Li DNA and Na DNA samples. This observation provides additional evidence that the scatter in the DNA yields is intrinsic to the DNA aggregatea nd not due to unrecognized measurement errors.
FIG. 4.
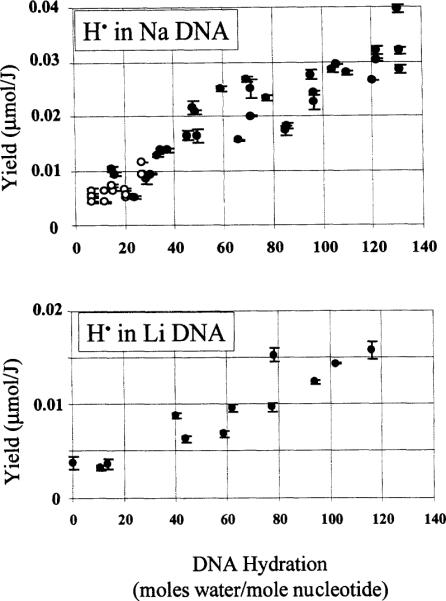
Yield of hydrogen atoms in Na and Li DNA as a function of DNA hydration. Each data point represents one dose-response curve determined from four or five data points. The error bars are ± 1 SD of the slope of the dose-response curve. The yields were obtained by integrating only the low-field line and multiplying by 2. Hydrogen-atom yields reflect hydrogen atoms in the quartz sample holder and in the DNA sample. Open and closed circles are as defined in the legend to Fig. 2.
The yields of H. should provide an estimate of the average boundary for electron transfer. The DNA hydration, above which H. begins to grow in, is the hydration at which crystalline ice begins to form, and damage transfer begins to be limited. An accurate calculation of this threshold is compromised to some degree by a background of H. trapped in the quartz sample holder. This background H. signal is present in all of our EPR spectra. The quartz H. yield is independent of Γ (N. Mroczka and W. A. Berhard, unpublished data).
In Na DNA, the H. yield is consistently above the quartz H. baseline at hydrations of >24 waters per nucleotide. Although Γ = 24 cannot be given as a precise boundary, the data indicate that this boundary occurs at a DNA hydration greater than 20 waters per nucleotide. The two relatively large H. yields at Γ ~ 15 arise from samples dehumidified from a relatively dilute state (Γ > 120) and thus may represent a different packing order. In Li DNA, the threshold is less accurately determined, due to fewer data in the 15 ≤ Γ ≤ 35 hydration regime. The threshold for Li DNA is estimated to be ~ 20 waters per nucleotide.
Positive Slope of the Curve for Free Radical Yield as a Function of Γ (Γ < 15)
With the reduced scatter in the Na DNA film data (Fig. 3), the relationship between free radical yield and DNA hydration is more readily discerned. It is apparent that, at low Γ, the free radical yield increases with increasing hydration. As Γ increases from ~5 to 15, the free radical yield doubles, reaching a maximum at Γ = 15; further increases in hydration are accompanied by a decreasei n free radical yield. A similar dependence of free radical yield upon hydration was observed by Sevilla and coworkers (6).
The increasing free radical yield with increasing hydration at Γ < 15 indicates that not only does the tightly bound water transfer radicals to the DNA, but it must also enhance radical trapping by the DNA. In this same region of hydration, there are qualitative changes in the EPR spectrum which support this notion; namely, the EPR doublet associated with the reduced pyrimidines becomes better resolved (3, 4). Deconvolution of the DNA EPR spectra indicates that, with increasing DNA hydration, the relative amount of reduced cytosine increases while the relative amount of reduced adenine decreases, disappearing at higher hydrations (Γ > 14) (7).
The hydration-dependent effects on DNA free radical yields [both in our data and in ref. (6)] may be due to conformational changes in the DNA molecule as proposed by Swarts et al. (10, 11). As discussed above, Na DNA undergoes a conformational change at low Γ. While it is plausible that different conformational states of DNA (e.g. B-like or A) possess different susceptibilities to direct-type damage, we suggest that the more important impact of hydration is due to changes in molecular packing.
The aqueous shell of DNA stabilizes radical damage on the DNA bases (6), allowing for increased yields as well as different product distributions. We suggest that this increase in stability is derived from an increased dielectric response to altered charge states. The dielectric response entails a repolarization of the medium and, more importantly, proton transfer. The expanded hydrogen-bonding network can facilitate proton shuttling interactions. Proton shuttling provides a means of stabilizing certain reduced and oxidized species (1, 21). Thus, as additional waters are added to the solvation shell, proton transfer and other forms of dielectric relaxation increase the probability of radical trapping by decreasing the probability of radical combination.
It is interesting to note that a similar increase in free radical yield, at low levels of hydration, is not observed in poly(A)·poly(U) powders or films. In these samples, the free radical yield remains independent of solvation shell hydration (5). One explanation is that the conformation of RNA polymers is relatively constant (A or the analogous A′ form) (34). Another possibility is that the adenine and uracil radicals are not as readily protonated and/or deprotonated as the cytosine and guanine radicals [the more prominent radical species in DNA (54–56)] and therefore are not as susceptible to changes in DNA hydration and packing. This proposal is consistent with the relative pKa's of the DNA radical species (57). Finally, RNA packing may be more flexible than DNA packing in responding to changes in hydration at low levels of hydration. Currently there is insufficient information to distinguish the relative importance of these different mechanisms.
Given the dramatic changes in free radical yield at low Γ (a factor of 2 between Γ < 5 and Γ > 20), it is apparent that heterogeneity in DNA conformation and/or DNA hydration could give rise to variable yields in this hydration regime. Indeed, at low Γ, samples could consist of DNA in various conformational states and in variable hydration states. Thus packing variability, coupled with conformational variability, could account for the scatter observed in earlier experiments (4). The reproducibility of the data in the present study suggests that Na DNA films possess relatively uniform hydration and conformation (at a given hydration state) in the Γ < 15 region. This system therefore provides evidence that filling in the primary solvation shell has two main consequences; it is a source of damage transfer, and it increases radical trapping (most likely by enhancing proton transfer).
Peak in the Curve for Free Radical Yield as a Function of Γ and the Negative Slope at Γ > 15
The curve for free radical yield as a function of Γ peaks at Γ ~ 15 waters per nucleotide (Fig. 3). With increasing hydration beyond Γ ~ 15, the free radical yield drops precipitously. A similar drop at Γ ~ 20 was observed by Wang et al. (6). They attributed this decline in free radical yield to the “freezing back” of DNA solvation shell waters. At DNA hydrations beyond that which gives the maximal yield, the hexagonal ice lattice of the bulk water purportedly encompasses some of the pre-existing solvation shell waters, effectively reducing the DNA-water target mass. Hydration-dependent changes in the hydrogen-bonding network of DNA as the outer hydration shells are filled may also contribute to the observed hydration dependence of free radical yields.
The hydrationl evel at which we observe a maximal free radical yield (Γ ~ 15) is smaller than that observed by Sevilla and coworkers (Γ ~ 20). This discrepancy could be due to differences in experimental conditions, e.g. freezing rate and working temperature (4 K compared to 77 K). Another possibility is that differences in sample preparation resulted in significant differences in DNA packing. Sevilla and coworkers studied lyophilized DNA “fluff balls”. The degree of heterogeneity is also important in analyzing data for free radical yield as a function of Γ. In our preparation of DNA fluff balls, we observed macroscopic packing heterogeneity, specifically, fibrous as well as film-like regions. Packing heterogeneity can influence DNA hydration. The measured value of DNA hydration is a global value, averaged over an ensemble of local values. The global (average) hydration will not necessarily reflect the mean hydration. In general, increased packing heterogeneity is expected to result in increased dispersion in the extent of local DNA hydration.
The observed peak can also be explained by hydration-dependent changes in DNA packing. The relative importance of this packing becomes apparent when one attempts to use the data of Fig. 3 to characterizeth e target mass.
Target Mass Γ and Target Mass Free Radical Yields
The target mass of the DNA aggregate comprises the DNA molecule as well as the waters that are involved in damage transfer to the DNA. The peak in the curve for free radical yield as a function of Γ suggests that, at a Γ of ~15, most or all of the waters are involved in damage transfer. It is therefore of interest to explore whether these waters remain associated with the target mass throughout the hydration regime of 15 < Γ < 140. To test this, we modeled the expected free radical yields using the following criteria: (1) DNA plus 15 hydration waters constitute the target mass; (2) the free radical yield of the target mass equals the free radical yield at Γ ~ 15 (0.46 μmol/J); and (3) additional waters, beyond Γ ~ 15, do not contribute to the target mass free radical yield. Using these criteria, the dashed curve in Fig. 3 was generated.
One aspect of the third criterion requires that the yield of HO. in ice be negligible and that the overlap of the HO. spectrum with that of DNA be small (the spectrum of H. does not overlap that of DNA radicals at all). Figure 5 shows the 4 K Q-band EPR spectra of HO. derived from crystalline ice and from 6 M CsF glass. For determination of free radical yield, the DNA spectrum (4, 58) is normally integrated over a field width of ~16 mT, while the HO. spectrum encompasses >90 mT. To determine the relative contribution of HO. to the DNA spectrum, the HO. was integrated over 100 mT and 16 mT (with appropriate baseline subtractions). In both the CsF glass and crystalline ice spectra, the central 16-mT region constitutes ~6% of the total intensity. With an estimated (from H. yields) yield of HO. of ~0.05 μmol/J, the yield of HO. that contributes to the DNA spectrum is therefore of the order of 0.003 μmol/J. This amount of HO. signal intensity does not affect our analyses significantly.
FIG. 5.

Q-band EPR spectra of HO. derived from crystalline ice and from 6 M CsF glass. In both spectra, the H. lines are apparent (arrows). For the quantitative analysis of HO., the H. lines were removed, and an approximated HO. signal in the region was added (the smooth noise-free curve that overlays each of the H. lines). The exact spectral characteristics in this region are unknown, but they do not significantly affect the quantitative analysis. The double-sided arrow denotes the 16-mT region employed for integration of the DNA signal.
The modeled yields shown by the dashed line in Fig. 3 are significantly higher than the observed free radical yields for Γ > ~20–25. A freezing back of DNA hydration waters would be consistent with the smaller than expected free radical yields. It is therefore of interest to calculate how many waters appear to remain in the DNA target mass. To calculate the apparent target mass hydration, a target mass free radical yield was assumed (as above, free radical yield = 0.46 μmol/J), while the sample free radical yield was derived from a power regression of the experimentally observed free radical yields. This model predicts that the number of hydration waters in the target mass decreases progressively with increasing global hydration, approaching an asymptote of roughly 4 waters per nucleotide. It is unrealistic, however, that the bulk-ice phase would penetrate so deeply into the tightly bound DNA solvation shell waters. Although freeze-back of target mass waters may play a role, it appears that other factors are involved. That at least one of the above criteria is incorrect can be seen by testing various combinations of target mass and target mass free radical yields.
Figure 6 compares the experimental data with several models for the hydration dependence of free radical yields. Each model assumes a different combination of target mass hydration [Γ(TM)] and target mass free radical yield [free radical yield(TM)], and otherwise follows the above three criteria. At higher levels of hydration (Γ > 50), smaller values of free radical yield(TM) (compared to those at lower levels of hydration, Γ < 15) are needed to best fit the data. In general, the same curve can be obtained from an infinite set of values for free radical yield(TM) and Γ(TM); for greater values of free radical yield(TM), Γ(TM) is less. Choosing a reasonable value for Γ(TM) serves as a constraint. Without additional constraints, however, we cannot determine unique values of free radical yield(TM) and Γ(TM). Clearly, none of these models fit our data throughout the entire hydration regime. This suggests that either the target mass hydration or target mass yield must decrease with increasing hydration. For DNA of Γ > 50, if one assumes 24 ≥ Γ(TM) ≥ 16, then free radical yield(TM) ranges from 0.25 to 0.31 μmol/J. For fully hydrated DNA (Γ ~ 15–20), if one assumes Γ(TM) ~ 16, then free radical yield(TM) is 0.46 μmol/J. The variations in free radical yield(TM) and Γ(TM) are, by our model, primarily a consequence of changes in packing.
FIG. 6.
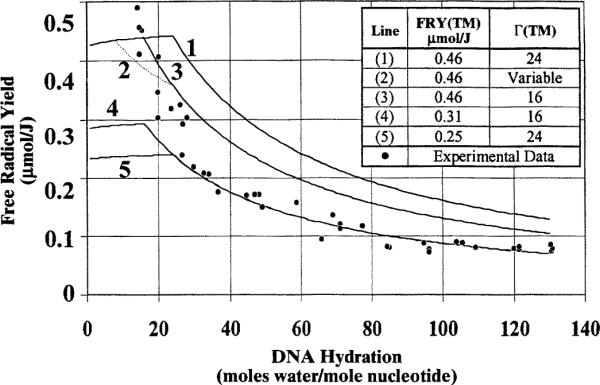
Theoretical models of free radical yield as a function of DNA hydration. Theoretical models are shown in conjunction with the experimental data for Γ ≥ 15 and the corresponding fit to the data. The theoretical free radical yields are calculated in a similar fashion to that described in the legend to Fig. 3. The target mass hydration [Γ(TM)] and target mass free radical yield [FRY(TM)] must be assumed; these are shown on the figure. Models in which the target mass hydration remains uniform are shown as solid lines. A model in which the boundary for electron transfer occurs at Γ = 8, and the boundary for hole transfer that occurs at Γ = 24 is also shown (dashed line, curve 2). At γ > 24, these boundaries are equal to a target mass of 16 waters per nucleotide. The assumed target mass free radical yield is listed. The slight increase in free radical yield, evident in all curves from Γ = 0 to the onset of the target mass boundary, results from the assumption of a 6% dry weight due to excess NaCl. As hydration is increased, the excess salt constitutes a relatively smaller percentage of the total mass. The lower set of curves (4 and 5) best fit the experimental data for hydrations greater than 30 waters per nucleotide. The higher set of curves (numbered 1 to 3) reflect the greater yields at low levels of hydrations. With the simple weighted sum model, no one curve fits all of the experimental data well.
The concept of target mass is complicated further by the possibility that the boundary for electron transfer is not the same as for hole transfer. The yields of H. (discusseda bove) suggest that the boundary for electron transfer to the DNA molecule is roughly 24 waters per nucleotide. The target mass for hole transfer, however, cannot be elucidated readily from our data. Recently reported work suggests that this boundary is well below 24 waters per nucleotide (8, 11), possibly as low as 8 (9). As discussed in the Introduction, the calculated boundaries for electron and hole transfer may be subject to underestimation, given the possibility of non-homogeneous packing (as well as other experimental errors). Nevertheless, the data do imply different boundaries for electron and hole transfer. For comparison, Fig. 6 shows how curve 3 changes to curve 2 using these two different boundaries. It is notable that the existence of different boundaries decreases the slope of the initial drop in free radical yield, giving a poorer fit to our data. This may argue against widely separated boundaries. On the other hand, from the proposed differences in electron and hole transfer boundaries, the yield of reduced DNA radicals would be expected to be 1.6 times greater than that of oxidized free radicals (for Γ > 24). This agrees well with earlier work in the deconvolution of the DNA spectrum (7, 56, 59–61). Other factors may also contribute to this observed imbalance [particularly since it is observed at low levels of hydration as well as high levels of hydration (7)]; these include hole–hole combination (7), the presence of undetected oxidized species (59) [i.e. sugar radicals (62)], and deconvolution errors (7).
A Model Based on DNA Packing
Na DNA films are clear, flexible and relatively resistant to compression and tear. These macroscopic properties are maintained at low levels of hydration, Γ < ~15. At low levels of hydration, our model asserts that the interlocking strands of DNA form a mesh that is resistant to changes in interhelix spacing. As the solvation shell becomes filled (5 > Γ > 15–20), the hydration is relatively homogeneous; i.e., there is little dispersion in the local hydration. If hole and electron transfer from the hydration layer to DNA were the only effect of hydration, then the curves for free radical yield as a function of Γ would be flat. Instead free radical yield increases as Γ increases from 5 to 15. We believe this increase is primarily a consequence of improved dielectric relaxation (mainly proton transfer) afforded by the solvation layer and not conformational change. Figure 7 portrays the relatively fixed DNA lattice filling in with water as Γ increases from 5 to ~15 to 20.
FIG. 7.
Postulated hydration-dependent packing changes in Na DNA films. As water is added to the DNA film, the interhelix separation remains relatively constant while the solvation shell is filled. Eventually, the volume of hydration water will exceed the volume of the interstitial spaces, causing the lattice to expand. This lattice expansion is consistent with the observation of sample expansion as the DNA is hydrated (12, 13, 37). Upon further hydration, the lattice is expected to begin to dissociate; upon freezing, this DNA precipitates into a new form of aggregate with a packing different from that of the film.
In the range ~20 < Γ < 30, the films begin to become translucent. It is in this hydration range in which the experimental free radical yields begin to conflict with the simple model of bulk ice growing in (dashed line in Fig. 3). DNA films are known to swell in this range (12, 13, 37). As depicted in Fig. 7, the interhelix spacing increases as Γ increases from 20 to ~30. As described in Fig. 1, increasing the interhelix spacing distance should decrease trapping, since electrons and holes are less likely to be scavenged onto separate DNA molecules, and are therefore more likely to combine. This mechanism could account for the lower than expected free radical yields (Fig. 3).
With increasing hydration, beyond Γ ~ 30, further separation of the DNA strands is possible, though our data do not provide strong evidence for this (Fig. 6). With increasing hydration, crystalline ice begins to account for a significant amount of the sample mass. It is not clear whether this ice is distributed evenly throughoutt he DNA sample, and thus it is not clear whether the local hydration of DNA is uniform.
The film structure is expected to be disrupted at higher levels of hydration (somewhere in the range of 60 > Γ > 140), giving rise to a gel phase which, upon freezing, is more similar in packing to that of frozen dilute solutions than to that of frozen films. Since there is a large body of qualitative information on irradiated DNA in frozen solutions, it is of considerable interest to determine the target mass and free radical yield of such gel-like aggregates. The ranges presented here (Fig. 6), 0.25 < free radical yield(TM) < 0.31 μmol/J, for 24 ≥ Γ(TM) ≥ 16, are reasonable working estimates.
The above model is consistent with our film data, our previous results (4, 5), and the work of others (3, 6). The model can be tested further by measuring free radical yields in solid-state DNA over a wide range of different packing orders and, where possible, with direct determination of the packing order. This approach is facilitated by the fact that DNA displays an extraordinary diversity in packing. Studies are under way in our laboratory that explore more fully how the wide range of DNA packing affects free radical yields.
CONCLUSIONS
Data have been presented here and elsewhere (2–7, 9–11) that strongly support the mechanism of damage transfer from the DNA solvation shell to the DNA molecule. The average boundary, beyond which electron transfer to DNA is not observed, is at a hydration of ~24 waters per nucleotide. DNA hydration also affects packing and conformation. Packing and conformation can, in turn, affect yields in DNA (Fig. 8). Although the free radical yields at relatively low levels of hydration in Na DNA films may be influenced by conformational changes, the large fluctuations in Li DNA films cannot be explained by conformational changes. Hence DNA packing is proposed to be the more important variable. Hydration-dependent changes in free radical yields observed in Na DNA are also attributed in part to changes in packing. We therefore conclude that packing is the dominant variable in determining yields of direct-type damage in hydrated DNA at low temperatures (<100 K).
FIG. 8.
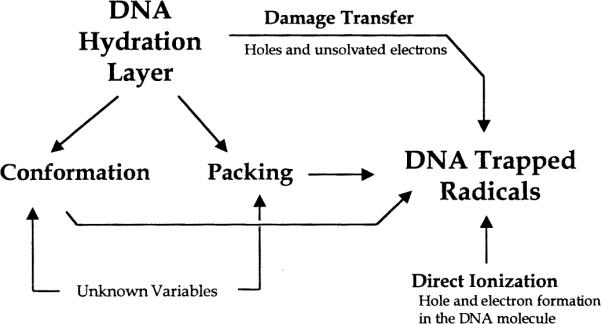
DNA hydration may affect the yield of radical trapping directly and indirectly. The indirect routes are a consequence of the covariance between hydration and conformation and packing.
ACKNOWLEDGMENTS
We thank Kermit R. Mercer for his invaluable technical assistance and Michael G. Debije for his thoughtful discussions. The investigation was supported by PHS Grant 2-R37-CA32546, awarded by the National Cancer Institute, DHHS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute.
REFERENCES
- 1.Mroczka NE, Mercer KR, Bernhard WA. The effects of lattice water on free radical yields in X-irradiated crystalline pyrimidines and purines: A low-temperature electron paramagnetic resonance investigation. Radiat. Res. 1997;147:560–568. [PubMed] [Google Scholar]
- 2.Gregoli S, Olast M, Bertinchamps A. Radiolytic pathways in γ-irradiated DNA: Influence of chemical and conformational factors. Radiat. Res. 1982;89:238–254. [PubMed] [Google Scholar]
- 3.Hüttermann J, Röhrig M, Köhnlein W. Free radicals from irradiated lyophilized DNA: Influence of water of hydration. Int. J. Radiat. Biol. 1992;61:299–313. doi: 10.1080/09553009214550981. [DOI] [PubMed] [Google Scholar]
- 4.Mroczka N, Bernhard WA. Hydration effects on free radical yields in DNA X-irradiated at 4 K. Radiat. Res. 1993;135:155–159. [PubMed] [Google Scholar]
- 5.Mroczka N, Bernhard WA. Electron paramagnetic resonance investigation of X-irradiated poly (U), poly (A) and poly (A):poly (U). Influence of hydration, packing and conformation on radical yield at 4 K. Radiat. Res. 1995;144:251–257. [PubMed] [Google Scholar]
- 6.Wang W, Becker D, Sevilla MD. The influence of hydration on absolute yields of primary ionic free radicals in γ-irradiated DNA at 77 K. I. Total radical yields. Radiat. Res. 1993;135:146–154. [PubMed] [Google Scholar]
- 7.Wang W, Yan M, Becker D, Sevilla MD. The influence of hydration on the absolute yields of primary free radicals in gamma-irradiated DNA at 77 K. II. Individual radical yields. Radiat. Res. 1994;137:2–10. [PubMed] [Google Scholar]
- 8.Becker D, La Vere T, Sevilla MD. ESR detection at 77 K of the hydroxyl radical in the hydration layer of gamma-irradiated DNA. Radiat. Res. 1994;140:123–139. [PubMed] [Google Scholar]
- 9.La Vere T, Becker D, Sevilla MD. Yields of .OH in gamma-irradiated DNA as a function of DNA hydration: Hole transfer in competition with .OH formation. Radiat. Res. 1996;145:673–680. [PubMed] [Google Scholar]
- 10.Swarts SG, Sevilla MD, Becker D, Tokar CJ, Wheeler KT. Radiation-induced DNA damage as a function of hydration. I. Release of unaltered bases. Radiat. Res. 1992;129:333–344. [PubMed] [Google Scholar]
- 11.Swarts SG, Becker D, Sevilla M, Wheeler KT. Radiation-induced DNA damage as a function of hydration. II. Base damage from electron loss centers. Radiat. Res. 1996;145:304–314. [PubMed] [Google Scholar]
- 12.Tao NJ, Lindsay SM. Structure of DNA hydration shells studied by Raman spectroscopy. Biopolymers. 1989;28:1019–1030. doi: 10.1002/bip.360280509. [DOI] [PubMed] [Google Scholar]
- 13.Falk M, Hartman JKA, Lord RC. Hydration of deoxyribonucleic acid II. An infrared study. J. Am. Chem. Soc. 1963;85:387–391. [Google Scholar]
- 14.van Lith D, Warman J, Haas MPD, Hummel A. Electron migration in hydrated DNA and collagen at low temperatures. Part 1. Effect of waterc oncentration. J. Chem. Soc. Faraday Trans. 1. 1986;82:2933–2943. [Google Scholar]
- 15.Lett JT, Alexander P. Crosslinking and degradation of deoxyribonucleic acid gels with varying water contents when irradiated with electrons. Radiat. Res. 1961;15:159–173. [PubMed] [Google Scholar]
- 16.Lett JT. Damage to DNA and chromatin structure from ionizing radiations, and the radiations sensitivities of mammalian cells. Prog. Nucleic Acid Res. 1990;39:305–352. doi: 10.1016/s0079-6603(08)60630-3. [DOI] [PubMed] [Google Scholar]
- 17.Lett JT. Damage the cellular DNA from particulater adiations, the efficacy of its processing and the radiosensitivity of mammalian cells. Emphasis on DNA double strand breaks and chromatin breaks. Radiat. Environ. Biophys. 1992;31:257–277. doi: 10.1007/BF01210207. [DOI] [PubMed] [Google Scholar]
- 18.Goodsell DS. Inside a living cell. Trends Biochem. Sci. 1991;16:203–206. doi: 10.1016/0968-0004(91)90083-8. [DOI] [PubMed] [Google Scholar]
- 19.Richmond TJ, Finch JT, Rushton B, Rhodes D, Klug A. Structure of the nucleosome core particle at 7Å resolution. Nature. 1984;311:532–537. doi: 10.1038/311532a0. [DOI] [PubMed] [Google Scholar]
- 20.Struck M-M, Klug A, Richmond T. Comparison of x-ray structures of the nucleosome core particle in two different hydration states. J. Mol. Biol. 1992;224:253–264. doi: 10.1016/0022-2836(92)90588-b. [DOI] [PubMed] [Google Scholar]
- 21.Bernhard WA, Barnes J, Mercer KR, Mroczka N. The influence of packing on free radical yields in crystalline nucleic acids: The pyrimidine bases. Radiat. Res. 1994;140:199–214. [PubMed] [Google Scholar]
- 22.Bernhard WA, Mroczka N, Barnes J. Combination is the dominant free radical process initiated in DNA by ionizing radiation: An overview based on solid-state EPR studies. Int. J. Radiat. Biol. 1994;66:491–497. doi: 10.1080/09553009414551511. [DOI] [PubMed] [Google Scholar]
- 23.Berman HM. Hydration of DNA. Curr. Opin. Struct. Biol. 1991;1:423–427. [Google Scholar]
- 24.Rill R. Spontaneous ordering of DNA. J. Biol. Chem. 1983;258:250–256. [PubMed] [Google Scholar]
- 25.Oleinick NL, Balasubramaniam U, Xue L, Chiu S. Nuclear structure and the microdistribution of radiation damage in DNA. Int. J. Radiat. Biol. 1993;66:523–529. doi: 10.1080/09553009414551561. [DOI] [PubMed] [Google Scholar]
- 26.Roti Roti JL, Wright WD, Taylor YC. DNA loop structure and radiation response. Adv. Radiat. Biol. 1993;17:53–120. [Google Scholar]
- 27.Sapora 0, Belli M, Maione B, Pazzaglia S, Tabocchini MA. The influence of genome structuraol rganization on DNA damage and repair in eukaryotic cells exposed to ionizing radiation. In: Fielden EM, O'Neill P, editors. Early Effects of Radiation on DNA. Vol. 54. Springer-Verlag; Berlin: 1991. pp. 141–154. NATO ASI Series H. [Google Scholar]
- 28.Spotheim-Maurizot M, Ruiz S, Sabattier R, Charlier M. Radioprotection of DNA by polyamines. Int. J. Radiat. Biol. 1995;68:571–577. doi: 10.1080/09553009514551561. [DOI] [PubMed] [Google Scholar]
- 29.Savoye C, Swenberg C, Hugot S, Sy D, Sabattier R, Charlier M, Spotheim-Maurizot M. Thiol WR-1065 and disulphide WR-33278, two metabolites of the drug ethyol (WR-2721), protect DNA against fast neutron-inducesd strand breakage. Int. J. Radiat. Biol. 1997;71:193–202. doi: 10.1080/095530097144319. [DOI] [PubMed] [Google Scholar]
- 30.Newton GL, Aguilera JA, Ward JF, Fahey RC. Effect of polyamine-induced compaction and aggregation of DNA on the formation of radiation-induced strand breaks: Quantitative models for cellular radiation damage. Radiat. Res. 1997;148:272–284. [PubMed] [Google Scholar]
- 31.Ward JF. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- 32.Warman JM, de Haas MP, Rupprecht A. DNA: A molecular wire? Chem. Phys. Lett. 1996;249:319–322. [Google Scholar]
- 33.van Lith D, de Haas MP, Warman JM. Highly mobile charge carriers in hydrated DNA and collagen formed by pulsed ionization. Biopolymers. 1983;22:807–810. doi: 10.1002/bip.360220302. [DOI] [PubMed] [Google Scholar]
- 34.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 35.Rupprecht A. Preparation of oriented DNA by wet spinning. Acta Chem. Scand. 1969;20:494–504. doi: 10.3891/acta.chem.scand.20-0494. [DOI] [PubMed] [Google Scholar]
- 36.Rich A, Kasha M. The n → π* transitions in nucleic acids and polynucleotides. J. Am. Chem. Soc. 1960;82:6197–6199. [Google Scholar]
- 37.Falk M, Hartman JKA, Lord RC. Hydration of deoxyribonucleic acid I. A gravimetric study. J. Am. Chem. Soc. 1962;84:3843–3846. [Google Scholar]
- 38.Falk M, Hartman JKA, Lord RC. Hydration of deoxyribonucleic acid III. A spectroscopic study of the effect of hydration on the structure of deoxyribonucleic acid. J. Am. Chem. Soc. 1963;85:391–394. [Google Scholar]
- 39.Brandes R, Kears D, Rupprecht A. A 2H-NMR study of the DNA hydration water in solid Li-DNA assemblies. Biopolymers. 1988;27:717–732. doi: 10.1002/bip.360270502. [DOI] [PubMed] [Google Scholar]
- 40.Chargaff E, Vischer E, Doniger R, Green C, Misani F. The composition of the desoxypentose nucleic acids in thymus and spleen. J. Biol. Chem. 1949;177:405–416. [PubMed] [Google Scholar]
- 41.Tamm C, Hodes ME, Chargaff E. The formation of apurinic acid from the desoxyribonucleic acid. J. Biol. Chem. 1952;195:49–63. [PubMed] [Google Scholar]
- 42.Livolant F. Cholesteric organization of DNA in vivo and in vitro. Eur. J. Cell Biol. 1984;33:300–311. [PubMed] [Google Scholar]
- 43.Rupprecht A, Forslind B. Variation of electrolyte content in wet-spun lithium- and sodium-DNA. Biochim. Biophys. Acta. 1970;204:304–316. doi: 10.1016/0005-2787(70)90148-6. [DOI] [PubMed] [Google Scholar]
- 44.Mercer KR, Bernhard WA. Design and operation of a variable temperature accessory for Q-band ESR. J. Magn. Reson. 1987;74:66–71. [Google Scholar]
- 45.Bradshaw WW, Cadena DG, Crawford GW, Spetzler HAW. The use of alanine as a solid dosimeter. Radiat. Res. 1962;17:11–21. [PubMed] [Google Scholar]
- 46.Regulla DF, Deffner U. Dosimetry by ESR spectroscopy of alanine. Int. J. Appl. Radiat. Isot. 1982;33:1101–1114. [Google Scholar]
- 47.Spalletta R, Bernhard WA. Free radical yields in A:T polydeoxynucleotides, oligodeoxynucleotides, and monodeoxynucleotides at 4 K. Radiat. Res. 1992;130:7–14. [PubMed] [Google Scholar]
- 48.Saenger W, Hunter WH, Kennard O. DNA conformation is determined by economics in the hydration of phosphate groups. Nature. 1986;324:385–388. doi: 10.1038/324385a0. [DOI] [PubMed] [Google Scholar]
- 49.Lindsay SM, Lee SA, Powell JW, Weidlich T, DeMarco C, Lewen GD, Tao NJ. The origin of the A to B transition in DNA fibers and films. Biopolymers. 1988;27:1015–1043. doi: 10.1002/bip.360270610. [DOI] [PubMed] [Google Scholar]
- 50.Shakked Z, Guerstein-Guzikevich G, Eisenstein M, Frolow F, Rabinovich D. The conformation of the DNA double helix in the crystal is dependent on its environment. Nature. 1989;342:456–460. doi: 10.1038/342456a0. [DOI] [PubMed] [Google Scholar]
- 51.Rill R. Liquid crystalline phases in concentrated aqueous solutions of Na+ DNA. Biophysics. 1986;83:342–346. doi: 10.1073/pnas.83.2.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leforestier A, Livolant F. Supramolecular ordering of DNA in the cholesteric liquid crystalline phase: An ultrastructural study. Biophys. J. 1993;65:56–72. doi: 10.1016/S0006-3495(93)81063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strzelecka TE. Solid state 31p NMR studies of DNA liquid crystalline phases. The isotropic to cholesteric transition. J. Am. Chem. Soc. 1987;109:4513–4518. [Google Scholar]
- 54.Bernhard WA. Sites of electron trapping in DNA as determined by ESR of one-electron reduced oligonucleotides. J. Phys. Chem. 1989;93:2187–2189. [Google Scholar]
- 55.Bernhard WA. Free radicals formed by electron gain in oligomers of DNA. Free Radic. Res. Commun. 1989;6:93–94. doi: 10.3109/10715768909073436. [DOI] [PubMed] [Google Scholar]
- 56.Sevilla MD, Becker D, Yan M, Summerfield SR. Relative abundances of primary ion radicals in γ-irradiated DNA: Cytosine vs. thymine anions and guanine vs. adenine cations. J. Phys. Chem. 1991;95:3409–3415. [Google Scholar]
- 57.Steenken S. Electron-transfer induced acidity/basicity and reactivity changes of purine and pyrimidine bases. Consequences of redox processes for DNA base pairs. Free Radic. Res. Commun. 1992;6:349–379. doi: 10.3109/10715769209049187. [DOI] [PubMed] [Google Scholar]
- 58.Milano MT, Bernhard WA. Migration of electrons and holes in crystalline d(CGATCG)-anthracycline complexes X-irradiated at 4 K. Radiat. Res. 1998;150:101–114. [PubMed] [Google Scholar]
- 59.Gräslund A, Ehrenberg A, Rupprecht A, Ström G, Crespi H. Ionic base radicals in γ-irradiated oriented non-deuterated and fully deuterated DNA. Int. J. Radiat. Biol. 1975;28:313–323. doi: 10.1080/09553007514551101. [DOI] [PubMed] [Google Scholar]
- 60.Hüttermann J, Voit K, Oloff H, Köhnlein W, Gräslund A, Rupprecht A. Specific formation of electron gain and electron loss centres in x-irradiated oriented fibers of DNA at low temperatures. Faraday Discuss. Chem. Soc. 1984;78:135–149. doi: 10.1039/dc9847800135. [DOI] [PubMed] [Google Scholar]
- 61.Yan M, Becker D, Summerfield S, Renke P, Sevilla MD. Relative abundance and reactivity of primary ion radicals in γ-irradiated DNA at low temperatures. 2. Single- vs. double-stranded DNA. J. Phys. Chem. 1992;96:1695–1702. [Google Scholar]
- 62.Close DM. Where are the sugar radicals in irradiated DNA? Radiat. Res. 1997;147:663–673. [PubMed] [Google Scholar]