

Abstract
Palladium-catalyzed decarboxylative asymmetric allylic alkylation (DAAA) of allyl enol carbonates as a highly chemo-, regio- and enantio-selective process for the synthesis of ketones bearing either a quaternary or a tertiary α-stereogenic center has been investigated in detail. Chiral ligand L4 was found to be optimal in the DAAA of a broad scope of cyclic and acyclic ketones including simple aliphatic ketones with more than one enolizable proton. The allyl moiety of the carbonates has been extended to a variety of cyclic or acyclic di-substituted allyl groups. Our mechanistic studies reveal that, similar to the direct allylation of lithium enolates, the DAAA reaction proceeds through an “outer sphere” SN2 type of attack on the π-allylpalladium complex by the enolate. An important difference between the DAAA reaction and the direct allylation of lithium enolates is that in the DAAA reaction, the nucleophile and the electrophile were generated simultaneously. Since the π-allylpalladium cation must serve as the counterion for the enolate, the enolate probably exists as a tight-ion-pair. This largely prevents the common side reactions of enolates associated with the equilibrium between different enolates. The much milder reaction conditions as well as the much broader substrate scope also represent the advantages of the DAAA reaction over the direct allylation of preformed metal enolates.
Keywords: Asymmetric, Catalytic, Alkylation, Enolates, Palladium
Introduction
The formation of carbon-carbon bonds represents the most fundamental process for the synthesis of complex molecules from simple ones. Among carbon-carbon bond forming strategies, enolate alkylation has received intensive attention for decades partially because carbonyl groups, or functional groups easily derived from carbonyl groups, are frequently found in many synthetic targets and they are rich in transformations to many other structural moieties.1 While significant advances in asymmetric alkylation of enolates have been achieved by using chiral auxiliary approaches, 2 most notably SAMP/RAMP,3 it is more attractive in terms of atom economy,4 to develop catalytic enantioselective approaches to meet the environmental and economical requirements of modern chemistry. Many efforts have been made over the past three decades.1b,5 Based upon the role of the chiral catalyst, most approaches can be classified into two categories, in one of which the catalyst interacts with the enolate to form a more nucleophilic chiral complex and in the other, it interacts with the alkylating agent to form a more electrophilic chiral complex. In the first category, one of the most well studied methods is phase transfer catalysis.6 To achieve a high degree of enantioselective control it is essential but difficult to form a well organized transition state involving the enolate, the catalyst and the alkylating agent.5 Thus, the enantioselectivity is often found to be very sensitive to a subtle structural change of the substrate and optimization of the reaction conditions is often required for each substrate.5 Another straightforward method in this category has been demonstrated by Koga et. al., in which a chiral polydentate amine was used to coordinate the metal countercation of the lithium enolate to achieve enantioselectivity in a catalytic fashion, but its scope remains untested.7 High enantioselectivities have also been achieved in the recently emerging organocatalyst promoted asymmetric α-alkylation of aldehydes, although by a strict definition, it doesn’t go though enolate intermediates but enamines.8 Besides their own limitations, such as the restriction to intramolecular reactions or the use of harsh oxidative reagents, extending these methods to ketones hasn’t been succeeded. In the second category, a chiral Cr(salen)Cl complex catalyzed asymmetric alkylation of tin enolates with various alkylation agents was developed by Jacobsen and Doyle.9 The reaction was proposed to follow a SN2 type of pathway; the halide in the catalyst served as a Lewis base to activate the tin enolate by the formation of an “ate” complex while the chiral cationic Cr moiety may assist the departure of the halide in the alkylating agent. Transition metal catalyzed enantioselective alkylation of safer and less expensive lithium enolates by activation of the electrophile, however, has almost exclusively focused on a special type of alkylating group - the allylic groups. They can be activated towards nucleophilic substitution by coordinating with a low valent metal complex to form a cationic π-allylmetal complex.10,11,12 Although good yields and high enantioselectivities have been reported, the scope of these reactions was quite narrow, at least partially due to the structural complexity of lithium enolates in solution.13 In some cases, a small change of the enolate structure can dramatically affect the yield or enantioselectivity of the reaction.10 For substrates with more than one enolizable proton, proton-transfer between the initially formed enolate and the newly formed product may lead to problems such as loss of regioselectivity and di- or poly- alkylation.14 These problems caused by the faster equilibration between enolates compared to the alkylation were elegantly avoided in the decarboxylative alkylation of allyl enol carbonates or allyl β-ketoesters, as independently reported by Saegusa15 and Tsuji.16 The proposed mechanism includes the coordination of the Pd (0) complex to the allyl moiety, ionization of the carbonate or ester leaving group, Pd-promoted decarboxylation to form the enolate in situ and recombination of the enolate and the π-allylpalladium to form the allylated product and regenerate the catalyst (Scheme 1).15, 16e Tsuji and co workers have demonstrated that under these extremely mild and neutral conditions, the reaction preceded with highly conserved regioselectivity.16c
Scheme 1.
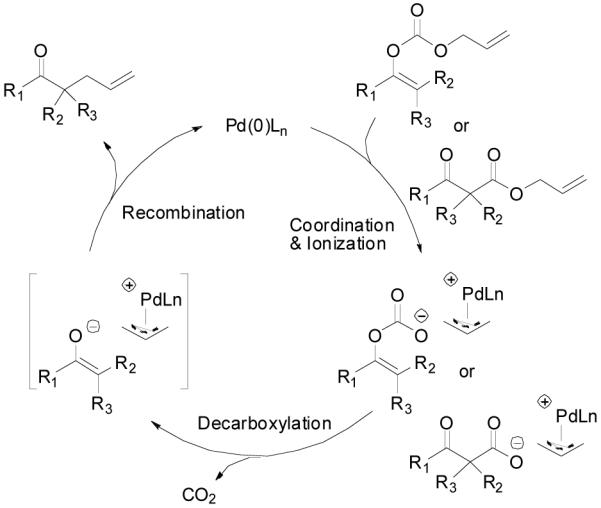
Mechanism of Palladium-Catalyzed Decarboxylative Allylic Alkylation of Enol Carbonates and α,α-Disubstituted β-Ketoesters.
At the time we started to question if we could use a chiral ligand to induce a good enantioselectivity of the reaction while keeping its regiospecificity, there was no enantioselective version of this reaction reported. As our research proceeded, Burger and Tunge reported the first palladium-catalyzed asymmetric DAAA of racemic 1,3-disubstituted allylic β-ketoesters to generate γ,δ-unsaturated ketones with a β-tertiary chiral center.17 It should be pointed out that in this report the newly formed asymmetric center is on the electrophile; the control of the stereoselectivity on the allyl moiety is easier than that on the prochiral nucleophile since the allyl moiety is directly attached to the chiral metal complex. In addition, as proposed by Tsuji et. al., the reaction probably doesn’t go through the ionization-decarboxylation-alkylation sequence but an alternative ionization-alkylation-decarboxylation pathway; that means that the nucleophile is not an unstabilized ketone enolate but a stabilized β-keto carboxylate.16f Stoltz et. al.18 and our group19 independently reported the first catalytic enantioselective DAAA reactions of allyl enol carbonates to form simple ketones bearing a quaternary stereogenic α-carbon center. Since then, many expansions and applications of this robust reaction have been reported. 20 We were specifically interested in the family of ligands derived from 2-diphenylphosphinobenzoic or 1-naphthoic acid and chiral scalemic diamines, which had been used successfully in inducing excellent enantioselectivity in numerous palladium-catalyzed AAA reactions (Figure 1).21 We reported herein a full account in the optimization, scope and mechanistic studies of the decarboxylative asymmetric allylic alkylation (DAAA) of various unstabilized ketone enolates.
Figure 1.
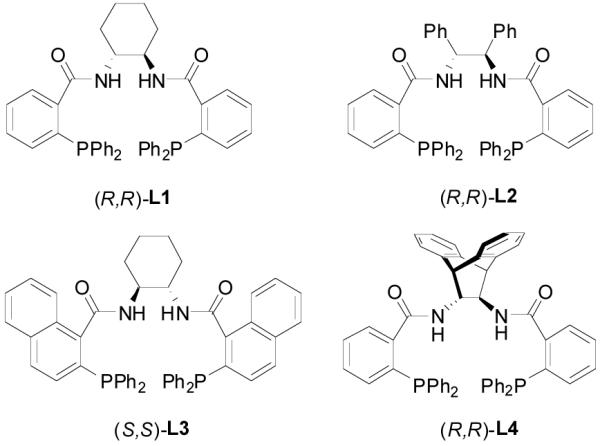
Ligands Used in Optimization.
Results and Discussion
The Preparation of Allyl Enol Carbonates
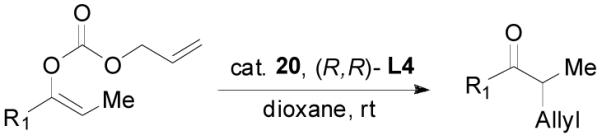
An allyl enol carbonate was typically prepared by quenching the enolate, generated from the deprotonation of a ketone, with allyl chloroformate at low temperature. The chemo- and regio-selective control of enolate formation has been well documented.1b For ketones with two enolizable centers, either kinetically or thermodynamically controlled enolate formation can be obtained by adjustment of base, solvent, reaction temperature, and, most importantly, the order of addition of the base and the ketone substrate. Most allyl enol carbonates of cyclic ketones were prepared by the method described by Houminer et. al.22 In a general procedure (eq. 1), 5 mmol of ketone in anhydrous tetrahydrofuran (THF) was slowly added into the solution of 6 mmol of sodium hexamethyldisilazane (NaHMDS) and 6.0 mmol of N,N,N’,N’-tetramethyl-1,2-ethylenediamine (TMEDA) in anhydrous THF at −78 °C. The reaction mixture was stirred for 1 h and transferred through a cannula into a solution of 6 mmol of allyl chloroformate in anhydrous THF at −78 °C and stirred for 5 min. The product was typically purified by flash column chromatography on silica gel; and most enol carbonates are stable for years at refrigerator temperature. Alternatively, the thermodynamically more stable enolates were generated by exposure to sodium hydride and TMEDA at higher temperature, and sequentially quenched with allyl chloroformate at low temperature (eq. 2).
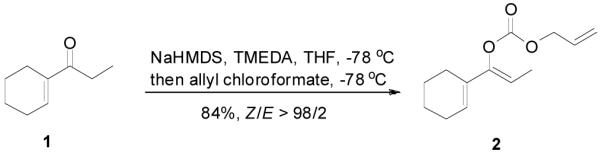 |
(1) |
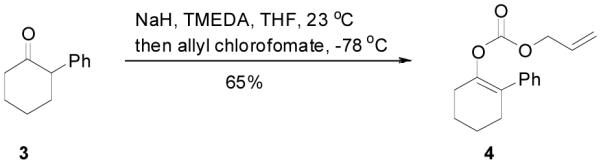 |
(2) |
The preparation of substituted allyl enol carbonates from the corresponding substituted allylchloroformates is, however, less convenient since substituted allylchloroformates are not commercial available and their preparation involves the use of highly toxic and strictly regulated phosgene. Besides, many chloroformates are not stable to heat and moisture; thus their purification and storage are problematic. Strongly acidic hydrogen chloride generated in the process also limits the range of compatible functional groups. Furthermore, a significant amount of diallyl carbonate is often formed lowering the yield of chloroformate, which is a disadvantage especially for precious allylic alcohols. On the other hand, allyl 1H-imidazole-1-carboxylates are easily prepared in high yields from the corresponding alcohol and carbonyldiimidazole (CDI) under mild conditions (eq. 3). They can be purified by flash column chromatography and most are stable for months when refrigerated. Compared with chloroformates, imidazolides are relatively “softer” eletrophiles, and thus, the reaction of sodium enolates with 5 gave exclusively the C-acylated product 6,23 even in a coordinating solvent DME, which presumably can stabilize the charge-separated ion-pairs, thus favoring the formation of the O-acylated product (eq. 4).24 However, by coordinating the more basic nitrogen on the imidazole with an azaphilic Lewis acid, such as BF3, to increase the polarity of the carbonyl group as well as to facilitate the leaving of the imidazole group, we were able to tune the complex to a “harder” electrophile, which favored the O-acylation and enol carbonate 7 was obtained exclusively (eq. 5).25 As listed in Table 1, a variety of substituted allyl enol carbonates were prepared in this manner in good to excellent yields.
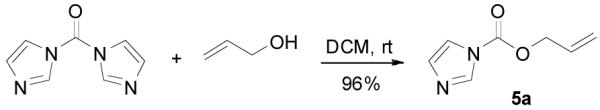 |
(3) |
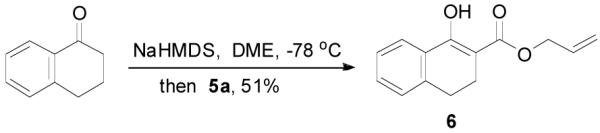 |
(4) |
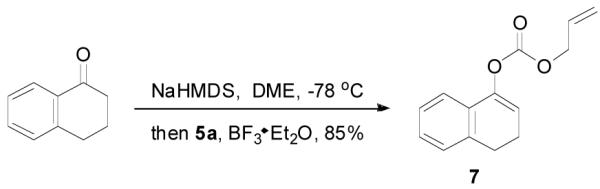 |
(5) |
Table 1.
Preparation of Various Substituted Allyl Enol Carbonates
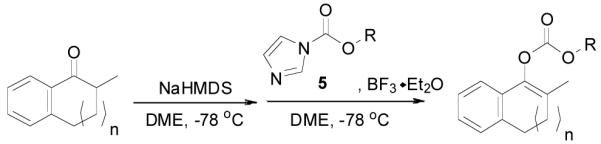 | ||||
|---|---|---|---|---|
| Entry | n | R | Product | Yield |
| 1 | 1 | 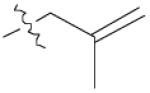 |
8 | 92% |
| 2 | 1 | 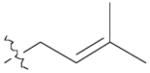 |
9 | 99% |
| 3 | 1 | 10 | 76% | |
| 4 | 1 | 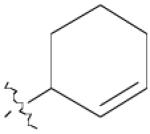 |
11 | 94% |
| 5 | 1 | 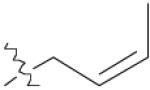 |
12 | 89% |
| 6 | 1 | 13 | 96% | |
| 7a | 0 | 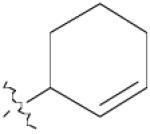 |
14 | 88% |
KOt-Bu was used as base.
Besides intrinsic electronic and steric effects, the double bond geometry of an acyclic ketone enolate is also controlled by the choice of base, solvent and reaction temperature, etc.1b,26 Normally the preformed enolate is geometrically stable at low temperature if there is no external proton source to allow the equilibration between different enolate structures; and it can be rapidly trapped by allyl chloroformate at low temperature. As an example, deprotonation of cyclohexyl ethyl ketone by (PhMe2Si)2NLi in THF at −78 °C followed by quenching the produced lithium enolate with allylchloroformate gave Z-15 in 56% yield and greater than 49:1 Z/E selectivity (by 1HNMR), with the formation of a small portion of β-ketoester (Table 2, entry 1). The double bond configuration was assigned by the difference between the chemical shifts of the vinyl proton on the tri-substituted enol carbonate. As proposed by Pascual et. al., the proton trans to the carbonate substitution (in the Z-isomer) has a lower chemical shift (δ = 5.06) than the one cis to the carbonate (in the E-isomer, δ = 5.17).27 In the presence of lithium diisopropylamide (LDA)/TMEDA as base, the ratio of E-15 to Z-15 increased to 1:1.6 (entry 2). Switching from LDA to lithium 2,2,6,6-tetramethylpiperidinide-LiBr (LiTMP-LiBr), 28 the E-15 was obtained as the major isomer with a 3:1 E/Z ratio (entry 3). Several E-enol carbonates were prepared in this manner in good yields and high E/Z selectivity (Scheme 2).
Table 2.
The Control of Double Bond Geometry by the Base in the Formation of Enol Carbonate
 | |||
|---|---|---|---|
| entry | base | yield | Z/E |
| 1 | (PhMe2Si)2NLi | 56% | >49/1 |
| 2 | LDA, TMEDA | 91% | 1.6/1 |
| 3 | LiTMP-LiBr-TMEDA | 87% | 1/3 |
Scheme 2.
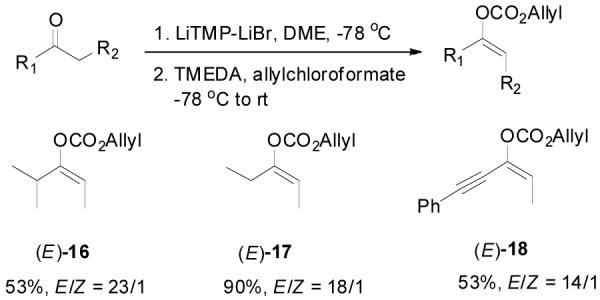
The Preparation of E-Enol Carbonates.
Palladium-Catalyzed DAAA of Allyl Enol Carbonates to Generate Ketones Possessing an α-Quaternary Center
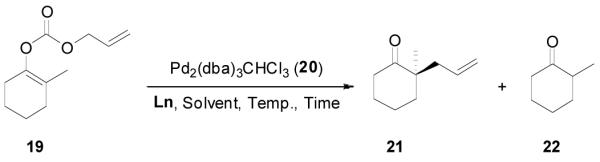
Using 2-methylcyclohexanone as a model we systematically investigated the DAAA reaction of its allyl enol carbonate 19 by screening chiral ligands, solvents, reaction temperature and substrate concentration. Some results are summarized in Table 3. Among the four chiral ligands listed in Figure 1, L4 gave the best ee and yield (entry 1–4). Solvent had a small effect on the ee of the reaction when L4 was used; and the highest ee (85%) was obtained in toluene (entry 4–8). The ee was slightly increased to 87% (entry 9) when the reaction temperature decreased from 23 °C to 4 °C, but it dropped to 83% at −10 °C (entry 10). The concentration of the substrate didn’t have a significant effect on the ee and yield of the product (entry 11 and 12). The reaction performed in the presence of 2 mol% Pd gave essentially the same results as that catalyzed by 5 mol% Pd (entry 13).
Table 3.
Selected Optimization Studies
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | ligand | solvent | temp | conc. | eeb | yieldc of 21 | yieldc of 22 |
| 1 | (R,R)-L1 | Toluene | 23 °C | 0.1 M | 31% | 73% | 0 |
| 2 | (R,R)-L2 | Toluene | 23 °C | 0.1 M | 61% | 73% | 2% |
| 3 | (R,R)-L3 | Toluene | 23 °C | 0.1 M | 60% | 85% | 1% |
| 4 | (R,R)-L4 | Toluene | 23 °C | 0.1 M | 85% | 88% | 0 |
| 5 | (R,R)-L4 | CH2Cl2 | 23 °C | 0.1 M | 84% | 64% | 26% |
| 6 | (R,R)-L4 | 1,4-Dioxane | 23 °C | 0.1 M | 80% | 99% | 0 |
| 7 | (R,R)-L4 | DME | 23 °C | 0.1 M | 84% | 87% | 7% |
| 8 | (R,R)-L4 | THF | 23 °C | 0.1 M | 81% | 85% | 1% |
| 9 | (R,R)-L4 | Toluene | 4 °C | 0.1 M | 87% | 91% | 2% |
| 10 | (R,R)-L4 | Toluene | −10 °C | 0.1 M | 83% | 84% | 6% |
| 11 | (R,R)-L4 | Toluene | 4 °C | 0.3 M | 85% | 94% | 4% |
| 12 | (R,R)-L4 | Toluene | 4 °C | 0.025M | 87% | 92% | 0% |
| 13d | (R,R)-L4 | Toluene | 4 °C | 0.3 M | 85% | 88% | 5% |
Unless otherwise indicated, all reactions were performed on a 0.3 mmol scale at 23 °C employing 2.5 mol% 20 and 5.5 mol% ligand.
The ee values were determined by chiral GC.
The yields were determined by quantitative GC analysis using decane as internal reference.
1.0 mol% 20 and 5 mol% L4 were used.
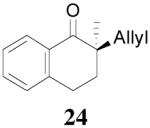
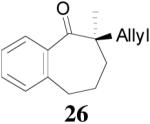
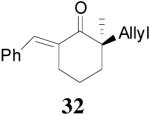
The asymmetric synthesis of 2-allyl-2-methylcyclohexan-1-one through palladium-catalyzed DAAA reaction of enol carbonate 19 selectively demonstrated the advantage of this method: extremely mild reaction conditions, exclusive regioselectivity, high catalyst efficiency and good enantioselectivity. Compared with our previous results carried out using preformed lithium or tin enolates,10 products obtained through the DAAA of enol carbonates under the above optimized conditions (2.5 mol% 20, 5.5 mol% (R,R)-L4, toluene, 23 °C) have much higher enantioselectivity in general (Table 4). For instance, only a poor 6% ee was obtained in the allylation of the preformed tin enolate of 2-methylbenzosuberone. By the DAAA method, however, the ee of 26 was dramatically improved to 91% (entry 2). More interestingly, R-24 was the major enantiomer (88% ee) in the allylation of the preformed lithium or tin enolate of 2-methyltetralone catalyzed by (S,S)-L1, while the same enantiomer was also found to be the major one in the DAAA reaction of the enol carbonate 23 catalyzed, however, by the (R,R)-ligand (entry 1). This means that completely opposite enantioselectivities were obtained by these two approaches. A similar observation was also found in the allylation of 2-methylindanone (entry 3). However, this phenomenon was not observed in the allylation of some other six-membered cyclic ketones, i.e., the same enantiomers of 30 and 32 were obtained by both approaches (entry 4 and entry 5). While the chiral scaffold differed in the DAAA (ie L4) vs the standard enolate process (ie L1), we have shown that the sense of asymmetric induction in each series is independent of the scaffold. The results are given for the best selectivities observed in each case.
Table 4.
Reaction of Various Allyl Enol Carbonatesa
| entry | substrate | product | yieldb eec |
Lit,d yield, ee |
|---|---|---|---|---|
| 1 | 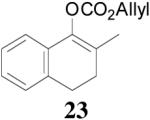 |
 |
88% 99.7% ee R-(+) e |
99% 88% ee R-(+) |
| 2 | 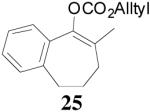 |
 |
94% 91% ee f (−) |
90% 6% ee (+) |
| 3 | 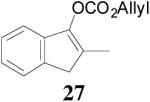 |
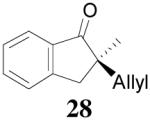 |
98% 76%ee (+) |
79% 38% ee (+) |
| 4 | 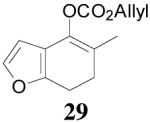 |
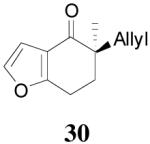 |
64% 82% ee (+) |
59% 45% ee (−) |
| 5 | 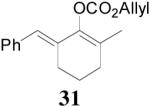 |
 |
99% 95% ee (+) |
98% 82% ee (−) |
All reactions were performed on a 0.3 mmol scale at 0.1 M in toluene at 23 °C for 20 h using 2.5 mol% 20 and 5.5 mol% ligand (R,R)-L4.
Isolated yields.
Ee values were determined by HPLC on a chiral stationary phase.
Conditions used were 2.5 mol% [(η3-C3H5)PdCl]2, 5.0 mol% (S,S)-L1, 2.0 equiv LDA, 1.0 equiv Me3SnCl, DME, r.t.10
The absolute configuration and the sign of the optical rotation ([α]D) of the product.9a
Ee was determined by GC on a chiral stationary phase.
One obvious difference between these two methods is that the DAAA reactions of enol carbonates are under neutral conditions and the AAA reactions of preformed lithium or tin enolates are under strong basic conditions. Since there are acidic amide protons in these ligands, under strong basic conditions the ligand must be deprotonated. Therefore, it might be the deprotonation of the ligand that caused the switch of enantioselectivity observed in some cases. To test this hypothesis, we pre-treated the Pd/L4 catalyst with LDA to fully deprotonate the catalyst before it was subjected to the solution of 23 (eq. 6). The reaction didn’t go to completion after 10 h, and the product 24 was obtained in only 31% yield, compared with the 88% yield in 5 min when the neutral catalyst was used. However, almost the same enantioselectivity was obtained with or without deprotonation of the ligand (eq. 6). Therefore, it is not likely that the deprotonation of the ligand is the reason causing the observed opposite enantioselectivity in Table 4, entries 1 and 3.
 |
(6) |
Similarly, our previous studies revealed that palladium-catalyzed AAA of the preformed enolate of 1-methyl-2-tetralone (33) was very sensitive to the choice of base. Actually, a completely opposite enantioselectivity was obtained by switching the base from LDA to cesium carbonate (Scheme 3).29 Presumably, the palladium enolate generated in situ from the decarboxylation of the enol carbonate should be more like the intermediate in the reaction of less covalently bonded cesium enolate than that in the reaction of more covalently bonded lithium enolate. Indeed, the DAAA reaction of the allyl enol carbonate of 1-methyl-2-tetralone (35) catalyzed by Pd-L4 complex (eq. 7) gave the same sense of chirality and nearly the same enantioselectivity as the AAA of the cesium enolate.
 |
(7) |
Scheme 3.

Countercation Effect in Palladium-Catalyzed AAA of 1-Methyl-2-tetralone.29
The observation that opposite enantioselectivity was obtained in the allylation of the lithium enolate of 2-methyl-1-tetralone and the DAAA reaction of its enol carbonate can be probably explained by the structural difference of the nucleophiles. As shown by the cartoons30 in Figure 2, the “naked” enolate approaches the π-allylpalladium-L4 complex by its Si face (A), to avoid the disfavored steric interaction between the “wall” of the ligand and the phenyl ring of the substrate (B). On the other hand, we have disclosed that two equivalents of LDA were necessary for good enantioselectivity in the AAA of lithium enolate.10 It possibly implies the dimer of one lithium enolate and one LDA is the nucleophile. The aggregation makes the O terminal very bulky so that it prefers to be located at the flap side of the catalyst and leaves its Re face exposed to the electrophile (C). The fact that enantioselectivity of the allylation of preformed enolates is very sensitive to structural change of the enolate may also be explained by the complexity of the lithium enolate in solution, which normally exists as a mixture of monomer, dimer and oligomers and the equilibria among them may change with the variation of substrate structure.31
Figure 2.

Cartoon Models for the Alkylation of Charge Separated Palladium Enolate.
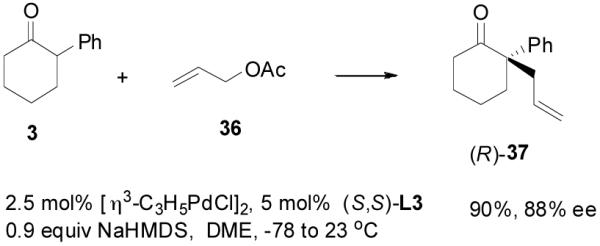
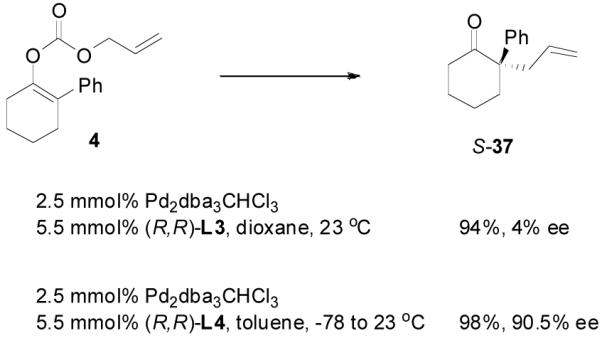
However, it is perhaps too simple to say that the DAAA of an enol carbonate is equivalent to the AAA reaction of a charge-separated enolate. For instance, the optimized conditions for the AAA of 2-phenylcyclohexan-1-one (3) involved the use of sub-stoichiometric amount of NaHMDS as base and L3 as ligand in a coordinating solvent 1,2-dimethoxyethane (DME, eq. 8).29 The DAAA reaction of 4 in the presence of the same catalyst, however, gave a nearly racemic product (eq. 9). Switching from L3 to L4 dramatically improved the ee of 37 to over 90% (eq. 9).
 |
(8) |
 |
(9) |
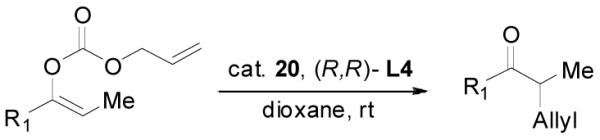
Palladium-Catalyzed DAAA of Substituted Allyl Enol Carbonates
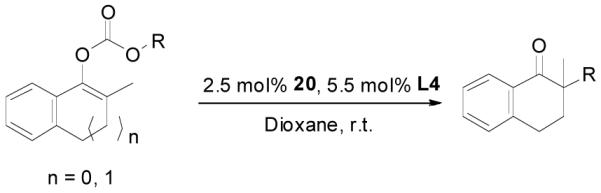
The palladium-catalyzed AAA of preformed lithium or tin enolates of ketones has been investigated in our previous work.10b In general, the yields and enantioselectivities were quite sensitive to the substitution changes on the allyl moiety. As a comparison, a variety of 1,1- or 1,2-disubstituted allyl enol carbonates of 2-methyl-1-tetralone were subjected to the optimized conditions (Table 5). The reaction of 2′-methallyl enol carbonate 8 went to completion within 4 h at room temperature to produce the desired product 38 in 89% yield and over 99% ee (Table 5, entry 1), similar to the results of the DAAA of the unsubstituted allyl enol carbonate 23 (Table 3, entry 1), but much better than the 47% ee obtained by the AAA of the preformed tin enolate. In another example, the reaction of cinnamyl enol carbonate 10 gave cleanly the linear product 39 quantitatively with excellent ee (92.5%), while the yield from the preformed enolate was only 11% (entry 2). The reaction of cis-crotyl enol carbonate 12 gave a 4:1 mixture of the linear product 40 and the branched product 41 (entry 3). The E isomer of 40 was the major one in a ratio of 11.7:1 to the Z-isomer, implying a fast π-σ-π equilibration compared to the decarboxylation and alkylation. Equally good enantioselectivities were achieved for the major product isomer from both the Z- and E-isomers of the allyl substrates (entry 3 and 4). Surprisingly, prenyl enol carbonate 9 remained intact for 13 h at 23 °C, while at the same temperature the prenylation of the preformed enolate was obtained in good yield (64%), albeit in poor ee (entry 5). It might be because the palladium-involved decarboxylation step is retarded by the steric hindrance of the π-prenylpalladium species. Increasing the reaction temperature to 60 °C did push the reaction of 9 to completion, but the only product found in the crude residue after concentration was 2-methyl-1-tetralone (entry 6). It was formed presumably through the β-hydride elimination of the π-prenylpalladium intermediate followed by reductive elimination of the hydrido-palladium enolate. Elimination from the π-allylpalladium complex to form diene can also occur via a base promoted process. Nevertheless, the palladium-catalyzed decarboxylative prenylation has been successfully applied in the synthesis of spirotryprostatin B, where the corresponding enolate was more stabilized, so the decarboxylation may be more facile.32 We cannot rule out a role for the hardness/softness of the enolate in the two cases although we favor the former explanation. In entry 7, 42 was isolated in 62% yield, over 20:1 dr and over 99% ee. The diastereoselectivity (5:1 dr) in the DAAA of 2-methyl-1-indanone (entry 8) was somehow lower, compared with that of 2-methyl-1-tetralone (entry 7), but the ee of the major diastereomer 43 remained high (98% ee).
Table 5.
Palladium-Catalyzed Decarboxylative AAA of Substituted Allyl Enol Carbonates.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | time | productsb | yield | ee (dr) | yield and ee (dr) from preformed enolatec |
| 1 | 8 | 4 h | 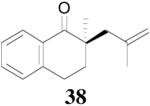 |
89% | >99% ee | 82%, 47% ee |
| 2 | 10 | 2 h | 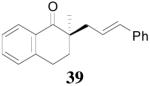 |
99% | 92.5% ee | 11%, NA |
| 3 | 12 | 4h |
40 (E:Z = 11.7:1) : 41 (4:1 dr) = 4: 1 |
94% | 93% ee | - |
| 4 | 13 | 2 h | 40 (E:Z = 20:1) : 41 (7:1 dr) = 7.4: 1 | 89% | 93% ee | 84%, 90% ee (40 : 41 >20:1) |
| 5 | 9 | 13 h |  |
0%e | - | 64%, 13% ee |
| 6d | 9 | 16 h |  |
>95%e | - | |
| 7 | 11 | 4 h | 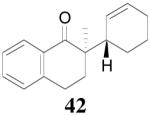 |
62% | 99% ee (20:1 dr) |
- |
| 8 | 14 | 4 h |  |
94% | 98% ee (5:1 dr) |
- |
All reactions were performed on a 0.2 mmol scale at 0.1 M in dioxane at 23 °C for 20 h using 2.5 mol% 20 and 5.5 mol% ligand (R,R)-L4; yields are Isolated yields; ee values were determined by HPLC on a chiral stationary phase.
The absolute and relative stereochemistry were assigned by analogy with compound 24 and 111.
Conditions used were 2.5 mol% [(η3-C3H5)PdCl]2, 5.0 mol% (S,S)-L1, 2.0 equiv LDA, 1.0 equiv Me3SnCl, DME, r.t.10b
The reaction was performed at 60 °C.
Conversion of the starting material by 1HNMR.
To Generate Ketones Possessing an α-Tertiary Center
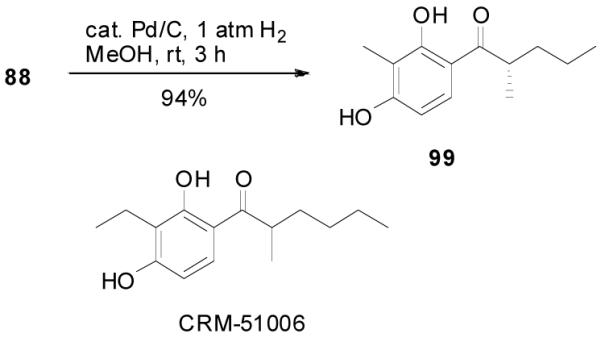
Since the reaction conditions are mild, we attempted to extend the reaction into the asymmetric syntheses of ketones bearing an α-tertiary center anticipating that no racemization of the α-center or dialkylation would happen. Under the optimized conditions described above, the reaction of 44 went to completion to afford R-45 in over 85% ee, albeit in a moderate yield (62%).33 A large portion of 1-tetralone (46, 29%) and a significant amount of 2,2-diallyl-1-tetralone (47, 9%) were obtained as side products (Table 6, entry 1). At lower temperature the yield of 46 increased to 39% and correspondingly the yield of the product decreased to 52% without change of the ee (entry 2). Raising the temperature from 23 to 50 °C in THF decreased the ee and the yield of the desired product and increased the yield of the diallylated product (entry 3 and 4). In the presence of less catalyst (2 mol%), diallylation was more severe; 47 was isolated in 38% yield and the yield of the desired product was only 22% (entry 5). The best results were achieved when the reaction was conducted in dioxane at ambient temperature (entry 6).
Table 6.
Selected Optimization Studies for DAAA of Tri-substituted Enol Carbonates.a
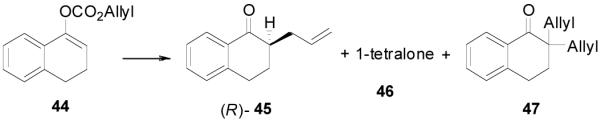 | ||||||
|---|---|---|---|---|---|---|
| entry | T °C | solvent | eeb | 45 | 46 | 47 |
| 1 | 23 | PhMe | 95% | (62)c | (29) c | 9% |
| 2 | 4 | PhMe | 95% | 53(52)c | 36(39)c | 8% |
| 3 | 23 | THF | 94% | 51(51)c | 25(35)c | 5% |
| 4 | 50 | THF | 89% | 46(44)c | 13(20)c | 19% |
| 5d | 50 | THF | 89% | 22(18)c | 7(10)c | 38% |
| 6 | 23 | dioxane | 97% | 81(81)c | 6 | <2% |
All reactions were performed on a 0.3 mmol scale at 0.1 M for 20 h at 23 °C using 2.5 mol% 20 and 5.5 mol% (R,R)-L4; yields were measured by quantitative GC analysis using decane as internal reference.
Ee values were determined by chiral HPLC.
Isloated yields.
1.0 mol% 20 and 5.0 mol% (R,R)-L4 were used.
The reaction scope has also been explored and the results are listed in Table 7. For comparison of this asymmetric process to one using an achiral ligand, the DAA of cyclohexanone with dppe was explored (eq. 10). To our surprise, this reaction didn’t give the desired mono-allylated product 50; instead, 2,2-diallylcyclohexanone 49 was isolated as the major product. However, when L4 was used as ligand there was little diallylated product and the mono-allylated products were isolated in good yields and moderate enantioselectivities (entry 1).34 Generally, better yield was obtained in dioxane than in toluene, but slightly better enantioselectivity was achieved in toluene than in dioxane (entry 1 vs. 2; entry 3 vs. 4 and entry 7 vs. 8). Substrate 51 containing a sulfur atom is compatible with the Pd catalyst and product 52 was obtained in excellent yield and enantioselectivity (entry 6). The positions of substituents on the aryl ring didn’t have a significant effect on the reaction and excellent ee’s were obtained (entry 8 and 9). The ring size of ketones, however, has a significant impact on the enantioselectivity of the reaction. For instance, 2-allylindanone 58 was obtained in lower ee (81% ee, entry 7) than 2-allyl-1-tetralone 45 (97% ee) while 2-allylbenzosuberone 60 was obtained in better ee (99% ee, entry 9).
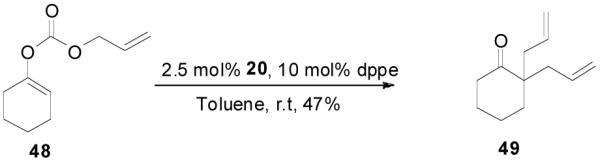 |
(10) |
Table 7.
Summary of the Scope of the Reactiona
| substrat e |
producte | solvent | yieldb | ee | |
|---|---|---|---|---|---|
| 1 | 48 |
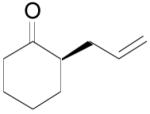 (R)-(+)-50 |
toluene | 78% | 78%c d |
| 2 | 48 | (R)-(+)-50 | dioxane | 90% | 70%c |
| 3 | 51 | 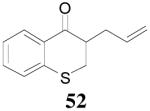 |
toluene | 89% | 93% |
| 4 | 51 | 52 | dioxane | 93% | 87% |
| 5 | 53 | 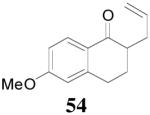 |
dioxane | 90% | >99% |
| 6 | 55 | 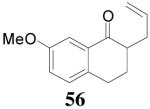 |
dioxane | 97% | 97% |
| 7 | 57 | 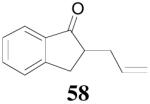 |
toluene | 87% | 81% |
| 8 | 57 | 58 | dioxane | 94% | 73% |
| 9 | 59 | 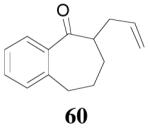 |
dioxane | 93% | 99% |
| 10 | 61 | 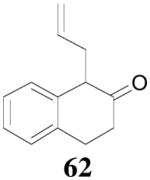 |
toluene | 85% | 0% |
| 11 | 63 | 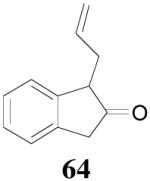 |
toluene | 90% | 0% |
Unless otherwise indicated, all reactions were performed on a 0.3 mmol scale at 0.1 M for 20 h at 23 °C using 2.5% 20 and 5.5% ligand L4 or 10% dppe; the ee values were determined by chiral HPLC.
The yields were isolated yields.
The ee values was determined by the analysis of its derivative by chiral GC.
The [α]D has the same sign as reported for the R configuration.35
Surprisingly, the DAAA of 2-tetralone and 2-indanone gave essentially racemic mono-allylated products 62 and 64 (entry 10 and 11), in stark contrast with the 80% ee obtained in the allylation of 1-methyl-2-tetralone under the same reaction conditions (eq. 10). This is hard to be explained by the slight structural difference between 2-tetralone and 1-methyl-2-tetralone. Instead, racemization through the equilibration of enolates is possibly a more plausible reason for the complete loss of ee in the product. The equilibration of enolates, which was largely avoided in the DAAA of 1-tetralone, was more severe for 2-tetralone, probably because the later was more acidic, thus the ion-pair of its palladium enolate was much looser so that the enolate can leak out more easily from the solvent cage to equilibrate with the product.
Palladium-Catalyzed DAAA of Allyl Enol Carbonates of Acyclic Ketones
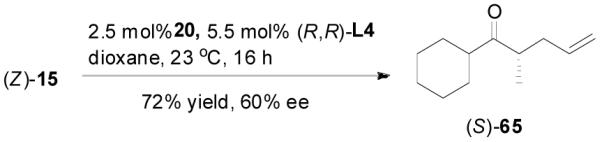
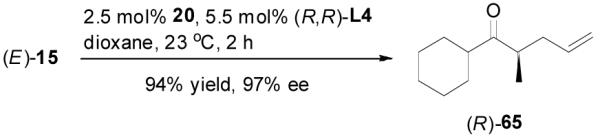
The alkylation of five to seven-membered cyclic ketones is simplified by the fact that the geometry of the enolate is fixed due to the strain in the formation of the Z-cyclic enolate. For acyclic enolates the influence of double bond geometry on the stereochemistry of reactions needs to be considered. Both E and Z isomers of 15 were prepared and submitted to the optimized palladium-catalyzed DAAA conditions. Interestingly, E-15 was more reactive than its isomer and its reaction went to completion in 2 h with 65 being isolated in 94% yield (eq. 11), while Z-15 remained mostly unchanged after 4 h as detected by GC analysis. The product was isolated in a lower yield (72%) after 16 h (eq. 12). In both cases 65 was the only product, however, the sign of its optical rotation was reversed. Similarly the DAAA of 66 with a 5/95 Z/E ratio went to completion in 6 h to afford 67 in quantitative yield and 93% ee (eq. 13), while only a trace amount of product was detected in 16 h in the reaction of 66 with a 94/6 Z/E ratio (eq. 14).
 |
(11) |
 |
(12) |
 |
(13) |
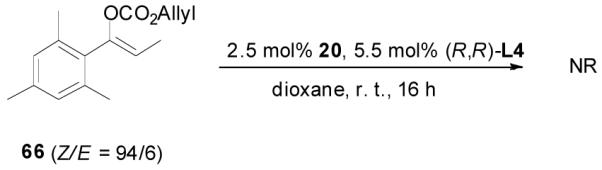 |
(14) |
These results indicated that the possible equilibrium between the E-enolate and the Z-enolate in the presence of a proton source (the product) was avoided and the π-allylpalladium reacted with the enolate from the same face. The different enantioselectivity may be interpreted by models as shown in Figure 3. The E-enolate approaching the π-allylpalladium complex from its Si face (transition state A), with both the cyclohexyl and the methyl groups occupying the “flap” side of the catalyst, should be more favored than that if it reacts from its Re face (B). Thus, R-65 was the predominating enantiomer in the reaction of E-15. In the allylation of the Z-enolate, no matter which side the enolate approaches the π-allylpalladium complex, either the methyl group (C) or the cyclohexyl group (D) has to have some steric interaction with the catalyst. Approach C should be more favored than D, but the energy difference between C and D is smaller than that between A and B, and thus a poorer ee was obtained in the DAAA of Z-15.
Figure 3.

Cartoon Models for the Explanation of the Reactivity and the Selectivity of the E- and Z-15.
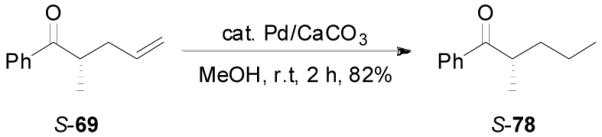
Z-Enol carbonates are easily accessible if one side of the ketone is sterically bulky, such as phenyl ketones. Several such enol carbonates were made by deprotonating ketones with NaHMDS/TMEDA at −78 °C followed by the quenching with allyl chloroformate. All carbonates were isolated with over 98/2 E/Z-selectivity. They were subjected to the palladium-catalyzed DAAA reactions in the presence of L4 ligand in dioxane at ambient temperature and the results are listed in Table 8. Carbonates bearing a small R2 group such as CH3, C2H5, C5H11 and CH2Ph reacted smoothly to generate the corresponding ketones in excellent yields and ee’s (entry 1–4).36 However, the reaction of carbonate 76 with R2 = i-Pr was sluggish and the desired product 77 was isolated in poor yield and ee (30%, 32% ee, entry 5). The absolute configuration of 69 was assigned to be S- by the hydrogenation of 69 to compound 78, which had an optical rotation of +25.7 (EtOH), opposite to the reported optical rotation of R-78 [−15.8 (EtOH), eq. 15].37
Table 8.
DAAA of Allyl Enol Carbonates of Various Phenyl Ketones
 | ||||||
|---|---|---|---|---|---|---|
| entry | R2 | substrate (Z/E) |
product | time | yield | ee |
| 1 | Me |
68 (>98/2) |
S-69 | 3 h | 96% | 94% |
| 2 | Et |
70 (>98/2) |
71 | 2 h | 94% | 94% |
| 3 | n-Pent |
72 (>98/2) |
73 | 16 h | 93% | 92% |
| 4 | Bn |
74 (>98/2) |
75 | 1 h | 75% | 88% |
| 5 | i-Pr |
76 (>98/2) |
77 | 24 h | 30% | 32% |
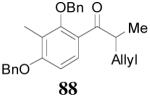
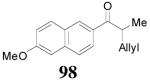
The reaction is compatible with halogen substitutions at either ortho-, meta-, or para- position of the phenyl ring and no significant difference was observed in these reactions compared with that of the unsubstituted one (Table 9, entry 1–3). Remarkably, sterically hindered ortho-substitution on the phenyl ring was tolerable; 86 and 88 were isolated in high yields and excellent ee’s (entry 4 and entry 5). Compound 88 was cleanly converted to 99, an analogue of the anti-tumor compound CRM-51006,38 by catalytic hydrogenation in methanol to saturate the double bond as well as to remove the benzyl groups (eq. 16). Replacement of the electron-rich methoxyl group in 85 with an electron-deficient CF3 group,
 |
(15) |
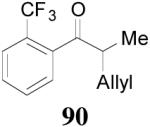
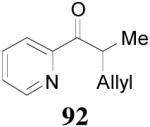
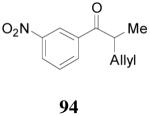
apparently shortened the reaction time to 2 h, possibly by facilitating the decarboxylation; the product 90 was obtained in a slightly lower ee (92%) (entry 6). The substrates possessing a more electron-deficient 2′-pyridyl (91) or 3′-nitrophenyl group (93) were even more reactive, but the corresponding products 90 and 92 were obtained in lower ee’s (73% ee and 83% ee respectively, entry 7 and 8). However, the reaction of substrate 95 bearing a more electron-rich but smaller 2′-furyl ring didn’t give better enantioselectivity (88% ee, entry 9) than that of 69 (94% ee, entry 1), implying the electronic property of the enolate may not be the only factor influencing the enantioselectivity of the reaction.
Table 9.
DAAA of Allyl Enol Carbonates of Various Acyclic Aryl Ethyl Ketones
 | |||||
|---|---|---|---|---|---|
| entry | substrate | product | time | yield | ee |
| 1 |
79 (>98/2) |
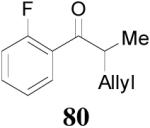 |
1 h | 80% | 94% |
| 2 |
81 (>98/2) |
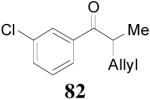 |
1 h | 97% | 93% |
| 3 |
83 (>98/2) |
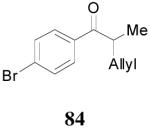 |
1 h | 94% | 93% |
| 4 |
85 (>98/2) |
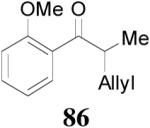 |
16 h | 99% | 98% |
| 5 |
87 (>98/2) |
 |
3 h | 78% | 94% |
| 6 |
89 (>98/2) |
 |
2 h | 94% | 92% |
| 7 |
91 (>98/2) |
 |
1 h | 95% | 73% |
| 8 |
93 (>98/2) |
 |
1 h | 83% | 82% |
| 9 |
95 (>98/2) |
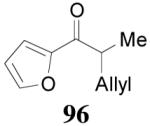 |
4 h | 89% | 88% |
| 10 |
97 (>98/2) |
 |
1 h | 90% | 95% |
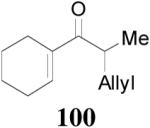
Beside aryl ketones, the DAAA reactions of alkenyl ketones were also investigated. Enol carbonate Z-2 was prepared in the same way as aryl ketone enol carbonates. Its DAAA reaction under the standard conditions went to completion in 5 h to afford ketone 100 in 94% yield and 88% ee (Table 10, entry 1). Comparison of the enantioselectivity in the DAAA of 68 (94% ee, Table 8, entry 1), Z-2 (88% ee) and Z-15 (60% ee, Scheme 4) may suggest that the enantio-recognition step involves not only steric effects of R1 and R2, but also some electronic effects such as π-stacking interactions between the substrate and the ligand. The higher reactivity of aryl enol carbonate and alkenyl enol carbonate than alkyl enol carbonate indicates that the decarboxylation step is probably the rate determining step since the lower pKa of the aryl and alkenyl ketones (presumably the inductive effect of sp2 substituents) should facilitate the decarboxylation. Similarly the reaction of enol carbonate 101, obtained as a 25/1 Z/E isomer mixture, reached full conversion in 15 min to form the corresponding product 102 in
 |
(16) |
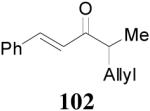
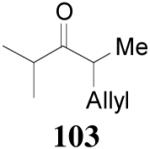
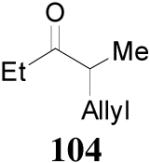
93% yield and 91% ee (entry 2). The DAAA of Z-enol carbonate of alkyl ketones was less successful. The reaction of 16 (Z/E > 98:2) went to completion in 16 h, detected by GC analysis; however, the ee of the product 103 was only moderate (46% ee) (entry 3). Fortunately, this limitation was overcome by the use of the E-isomer; the ee jumped to 94% (entry 4). Similarly, excellent conversions and good ee’s of the E-enol isomers were obtained when R1 was as simple as an ethyl group (17, entry 5), or when R1 was a sterically unhindered linear alkynyl group (18, entry 6).
Table 10.
DAAA of Allyl Enol Carbonates of Various Acyclic Alkyl, Alkenyl and Alkynyl Ketones
 | |||||
|---|---|---|---|---|---|
| entry | Substrate (Z/E) |
product | time | yield | ee |
| 1 |
2 (>98/2) |
 |
5 h | 94% | 88% |
| 2 |
101 (25/1) |
 |
0.3h | 93% | 91% |
| 3 |
16 (>98/2) |
 |
16 h | 99%a | 46% |
| 4 |
16 (1/23) |
103 | 16 h | 99%a | 94% |
| 5 |
17 (1/18) |
 |
16 h | 99%a | 88% |
| 6 |
18 (1/14) |
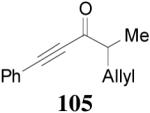 |
2 h | 91% | 81% |
Conversion based on GC analysis.
Scheme 4.

The Crossover Experiment Catalyzed by Pd/L4, PHOX-i-Pr or dppe Ligand.
Mechanism Studies of Palladium-Catalyzed DAAA Reaction of Enol Carbonates
The highly conserved regioselectivity in the decarboxylative alkylation of enol carbonates suggests that proton-shuffling between the product and the palladium enolate intermediate is negligible. However, the crossover experiment conducted by Stoltz et. al. showed a complete scrambling of the allyl and the ketone moieties.39 To rule out that this is not specific to certain substrates, certain ligands or certain solvents, we performed a similar crossover experiment using deuterium labeled allyl enol carbonate of 2-methyltetralone 106 and 107 in the presence of L4, PHOX-i-Pr or dppe as ligand in dioxane (Scheme 4). The reaction went to completion within 5 min in all cases. Mass spectral analysis of the products from each reaction clearly showed that four peaks (m/z 200, 202, 203 and 205) of nearly equal intensity were found implying a complete scrambling of the six possible products. This experiment confirmed complete crossover occurred. Such scrambling suggests that any ion-pair undergoes ion exchange faster than collapse to form products.
If the scrambling in the above crossover experiment occurred at the stage of palladium enolate, which means that palladium enolates are completely charge-separated, then, it is hard to explain why the decarboxylative alkylation reactions of enol carbonates are highly regioselective and the possible diallylation is largely minimized compared with the alkylation of preformed lithium enolates. If the palladium enolate is completely charge-separated, in the presence of an acidic additive the basic “naked” enolate should be protonated by the additive. To regenerate the Pd(0) catalyst, the anion of the additive is required to react with the π-allylpalladium and to simplify the reaction, the alkylation of the additive is best if made irreversible. Taking into account the convenience to tune the acidity of the additive, we decided to use 1,3-dicarbonyl compounds as additives. The pKa of a simple ketone such as propiophenone is about 24.4 in DMSO, and the pKa of dimethyl methylmalonate is about 18 in DMSO, so it is acidic enough to protonate the enolate of propiophenone. We subjected enol carbonate 68 to the DAAA reaction in the presence of one equivalent of dimethyl methylmalonate and found no change of the reaction (Table 11, entry 2 vs entry 1). In the presence of one equivalent of the more acidic dimethylmalonate (pKa = 15.9), there was a slight increase in the amount of the protonated product 108 together with the same amount of 109 (entry 3). The addition of one equivalent of acetoacetone (pKa = 13.3), eventually produced a large amount of proton transfer products; the yield of propiophenone was increased to 83% and allyl acetoacetone was obtained in 91% yield (entry 4). With the other conditions unchanged, the proton-transfer process was more severe in THF than in dioxane (entry 5 vs 3 and entry 6 vs. 4).
Table 11.
The Acidic Additive Effect in the Palladium-Catalyzed DAAA Reaction
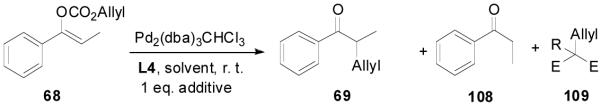 | ||||||
|---|---|---|---|---|---|---|
| solvent | additive | Time min |
69% (ee%) |
108 % |
109 % |
|
| 1 | dioxane | none | 120 | 94 (94) |
6 | - |
| 2 | dioxane | MeCH(CO2Me)2 | 30 | 94 (94) |
6 | 0 |
| 3 | dioxane | CH2(CO2Me)2 | 15 | 90 (93) |
10 | 9 |
| 4 | dioxane | CH2(COMe)2 | 15 | 17 (81) |
83 | 91 |
| 5 | THF | CH2(CO2Me)2 | 10 | 71 (87) |
27 | 22 |
| 6 | THF | CH2(COMe)2 | <5 | 8 (83) |
92 | 76 |
There lacks pKa values of enol carbonates in DMSO, however, the pKa of HCO3− is 10.3 in water, which is lower than that of dimethyl malonate (pKa = 12.9 in water) but higher than that of acetoacetone (pKa = 9.0 in water). Therefore, it is probable that the protonation didn’t occur on the enolate but on the carbonate. The carbonate anion is more stabilized than the enolate anion due to the delocalization of the anion on two oxygen atoms, so the palladium carbonate should be more charge separated than that of the palladium enolate. It is probably also the stage that scrambling of the electrophile and nucleophile may occur.
There are two possible mechanistic pathways for the recombination of the π-allylpalladium complex and the enolate. One is an “inner sphere” mechanism: the nucleophile coordinates to the metal followed by a bond-forming reductive elimination (Scheme 5, A). The other mechanism is an “outer sphere” one: the nucleophile directly adds to one of the carbons of the allyl moiety and substitutes the Pd complex (Scheme 5, B). Arbitrarily, “soft nucleophiles” with pKa < 20 favored the “outer sphere” pathway and “hard nucleophiles” with pKa > 20 favored the “inner sphere” pathway.40 Previous stereochemistry studies suggested that unstabilized ketone enolates, including potassium,41 lithium,42 tin,43 zinc,44 and boron enolates,44 attack π-allylpalladium complexes through the “outer sphere” mechanism. Recently, Braun et. al. also proved that the palladium-catalyzed allylic alkylation of unstabilized lithium enolate was a net retention-of-configuration process favoring the “outer sphere” mechanism.12 In the palladium-catalyzed DAAA of enol carbonates, the palladium enolate species are formed in situ as nucleophiles and presumably they may behave similarly as other metal enolates to attack π-allylpalladium through an “outer sphere” pathway. On the other hand, since there is no other counter cation, it is quite reasonable to believe that the palladium enolate may covalently bond or at least form a contact ion-pair, especially in non-polar solvents such as toluene. This was implied by Tsuji’s early work, which showed that the major competitive pathway of the decarboxylative allylic alkylation of enol carbonates was the formation of conjugated enone, which was proposed involving a β-hydride elimination of the covalently bonded palladium enolate.45 Stoltz and Goddard et. al. recently reported a theoretical calculation on the mechanism of palladium-catalyzed DAAA reaction in the presence of PHOX-t-Bu ligand. The authors claimed that the penta-coordinated π-allylpalladium enolate complex was as stable as the charge separated palladium enolate and the product was formed by the reductive elimination of the η1-allylpalladium enolate complex through a seven-membered ring transition state.46
Scheme 5.
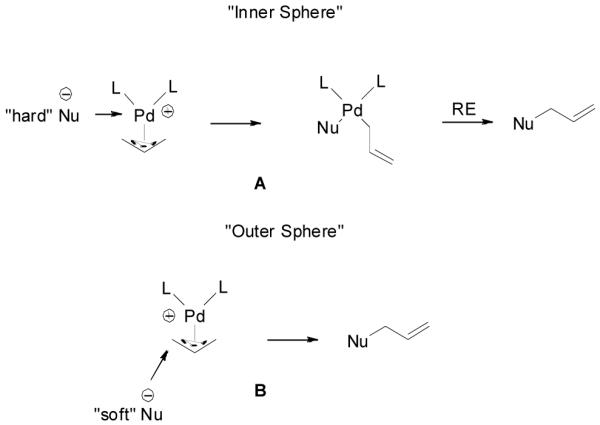
“Inner Sphere” and “Outer Sphere” Nucleophilic Addition to π-Allylpalladium.
Although it is highly likely that the π-allylpalladium complex is in some degree covalently bonded with the enolate, it does not necessarily mean that the covalently bonded palladium enolate is involved in the transition state of the alkylation step. To shed some light on this question, we synthesized enol carbonate 110 and subjected it to the “standard” conditions we used in the palladium-catalyzed DAAA reactions (Pd2dba3CHCl3, L4, dioxane, rt). A nearly perfect kinetic resolution was observed; one enantiomer of 110 reacted rapidly within 1 h to afford the single diastereomer 111 in 39% yield (78% based on 50% theoretical yield) and 99% ee (Scheme 6). The other enantiomer was not touched even after 12 h at rt and it was recovered in 37% yield (74% based on 50% theoretical yield) and 99.5% ee. A small amount of O-alkylation product 112 was also isolated (6%). The relative stereochemistry of 111 was assigned by 1HNMR and X-ray crystallography (Figure 4). The tetralone moiety and the phenyl group were cis. Since the ionization of 110 should most likely follow the same anti-addition mechanism as other allyl carbonates, the retention of configuration of the product strongly suggests that the alkylation step should also be an anti-nucleophilic addition. Thus, the alkylation is an “outer-sphere” process, not an “inner-sphere” one.
Scheme 6.

Palladium-Catalyzed DAAA Reaction of cis-110.
Figure 4.
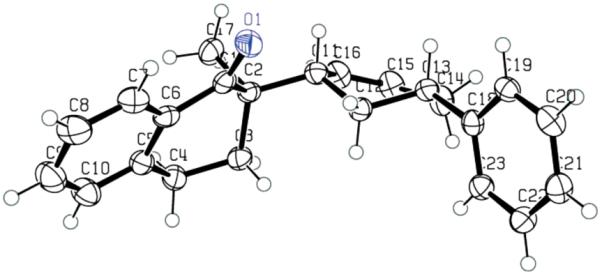
The X-ray Structure of 111.
Similar results were observed in the reaction of the trans-isomer 114; only the trans-diastereomer 113 was obtained. Compared with the reaction of 110, the reaction of 114 was significantly slower and was stopped after 8 h. Besides, a significant amount of the O-alkylation product 116, 2-methyl-1-tetralone (115) was also isolated. The reaction rates of the two enantiomers of 114 were also significantly different so that only one reacted in the given reaction time (eq. 17).
 |
(17) |
As a comparison, the preformed lithium enolate prepared by the deprotonation of 2-methyl-1-tetralone (115) by LDA was reacted with allyl acetate 117 in the presence of the same palladium catalyst (eq. 18). Only the cis-product 111 was obtained in 78% yield and 87% ee. This is in agreement with Braun’s observation that the addition of lithium enolate to π-allylpalladium was an “outer sphere” anti-attack.11 Interestingly, in contrast to the reaction of enol carbonate 110, the opposite enantiomer (-)-111 was obtained employing the same enantiomer of ligand L4.
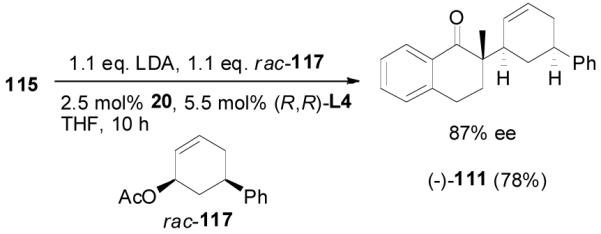 |
(18) |
Based on the above information, we proposed a mechanism as shown in Scheme 7. After the initial ionization, a Pd carbonate intermediate is formed. The decarboxylation step is probably the rate-determining step especially for simple unsubstituted allyl enol carbonates of sterically less hindered ketones. Since the carbonate anion is stabilized by delocalization, it is probably at this stage scrambling of the π-allylpalladium cation and the carbonate anion as well as protonation of the carbonate by an acidic proton source may occur. The decarboxylation must be assisted by the Pd complex;15 therefore, the enolate is generated in the presence of a π-allylpalladium complex very close to it. Since there is no other counter-cation in the system, the unstabilized enolate anion and the cationic π-allylpalladium complex probably form a tight ion-pair caged by solvent molecules. This may explain why less proton-transfer related by-products were found in dioxane than in THF, since dioxane is better at stabilizing caged ion-pairs.47 The palladium enolate ion-pair probably also equilibrates with the covalently bonded palladium enolate, but the alkylation occurs only through an “outer sphere” SN2 attack of the enolate to the π-allylpalladium complex.
Scheme 7.

A Plausible Mechanism for Palladium-Catalyzed DAAA of Enol Carbonates.
Conclusion
In the presence of enantiopure chiral ligand L4, palladium-catalyzed DAAA of enol carbonates is a quite general method for the asymmetric synthesis of cyclic and acyclic ketones with either a quaternary or a tertiary α-center. The reaction conditions are extremely mild; thus, it has quite broad functional group compatibility. Compared with the allylic alkylation of preformed lithium enolates, the nucleophile is structurally much simpler since there is no other counterion in the reaction. This makes the DAAA reaction much more tolerant to the structural diversity of the substrate. As a result, excellent yield and enantioselectivity were achieved in a broad range of substrates. Our studies suggested that solvent plays important roles in this reaction; for reactions to generate ketones bearing quaternary chiral centers, the non-polar solvent toluene is the solvent of choice leading to the highest enantioselectivity. For reactions to generate ketones with a tertiary chiral center, dioxane is the best solvent to minimize the equilibrium between the product and the enolate intermediate, and hence minimize the protonated and the diallylated side products.
The DAAA of the enol carbonates of aromatic ketones is faster than that of aliphatic ketones, presumably because the pKa of aromatic ketones is 2 or 3 units lower than the aliphatic ketones and thus decarboxylation of conjugated ketones is faster. The opposite enantiomers were obtained in the reaction of the E-enolate and the Z-enolate. This suggests that there is no equilibrium between the E-enolate and the Z-enolate through the C-bound palladium enolate. We have demonstrated that the E-enolate is more reactive than the corresponding Z-enolate and better enantioselectivity can be obtained in the allylation of the E-enolate as well. For a ketone with a big R1 group and a small R2 group, the Z-enol carbonate is more readily prepared. In this case, good enantioselectivity can be obtained since most important factor is the interaction between R1 and the chiral catalyst. For a ketone possesses a small R1 group and a small R2 group, the E enol carbonates can be readily synthesized by the use of LiTMP-LiBr as base. Generally high enantioselectivities were achieved for the allylation of those ketones via their E-enol carbonates.
Mechanistic studies revealed that the palladium enolate forms a tight ion pair in less polar solvents, such as toluene and dioxane. The scrambling of the enolate moiety and the allyl moiety before the alkylation step probably occurs at the palladium carbonate stage, not at the palladium enolate stage. The C-C bond formation is more likely an “outer sphere” SN2 substitution than the alternative “inner sphere” reductive elimination of the palladium enolate.
Experimental Section
All reactions were carried out under an atmosphere of nitrogen or argon in oven-dried glasswares with magnetic stirring, unless otherwise indicated. Tris(dibenzylideneacetone)palladium(0) monochloroform complex, Pd2dba3·CHCl3 was prepared by the procedure of Ibers.48 Ligands were prepared by literature procedures.49 All other reagents were used as obtained unless otherwise noted. Flash Chromatography was performed with EM Science silica gel (0.040–0.063μm grade).1,4-Dioxane for palladium-catalyzed reactions were freshly distilled from sodium. Other solvents were dried by J. C. Meyer’s Solvent Purification System. Proton nuclear magnetic resonance (1H-NMR) data were acquired on a Mercury 400 (400 MHz) or on a Varian Unity Inova-500 (500 MHz) spectrometer. Chemical shifts are reported in delta (δ) units, in parts per million (ppm) downfield from tetramethylsilane. Carbon-13 nuclear magnetic resonance (13C-NMR) data were acquired at 100 MHz on a Mercury 400 or at 125 MHz on a Varian Unity Inova 500 spectrometer. Chemical shifts are reported in ppm relative to the center line of a triplet at 77.1 ppm for chloroform-d. Infrared (IR) data were recorded as films on sodium chloride plates on a Perkin-Elmer Paragon 500 FT-IR spectrometer. Absorbance frequencies are reported in reciprocal centimeters (cm−1). Elemental analyses (Anal.) were performed by M.-H.-W. Laboratories of Pheonix, AZ. Chiral HPLC analyses were performed on a Themo Separation Products Spectra Series P-100 or 200 and UV100 (254 nm) using Chiralcel® columns (OD-H, AD-H, IA, IB or IC) eluting with heptane / iso-propanol mixtures indicated. Optical rotations were measured on a Jasco DIP-1000 digital polarimeter using 5 cm cells and the sodium D line (589 nm) at ambient temperature in the solvent and concentration indicated.
Typical Procedure for the Preparation of the Kinetically Controlled Z-Allyl Enol Carbonates
(Z)-Allyl 1-cyclohexenyl-1-propenyl carbonate (2)
To a clean dry 100 mL flask with a magnetic stirring bar was loaded sodium hexamethyldisilazane (Aldrich, 97%, 0.86 g, 4.5 mmol) stored in a glove box under nitrogen. The flask was cooled in a dry-ice-acetone bath for 5 min and THF (20 mL) was transferred to this flask with a syringe. The suspension was warmed to 0 °C with stirring to make a clear solution and TMEDA (freshly distilled from calcium hydride, 0.68 mL, 4.5 mmol) was added. The mixture was cooled to −78 °C again and a solution of 1-cyclohexenyl-1-propanone (1, 520 mg, 3.76 mmol) in 5 mL of THF was added slowly over 5 min. The reaction mixture was stirred for 1 h, and it was transferred through a cannula into a solution of allyl chloroformate (0.48 mL, 4.5 mmol) in 5 mL of THF at −78 °C. The reaction was stirred for 5 min and it was quenched with saturated aqueous ammonium chloride solution (10 mL). The mixture was extracted with diethyl ether (20 mL) twice. The combined organic layer was washed with brine (20 mL), then it was dried over anhydrous magnesium sulfate. After filtration and concentration in vacuo, the residue was purified by column chromatography on silica gel to afford the title compound (0.70 g, 84%) after purification by flash column chromatography eluting with 5% diethyl ether in petroleum ether. Colorless oil; Rf = 0.41 (Diethyl ether/petroleum ether 1:9); IR (film): , 2931 (s), 2861 (s), 1760 (s), 1664 (m), 1448 (s), 1364 (s), 1304 (s), 1248 (s), 1216 (s), 977 (s), 928 (s), 805 (s), 781 cm−1 (s); 1H NMR (400 MHz, CDCl3): δ= 5.97 (ddt, J1 = 17.1 Hz, J2 = 10.4 Hz, J3 = 5.8 Hz, 1H), 5.87 (bs, 1H), 5.40 (dq, J1 = 17.2 Hz, J2 = 1.4 Hz, 1H), 5.35 (qd, J1 = 7.0 Hz, J2 = 0.8 Hz, 1H), 5.30 (dq, J1 = 10.5 Hz, J2 = 1.2 Hz, 1H), 4.69 (dt, J1 = 5.8 Hz, J2 = 1.4 Hz, 2H), 2.13 (m, 4H), 1.71–1.54 (m, 4H), 1.64 (dd, J1 = 7.2 Hz, J2 = 0.6 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ= 153.1, 148.7, 131.7, 130.4, 123.7, 119.2, 110.2, 69.0, 25.4, 24.8, 22.6, 22.2, 11.2. Anal. Calcd. for C13H18O3: C, 70.24; H, 8.16; Found: C, 70.50; H, 7.97.
Typical Procedure for the Preparation of the Kinetically Controlled E-Allyl Enol Carbonates
Allyl (E)-pent-2-en-3-yl carbonate (E-17)
To a clean dry 100 mL flask was loaded TMP-HBr (500 mg, 2.25 mmol, 1.3 eq) and 25 mL of DME. To this rapidly stirred suspension at 0°C and under nitrogen was added a 2.4 M solution of n-BuLi/hexanes (1.70 mL, 4.15 mmol, 2.4 eq) dropwise over a 3-min period. After being stirred for additional 15 min, the resulting pale yellow solution was cooled to −78°C and a solution of 3-pentanone (153 mg, 1.78 mmol, 1 eq) in 1.5 mL of DME was added dropwise. After 15 min, TMEDA (0.62 mL, 4.15 mmol, 2.4 eq) was added dropwise over a 3-min period. After another 15 min, a pre-cooled (−78°C) solution of allylchloroformate (241 mg, 2.00 mmol, 1.1 eq) in 1.4 mL of DME was added at −78°C. After 30 min at −78°C the reaction was removed from the bath and was allowed to warm to room temperature. The reaction was quenched with 5% KH2PO4 aqueous solution and transferred into a separatory funnel. It was extracted twice with 25 mL of diethyl ether. The combined organic layer was washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was purified by silica gel column chromatography eluting with 2–3% diethyl ether in petroleum ether to afford 265 mg of colorless oil (90%). The E/Z ratio was determined by 1H-NMR to be 18:1 [by the signal of the CH3 at δ= 1.64 (d, J = 7.2 Hz, major) and 1.59 (dt, J = 6.9, 1.4 Hz, minor)]. Rf = 0.35 (diethyl ether/ petroleum ether 1:9); IR (film): , 1756 (s), 1694 (m), 1459 (m), 1386 (m), 1365 (m), 1276 (s), 1245 (s), 1167 (s), 1031 (w), 983 (m), 942 (m), 916 (m), 783 (w), 734 cm−1 (m); 1H NMR (500 MHz, CDCl3): δ= 5.95 (ddt, J1 = 17.2 Hz, J2 = 10.5 Hz, J3 = 5.7 Hz, 1H), 5.37 (dq, J1 = 17.2 Hz, J2 = 1.4 Hz, 1H), 5.28 (dq, J1 = 10.5 Hz, J2 = 1.1 Hz, 1H), 5.24 (q, J = 7.2 Hz, 1H), 4.64 (m, 2H), 2.31 (q, J = 7.6 Hz, 2H), 1.64 (d, J = 7.2 Hz, 3H), 1.04 (d, J = 7.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ= 153.8, 150.9, 131.5, 119.1, 111.8, 68.7, 21.8, 11.5, 11.4. The structure as further confirmed by NOE experiment.
Typical Procedure for the Preparation of the Thermodynamically Controlled Allyl Enol Carbonates
Allyl 2-phenylcyclohex-1-enyl carbonate (4)
To a clean dry 50 mL three-neck flask with a magnetic stirring bar and a refluxing condenser was loaded a suspension of sodium hydride (60% in mineral oil, 0.44 g, 11.0 mmol) and was purged with nitrogen. The sodium hydride was washed three times with anhydrous hexane under nitrogen. Freshly distilled TMEDA (1.51 mL) and 5 mL of anhydrous THF were added to the flask and the suspension was added to a solution of 2-phenylcyclohexanone (1.74 g, 10.0 mmol) in 5 mL of THF at room temperature. The mixture was heated to reflux for 1 h. After the enolate solution was cooled by an ice-water bath for 5 min, it was transferred through a cannula into a solution of allyl chloroformate (1.07 mL, 10 mmol) in 5 mL of THF at 0 °C. After stirring for another 15 min, saturated aqueous ammonium chloride (10 mL) was poured into the reaction flask. The mixture was extracted with diethyl ether (20 mL) twice. The combined organic layers were dried over anhydrous magnesium sulfate. After filtration and concentration the residue was purified by column chromatography eluting with 2% diethyl ether in petroleum ether to afford 1.87 g of the title compound (72%). Colorless oil; Rf = 0.37 (Diethyl ether/petroleum ether 1:9); IR (film): , 1750 (s), 1444 (m), 1367 (m), 1238 (s), 1178 (m), 1036 cm−1 (s); 1H NMR (400 MHz, CDCl3): δ = 7.4–7.2 (m, 5H), 5.78 (ddt, J = 17.2, 10.4, 5.5 Hz, 1H), 5.20 (ddd, J = 11.3, 1.5, 1.5 Hz, 1H), 5.17 (ddd, J = 4.6, 1.5, 1.5 Hz, 1H), 4.51 (dt, J = 5.7, 1.5 Hz, 2H), 2.42 (m, 2H), 2.33 (m, 2H), 1.84 (m, 2H), 1.76 (m, 2H); 13C NMR (100 MHz, CDCl3): δ = 152.9, 143.5, 138.9, 131.4, 128.2, 127.7, 126.9, 126.0, 118.6, 68.5, 30.2, 27.2, 22.9, 22.6; HRMS (EI): [M]+ calcd. For C16H18O3, 258.1256; Found: 258.1257.
Typical Procedure for the Preparation of the Substituted Allyl Enol Carbonates
2-Methylprop-2-en-1-yl 1H-imidazole-1-carboxylate
To a clean oven-dried 250 mL flask with a magnetic stirring bar was charged with 1,1′-carbonyldiimidazole (4.86 g, 30.0 mmol) and 100 mL THF under nitrogen. The flask was cooled in an ice-water bath. A solution of 2-methyl-prop-2-en-1-ol (1.44 g, 20.0 mmol) in 30 mL methylene chloride was added slowly and stirred for 2 h. Most solvent was removed in vacuo by a rotaevaporator and the crude product was purified by silica gel column chromatography eluted with 1:1 ethyl acetate/petroleum ether to afford 3.23 g of the title compound as a white solid (97%). M.p. = 36–38 °C; Rf = 0.18 (30% ethyl acetate in petroleum ether); IR(neat): ; 1H-NMR (400 MHz, CDCl3): 8.17–8.11 (m, 1H), 7.43 (dd, J = 3.5, 2.1 Hz, 1H), 7.07 (dd, J = 1.5, 0.9 Hz, 1H), 5.08 (m, 1H), 5.04 (m, 1H), 4.80 (s, 2H), 1.82 (m, 3H); 13C NMR (100 MHz, CDCl3): δ = 148.5, 138.3, 137.1, 130.7, 117.1, 114.9, 71.2, 19.4. HRMS (EI): M+ calcd. for C8H10N2O2 166.0742, found 166.0737.
2-Methyl-3,4-dihydronaphthalen-1-yl 2-methylprop-2-en-1-yl carbonate (8)
To the solution of NaHMDS (460 mg, 2.51 mmol) in 5 mL of DME at −78 °C was added 2-methyl-1-tetralone (320 mg, 2.0 mmol) in 2 mL of DME. The solution was stirred at −78 °C for 30 min. Meanwhile, another clean oven-dried 50 mL flask was charged with 2-methylprop-2-en-1-yl 1H-imidazole-1-carboxylate (399 mg, 2.0 mmol) and 5 mL DME. The solution cooled to −78 °C was added boron trifluoride etherate (0.30 mL, 2.4 mmol). The solution was transferred into the solution of enolate through a cannula under nitrogen and stirred for 30 min. One portion of 10 mL saturated aqueous ammonium chloride was poured into the reaction mixture followed by 10 mL diethyl ether. The mixture was taken out from the bath and allowed to warm to ambient temperature. The organic layer was separated and the aqueous layer was extracted once with 10 mL diethyl ether. The organic layer was combined and dried over magnesium sulfate. After filtration and concentration in vacuo the crude material was purified by silica gel column chromatography eluted with 10% diethyl ether in petroleum ether to afford the title compound as colorless oil (472 mg, 92%). Rf = 0.34 (Diethyl ether/petroleum ether 1:9); IR (film): 1760 cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.20–7.07 (m, 4H), 5.08 (m, 1H), 4.99 (m, 1H), 4.64 (m, 2H), 2.86 (t, J = 8.0 Hz, 2H), 2.39 (dt, J = 8.1, 1.0 Hz, 2H), 1.83 (t, J = 1.0 Hz, 3H), 1.80 (m, 3H); 13C NMR (100 MHz, CDCl3): δ = 153.2, 140.7, 139.3, 135.3, 130.9, 127.4, 127.1, 126.5, 124.5, 120.0, 113.7, 71.7, 29.0, 27.5, 19.4, 16.6. Anal. Calcd. for C16H18O3: C, 74.39; H, 7.02; Found: C, 74.50; H, 7.12.
Typical Procedure for the Palladium-Catalyzed DAAA Reaction of Allyl Enol Carbonates
2-Methyl-2-(2-methylallyl)-3,4-dihydronaphthalen-1(2H)-one (38)
Two clean oven-dried test tubes were individually charged with a magnetic stirring bar. One test tube was loaded with Pd2(dba)3CHCl3 (5.2 mg, 0.0075 mmol) and (R,R)-L4 ligand (9.2 mg, 0.0165 mmol); the other one was loaded with 8(52 mg, 0.20 mmol). They are sealed and connected with a double-end needle. The system was evacuated and flushed with argon three times at which point 1.0 mL of degassed anhydrous dioxane was added to both of the test tubes. After stirring for 20 min, the orange solution containing the catalyst was transferred into the test tube containing the carbonate substrate. The color of the reaction solution turned to light yellow immediately, and it turned back to orange within 4 h. The reaction mixture was concentrated in vacuo and the residue was purified by column chromatography to afford 38 mg of the title compound as a colorless oil (89%). Chromatography on silica gel, eluent: 3% diethyl ether in petroleum ether. Rf = 0.44 (Diethyl ether/petroleum ether 1:9); Colorless oil; IR (film): 1682 cm−1; [α]D25 = +45.3 (c = 1.77, CH2Cl2, >99% ee); HPLC (Chiralcel® AD-H column; 2000:1 Heptane / Isopropanol; flow rate = 1 mL of / min; t1 = 15.1 min (minor), t2 = 17.6 min (major)); 1H NMR (400 MHz, CDCl3): δ = 8.04 (m, 1H), 7.46 (dt, J =7.5, 1.5 Hz, 1H), 7.30 (m, 1H), 7.24 (m, 1H), 4.83(m, 1H), 4.68 (m, 1H), 2.99 (t, J = 6.5 Hz, 2H), 2.66 (dd, J = 13.5, 0.7 Hz, 1H), 2.23 (dd, J = 13.5, 0.7 Hz, 1H), 2.10 (ddd, J = 13.9, 6.8, 6.8 Hz, 1H), 1.88 (ddd, J = 13.9, 5.8, 5.8 Hz, 1H), 1.65 (m, 3H), 1.20 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 202.2, 143.2, 142.5, 133.0, 131.8, 128.6, 128.1, 126.7, 114.9, 45.0, 44.7, 33.6, 25.5, 24.5, 23.3. Anal. Calcd. for C15H18O: C, 84.07; H, 8.47; Found: C, 84.32; H, 8.55.
Supplementary Material
Acknowledgement
We thank the National Science Foundation and the National Institutes of Health, General Medical Sciences Grant GM13598, for their generous support of our programs. Mass spectra was provided by the Mass Spectrometry Regional Center of the University of California–San Francisco supported by the NIH Division of Research Resources. We thank Chirotech (now Dow) for their generous gifts of ligands and Johnson Matthey for gifts of palladium salts.
Footnotes
Supporting Information Available. Crystallographic data for compound 111 in cif format, experimental procedures and characterization data for all new compounds in pdf format. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Otera J. Modern Carbonyl Chemistry. Wiley-VCH; New York: 2000. [Google Scholar]; (b) Caine D. In: Comprehensive Organic Synthesis: Carbon-Carbon σ-Bond Formation. Trost BM, Ian Fleming, editors. Vol. 3. Pergamon; New York: 1991. [Google Scholar]
- 2.Seyden-Penne J. Chiral Auxiliaries and Ligands in Asymmetric Synthesis. John Wiley & Sons, Inc.; New York: 1995. [Google Scholar]
- 3.(a) Enders D. In: Asymmetric Synthesis: Stereodifferentiating Addition Reactions, Part B. Morrison JD, editor. Vol. 3. Academic; Orlando: 1984. p. 275. [Google Scholar]; (b) Seyden-Penne J. Chiral Auxiliaries and Ligands in Asymmetric Synthesis. John Wiley & Sons, Inc.; New York: 1995. [Google Scholar]; (c) Job A, Janeck CF, Bettray W, Peters R, Enders D. Tetrahedron. 2002;58:2253. [Google Scholar]; (d) Enders D, Voith M, Ince SJ. Synthesis. 2002:1775. [Google Scholar]
- 4.Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 5.Hughes DL. In: Comprehensive Asymmetric Catalysis I-III. Jacobsen EN, Pfaltz A, Yamamoto H, editors. III. Springer; Berlin: 1999. [Google Scholar]
- 6.(a) Hashimoto T, Maruoka K. Chem. Rev. 2007;107:5656. doi: 10.1021/cr068368n. [DOI] [PubMed] [Google Scholar]; (b) Ooi T, Maruoka K. Angew. Chem., Int. Ed. 2007;46:4222. doi: 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]; (c) Maruoka K. Asymmetric Phase Transfer Catalysis. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2008. [Google Scholar]
- 7.(a) Murakata M, Nakajima M, Koga K. J. Chem. Soc.-Chem. Commun. 1990:1657. [Google Scholar]; (b) Imai M, Hagihara A, Kawasaki H, Manabe K, Koga K. J. Am. Chem. Soc. 1994;116:8829. [Google Scholar]; (c) Yamashita Y, Odashima K, Koga K. Tetrahedron Lett. 1999;40:2803. [Google Scholar]
- 8.(a) Vignola N, List B. J. Am. Chem. Soc. 2004;126:450. doi: 10.1021/ja0392566. [DOI] [PubMed] [Google Scholar]; (b) Mukherjee S, List B. J. Am. Chem. Soc. 2007;129:11336. doi: 10.1021/ja074678r. [DOI] [PubMed] [Google Scholar]; (c) Sato T, Tomioka K. Heterocycles. 2009;77:587. [Google Scholar]; (d) Beeson TD, Mastracchio A, Hong JB, Ashton K, Macmillan DWC. Science. 2007;316:582. [PubMed] [Google Scholar]; (e) Jang HY, Hong JB, Macmillan DWC. J. Am. Chem. Soc. 2007;129:7004. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]; (f) Kim H, Macmillan DWC. J. Am. Chem. Soc. 2008;130:398. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]; (g) Nicewicz DA, Macmillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Doyle AG, Jacobsen EN. J. Am. Chem. Soc. 2005;127:62. doi: 10.1021/ja043601p. [DOI] [PubMed] [Google Scholar]; (b) Doyle AG, Jacobsen EN. Angew. Chem., Int. Ed. 2007;46:3701. doi: 10.1002/anie.200604901. [DOI] [PubMed] [Google Scholar]
- 10.(a) Trost BM, Schroeder GM. J. Am. Chem. Soc. 1999;121:6759. [Google Scholar]; (b) Trost BM, Schroeder GM. Chem. Eur. J. 2005;11:174. [Google Scholar]
- 11.(a) You SL, Hou XL, Dai LX, Zhu XZ. Org. Lett. 2001;3:149. doi: 10.1021/ol0067033. [DOI] [PubMed] [Google Scholar]; (b) Yan XX, Liang CG, Zhang Y, Hong W, Cao BX, Dai LX, Hou XL. Angew. Chem., Int. Ed. 2005;44:6544. doi: 10.1002/anie.200502020. [DOI] [PubMed] [Google Scholar]; (c) Zheng WH, Zheng BH, Zhang Y, Hou XL. J. Am. Chem. Soc. 2007;129:7718. doi: 10.1021/ja071098l. [DOI] [PubMed] [Google Scholar]; (d) Zhang K, Peng Q, Hou XL, Wu YD. Angew. Chem., Int. Ed. 2008;47:1741. doi: 10.1002/anie.200704629. [DOI] [PubMed] [Google Scholar]
- 12.(a) Braun M, Laicher F, Meier T. Angew. Chem., Int. Ed. 2000;39:3494. doi: 10.1002/1521-3773(20001002)39:19<3494::aid-anie3494>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; (b) Braun M, Meier T. Synlett. 2005:2968. [Google Scholar]; (c) Braun M, Meier T. Synlett. 2006:661. [Google Scholar]; (d) Braun M, Meier T, Laicher F, Meletis P, Fidan M. Adv. Synth. Catal. 2008;350:303. [Google Scholar]
- 13.Seebach D. Angew. Chem., Int. Ed. Engl. 1988;27:1624. [Google Scholar]
- 14.Trost BM, Keinan E. Tetrahedron Lett. 1980;21:2591. [Google Scholar]
- 15.Tsuda T, Chujo Y, Nishi S, Tawara K, Saegusa T. J. Am. Chem. Soc. 1980;102:6381. [Google Scholar]
- 16.(a) Shimizu I, Yamada T, Tsuji J. Tetrahedron Lett. 1980;21:3199. [Google Scholar]; (b) Shimizu I, Ohashi Y, Tsuji J. Tetrahedron Lett. 1983;24:3865. [Google Scholar]; (c) Tsuji J, Minami I, Shimizu I. Tetrahedron Letters. 1983;24:1793. [Google Scholar]; (d) Shimizu I, Minami I, Tsuji J. Tetrahedron Lett. 1983;24:1797. [Google Scholar]; (e) Tsuji J, Minami I. Acc. Chem. Res. 1987;20:140. [Google Scholar]; (f) Tsuji J, Yamada T, Minami I, Yuhara M, Nisar M, Shimizu I. J. Org. Chem. 1987;52:2988. [Google Scholar]
- 17.Burger EC, Tunge JA. Org. Lett. 2004;6:4113. doi: 10.1021/ol048149t.For a review seeTunge JA, Burger EC. Eur. J. Org. Chem. 2005;9:1715.
- 18.Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 19.Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:2846. doi: 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]
- 20.For reviews in transition metal catalyzed decarboxylative asymmetric allylic alkylation, seeYou S-L, Dai L-X. Angew. Chem. Int. Ed. 2006;45:5346.Braun M, Meier T. Angew. Chem., Int. Ed. 2006;45:6952. doi: 10.1002/anie.200602169.Stoltz BM, Mohr JT. Chem. Asian J. 2007;2:1476. doi: 10.1002/asia.200700183.
- 21.(a) Trost BM, Vanvranken DL. Chem. Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; (b) Trost BM. Chem. Pharm. Bull. 2002;50:1. doi: 10.1248/cpb.50.1. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Crawley ML. Chem. Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Fandrick DR. Aldrichimica Acta. 2007;40:59. [Google Scholar]
- 22.Harwood LM, Houminer Y, Manage A, Seeman JI. Tetrahedron Lett. 1994;35:8027. [Google Scholar]
- 23.(a) Moyer MP, Feldman PL, Rapoport H. J. Org. Chem. 1985;50:5223. [Google Scholar]; (b) Shone RL, Deason JR, Miyano M. J. Org. Chem. 1986;51:268. [Google Scholar]; (c) Tanaka T, Okamura N, Bannai K, Hazato A, Sugiura S, Tomimori K, Manabe K.i, Kurozumi S. Tetrahedron. 1986;42:6747. [Google Scholar]
- 24.(a) House HO, Auerbach RA, Gall M, Peet NP. J. Org. Chem. 1973;38:514. [Google Scholar]; (b) Olofson RA, Cuomo J, Bauman BA. J. Org. Chem. 1978;43:2073. [Google Scholar]; (c) Taylor RJK. Synthesis. 1985;4:364. [Google Scholar]; (d) Bairgrie LM, Leung-Toung R, Tidwell TT. Tetrahedron Lett. 1988;29:1673. [Google Scholar]; (e) Zhong M, Brauman JI. J. Am. Chem. Soc. 1996;118:636. [Google Scholar]
- 25.Trost BM, Xu JY. J. Org. Chem. 2007;72:9372. doi: 10.1021/jo7016313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heathcock CH, Buse CT, Kleschick WA, Pirrung MC, Sohn JE, Lampe J. J. Org. Chem. 1980;45:1066. [Google Scholar]
- 27.Pascual C, Meier J, Simon W. Helv. Chim. Acta. 1966;49:164. [Google Scholar]
- 28.(a) Hall PL, Gilchrist JH, Collum DB. J. Am. Chem. Soc. 1991;113:9571. [Google Scholar]; (b) Hall PL, Gilchrist JH, Harrison AT, Fuller DJ, Collum DB. J. Am. Chem. Soc. 1991;113:9575. [Google Scholar]
- 29.Trost BM, Schroeder GM, Kristensen J. Angew. Chem., Int. Ed. 2002;41:3492. doi: 10.1002/1521-3773(20020916)41:18<3492::AID-ANIE3492>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 30.Trost BM, Toste FD. J. Am. Chem. Soc. 1999;121:4545. [Google Scholar]
- 31.Seebach D. Angew. Chem., Int. Ed. 1988;27:1624. [Google Scholar]
- 32.Trost BM, Stiles DT. Org. Lett. 2007;9:2763. doi: 10.1021/ol070971k. [DOI] [PubMed] [Google Scholar]
- 33.The absolute configuration of 45 was assigned by the comparison of its optical rotation with that known in the literature.Imai M, Hagihara A, Kawasaki H, Manabe K, Koga K. Tetrahedron. 2000;56:179.
- 34.The direct catalytic asymmetric mono-allylation of lithium enolate of cyclohexanone was recently reported. See:Braun M, Meletis P, Fidan M. Synthesis. 2009;86:47.
- 35.Meyers AI, Williams DR, Erickson GW, White S, Druelinger M. J. Am. Chem. Soc. 1981;103:3081. [Google Scholar]
- 36.The absolute configuration of 69 was assigned as S by hydrogenating the double bond and comparing the optical rotation of the product ([α]D = +25.7) with the reported one for (R)-69 ([α]D = −15.8).Baldwin JE, Patrick JE. J. Am. Chem. Soc. 1971;93:3556.
- 37.Wagner PJ, Liu KC, Noguchi Y. J. Am. Chem. Soc. 1981;103:3837–3841. [Google Scholar]
- 38.Oh WK, Oh H, Kim BY, Kim BS, Ahn JS. J. Antibiotics. 2004;57:808. doi: 10.7164/antibiotics.57.808. [DOI] [PubMed] [Google Scholar]
- 39.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem., Int. Ed. 2005;44:6924. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 40.(a) Trost BM, Fullerton TJ. J. Am. Chem. Soc. 1973;95:292. [Google Scholar]; (b) Trost BM, Weber L. J. Am. Chem. Soc. 1975;97:1611. [Google Scholar]; (c) Trost BM, Verhoeven TR. J. Org. Chem. 1976;41:3215. [Google Scholar]; (d) Trost BM, Weber L, Strege PE, Fullerton TJ, Dietsche TJ. J. Am. Chem. Soc. 1978;100:3416. [Google Scholar]; (e) Trost BM, Verhoeven TR. In: Comprehensive Organometallic Chemistry. Wilkinson G, Stone FGA, Abel EW, editors. Vol. 8. Pergamon; Oxford: 1982. p. 799. [Google Scholar]; (f) Fiaud JC, Legros JY. J. Org. Chem. 1987;52:1907. [Google Scholar]; (g) Suzuki T, Fujimoto H. Inorg. Chem. 1999;38:370. [Google Scholar]
- 41.Akermark B, Jutand A. J. Organomet. Chem. 1981;217:C41. [Google Scholar]
- 42.(a) Trost BM, Keinan E. Tetrahedron Lett. 1980;21:2591. [Google Scholar]; (b) Fiaud JC, Malleron JL. J. Chem. Soc.-Chem. Commun. 1981:1159. [Google Scholar]
- 43.Trost BM, Self CR. J. Org. Chem. 1984;49:468. [Google Scholar]
- 44.(a) Negishi E, Matsushita H, Chatterjee S, John RA. J. Org. Chem. 1982;47:3188. [Google Scholar]; (b) Negishi EI, John RA. J. Org. Chem. 1983;48:4098. [Google Scholar]
- 45.Shimizu I, Tsuji J. J. Am. Chem. Soc. 1982;104:5844. [Google Scholar]
- 46.Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA. J. Am. Chem. Soc. 2007;129:11876. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]
- 47.Hogen-Esch TE, Smid J. J. Am. Chem. Soc. 1965;87:669. [Google Scholar]
- 48.Ukai T, Kawazura H, Ishii Y, Bonnet JJ, Ibers JA. J. Organomet. Chem. 1974;65:253. [Google Scholar]
- 49.(a) Trost BM, Vanvranken DL, Bingel C. J. Am. Chem. Soc. 1992;114:9327. [Google Scholar]; (b) Trost BM, Bunt RC, Lemoine RC, Calkins TL. J. Am. Chem. Soc. 2000;122:5968. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.