

Abstract

Total syntheses of the Lycopodium alkaloids nankakurines A and B have been accomplished in 6 and 7 steps, respectively, via a sequence that passes through a third Lycopodium alkaloid, luciduline, and forgoes the use of protecting groups on nitrogen. Key features include a short preparation of luciduline followed by a concise and stereoselective aminoallylation/ring-closing metathesis protocol to fashion the spiropiperidine ring common to nankakurines A and B.
Studies on the extracts of Lycopodium club mosses continue to reveal a rich assortment of new alkaloid metabolites with interesting architectures and diverse biological activities.1 In 2006, Kobayashi and co-workers disclosed the structure of nankakurine B (2, Figure 1) and a revised assignment for nankakurine A (1), each isolated in minute quantities from Lycopodium hamiltonii.2 This 2006 report corrected an earlier stereochemical assignment of the spiro stereocenter in nankakurine A (1), arriving at the structures shown below.3 In 2008, the relative and absolute stereochemical assignments for 1 and 2 were confirmed by Overman and co-workers through enantioselective total syntheses in 13 and 14 steps, respectively.4
Figure 1.
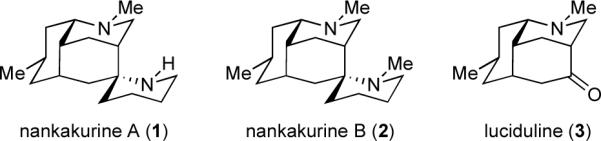
The Lycopodium alkaloids nankakurines A and B and their structural relationship to luciduline.
Low micromolar concentrations of nankakurine A (1) were found to induce the secretion of neurotrophic factors in a glial cell assay, which in turn promoted significant glial cell-mediated morphological differentiation and neurite outgrowth in rat adrenal cells.2 Because the extent of neurodegeneration in disorders such as Alzheimer's and Parkinson's diseases can be correlated with diminished levels of neurotrophic support,5 there has been recent interest in small molecule nonpeptidyl agents capable of eliciting a neurotrophic response.6 While the use of naturally occurring polypeptidyl neurotrophins has been complicated by their poor pharmacokinetic properties,7 one potential therapeutic advantage to small molecule nonpeptidyl neurotrophic agents might rest upon their improved capacity to cross the blood-brain barrier.8
The scarcity of nankakurine A (1) and nankakurine B (2) has not allowed for a more comprehensive evaluation of their neurobiological properties.9 This consideration, together with the irresistible synthetic challenge presented by the construction of their novel Lycopodium alkaloid framework, prompted us to launch an initiative aimed toward their total syntheses. One important requirement was that the synthetic design should be sufficiently succint as to allow for the rapid accumulation of potentially biologically active congeners. In this Letter, we report our successful progress related to these undertakings.
Nankakurines A (1) and B (2) represent a unique pair of Lycopodium alkaloids each featuring six stereogenic centers (one quaternary) and a tetracyclic ring system including an unusual spiropiperidine fusion. From the outset of our studies, we immediately took notice that nankakurine A (1) and nankakurine B (2) could be viewed as aza-spiroannulated versions of a third Lycopodium alkaloid, luciduline (3, Figure 1).10 Guided by pattern recognition,11 we envisioned that the spiropiperidine portions of 1 and 2 could well be forged via ring-closing metathesis of a bis-terminal olefin (cf. 5, Scheme 1) after stereoselective installation of two allylic moieties onto luciduline (3). Therefore, our early checkpoint became the synthesis of luciduline (3) for the purpose of utilizing it as an intermediate en route to nankakurine A (1) and nankakurine B (2).
Scheme 1.
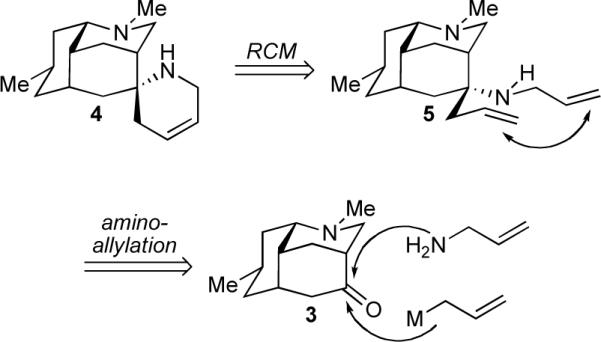
Selected Bond Formations toward the Nankakurines
While syntheses of 3 have been reported,12 a streamlined preparation was desired if large quantities were to be carried forward in an extended sequence. Ultimately, a three-step protocol capturing elements of the classic routes of Oppolzer12d and Evans12e, but with several key improvements, was defined. Aluminum-mediated Diels–Alder reaction of (±)-5-methylcyclohex-2-en-1-one (6)13 and 2-tert-butyldimethylsilyloxy-1,3-butadiene (7)14 afforded cis-decalone 8 accompanied by only minor amounts of the trans epimer (Scheme 2). This high cis:trans selectivity is noteworthy since some conditions employing enone 6 with other dienes have been reported to cause epimerization in favor of the trans-decalone.15 However, the most important consequence of the cycloaddition – and our primary motive for utilizing a 2-silyloxydiene – was that it directly furnished a silyl enol ether correctly positioned for use in a projected Mannich-type ring closure. Toward the latter goal, reductive amination of the ketone in 8 proceeded with high facial selectivity (dr > 20:1) to provide N-methyl amine 9. While the silyl enol ether in 9 could be liberated with dilute acid to afford a ketone which has been previously converted to 3 under forcing conditions,12e we anticipated that 9 would stand out as a more cooperative surrogate. Indeed, treatment of amine 9 with aqueous formaldehyde at room temperature readily gave luciduline (3) as the major product after simple acid-base extraction.16 Gram-scale quantities of material have been prepared through this short sequence.
Scheme 2.
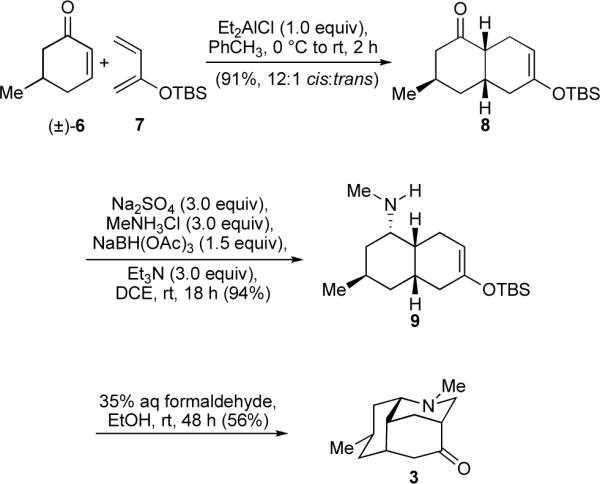
A Three-Step Total Synthesis of Luciduline
Attention was next given to the syntheses of nankakurines A (1) and B (2). We were presented first with the important task of securing the correct relative configuration at the aza-spiro stereocenter. In the early planning stages, we sought to make use of the cage-like topography of 3 to direct the stereochemical outcome of a two-stage nucleophilic addition sequence. Happily, the remaining building blocks could be incorporated neatly through a one-pot aminoallylation protocol (Scheme 3).17 Treatment of luciduline (3) with allylamine in benzene gave a ketimine product which, after replacement of solvent and addition of allylmagnesium bromide, provided diamine 5 as a separable 10:1 mixture in favor of the desired epimer formed by axial approach of the Grignard reagent from the more readily accessible convex face. Subsequent ring-closing metathesis of the terminal olefins in intermediate 5 using 5 mol% of Grubbs' second-generation catalyst18 gave aza-spirocycle 4. The catalyst proved to be remarkably tolerant of the amino functions provided that they were first converted to their corresponding ammonium salts in situ by addition of two equivalents of acid.19 In this manner, resorting to a lengthy protecting group strategy was averted. Hydrogenation of the alkene function in spiroamine 4 afforded nankakurine A (1) in excellent yield. Methylation of the secondary amine moiety in nankakurine A (1) through reductive amination provided nankakurine B (2). Spectroscopic data obtained for synthetic 1 and 2 were in agreement with those reported. Specifically, the 1H and 13C NMR spectra of the free basic forms of 1 and 2 in CD3OD were identical to those reported by Overman,4 while partially protonated forms gave spectra in agreement with those reported by Kobayashi.2,3 These observations reinforce the recent studies carried out by Overman and co-workers which suggest that the natural samples may have contained some amounts of the corresponding conjugate acids.4
Scheme 3.
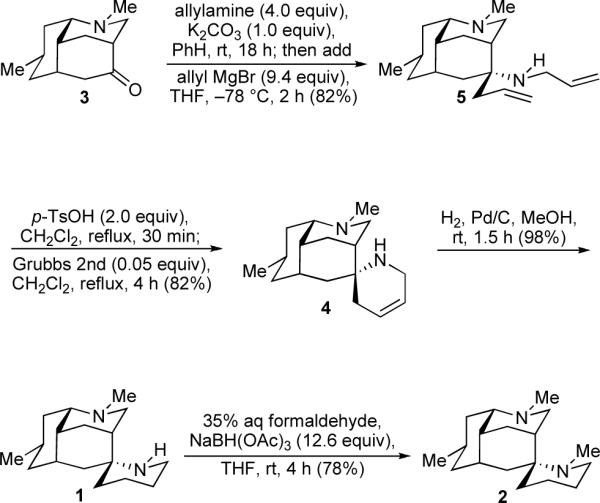
Syntheses of Nankakurines A and B
In summary, total syntheses of nankakurines A (1) and B (2) have been accomplished in 6 and 7 steps, respectively, via a sequence that passes through luciduline (3) and forgoes the use of protecting groups on nitrogen. Because optically pure (–)-6 is known through degradation of (+)-pulegone,20 the prospects for enantioselective syntheses are evident. The practical aspects of our synthetic strategy will enable gram-scale preparations of nankakurines A and B for a further assessment of their neurobiological properties. The outcomes of these current studies will be reported in due course.
Supplementary Material
Acknowledgment
We gratefully acknowledge the University of Vermont for financial support. This work was also generously supported by the Vermont Genetics Network through Grant Number P20 RR16462 from the INBRE Program of the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). We thank Dr. Rakesh K. Kohli, Director of the Mass Spectrometry Center at the University of Pennsylvania, for obtaining high-resolution mass spectra. We also thank Professor Martin Kuehne of the University of Vermont for helpful discussions related to alkaloid synthesis.
Footnotes
Supporting Information Available: Experimental procedures, characterization data, and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For reviews of the Lycopodium alkaloids, see: Hirasawa Y, Kobayashi J, Morita H. Heterocycles. 2009;77:679–729.. Kobayashi J, Morita H. In: The Alkaloids. Cordell GA, editor. Vol. 61. Academic Press; New York: 2005. p. 1.. Ma X, Gang DR. Nat. Prod. Rep. 2004;21:752–772. doi: 10.1039/b409720n.. Ayer WA, Trifonov LS. In: The Alkaloids. Cordell GA, Brossi A, editors. Vol. 45. Academic Press; New York: 1994. p. 233.. Ayer WA. Nat. Prod. Rep. 1991;8:455–463. doi: 10.1039/np9910800455.. MacLean DB. In: The Alkaloids. Brossi A, editor. Vol. 26. Academic Press; New York: 1985. p. 241.. MacLean DB. In: The Alkaloids. Manske RHF, editor. Vol. 14. Academic Press; New York: 1973. p. 348.. MacLean DB. In: The Alkaloids. Manske RHF, editor. Vol. 10. Academic Press; New York: 1968. p. 305..
- (2).Hirasawa Y, Kobayashi J, Obara Y, Nakahata N, Kawahara N, Goda Y, Morita H. Heterocycles. 2006;68:2357–2364. [Google Scholar]
- (3).Hirasawa Y, Morita H, Kobayashi J. Org. Lett. 2004;6:3389–3391. doi: 10.1021/ol048621a. [DOI] [PubMed] [Google Scholar]
- (4).Nilsson BL, Overman LE, Read de Alaniz J, Rohde JM. J. Am. Chem. Soc. 2008;130:11297–11299. doi: 10.1021/ja804624u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dawbarn D, Allen SJ. Neuropath. Appl. Neurobiol. 2003;29:211–230. doi: 10.1046/j.1365-2990.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- (6).The ongoing synthesis program of Danishefsky toward such molecules has been particularly fruitful: Wilson RM, Danishefsky SJ. Acc. Chem. Res. 2006;39:539–549. doi: 10.1021/ar068018n.. Waters SP, Tian Y, Li Y-M, Danishefsky SJ. J. Am. Chem. Soc. 2005;127:13514–13515. doi: 10.1021/ja055220x..
- (7).Kirik D, Georgievska B, Bjorklund A. Nat. Neurosci. 2004;7:105–110. doi: 10.1038/nn1175. [DOI] [PubMed] [Google Scholar]
- (8).(a) Luu B, de Aguilar J-LG, Girlanda-Junges C. Molecules. 2000;5:1439–1460. [Google Scholar]; (b) Hefti F. Annu. Rev. Pharmacol. Toxicol. 1997;37:239–267. doi: 10.1146/annurev.pharmtox.37.1.239. [DOI] [PubMed] [Google Scholar]
- (9).Nankakurine A (1) was isolated in 0.0003% yield (1.6 mg from 0.5 kg of plant material), while nankakurine B (2) was isolated in 0.0002% yield (1.1 mg from 0.5 kg of plant material). Neurite outgrowth could not be evaluated for nankakurine B (2) due to lack of compound (ref 2).
- (10).Ayer WA, Masaki N, Nkunika DS. Can. J. Chem. 1968;46:3631–3642. [Google Scholar]
- (11).Wilson RM, Danishefsky SJ. J. Org. Chem. 2007;72:4293–4305. doi: 10.1021/jo070871s. [DOI] [PubMed] [Google Scholar]
- (12).(a) Comins DL, Brooks CA, Al-awar RS, Goehring RR. Org. Lett. 1999;1:229–231. doi: 10.1021/ol990028j. [DOI] [PubMed] [Google Scholar]; (b) Schumann D, Naumann A. Liebigs Ann. Chem. 1984;8:1519–1528. [Google Scholar]; (c) Szychowsky J, MacLean DB. Can. J. Chem. 1979;57:1631–1637. [Google Scholar]; (d) Oppolzer W, Petrzilka M. Helv. Chim. Acta. 1978;61:2755–2762. [Google Scholar]; (e) Scott WL, Evans DA. J. Am. Chem. Soc. 1972;94:4779–4780. doi: 10.1021/ja00768a083. [DOI] [PubMed] [Google Scholar]
- (13).Chong B, Ji Y, Oh S, Yang J, Baik W, Koo S. J. Org. Chem. 1997;62:9323–9325. [Google Scholar]
- (14).Vedejs E, Eberlein TH, Mazur DJ, McClure CK, Perry DA, Ruggeri R, Schwartz E, Stults JS, Varie DL, Wittenberger S. J. Org. Chem. 1986;51:1556–1562. [Google Scholar]
- (15).For a detailed discussion of the problem, see: Charles Angell E, Fringuelli F, Minuti l., Pizzo F, Porter B, Taticchi A, Wenkert E. J. Org. Chem. 1985;50:4686–4690. and other papers in the series.
- (16).As did Ayer (ref 10), we found luciduline (3) to be unstable to chromatography. While a scaled-up run (4.55 g of 9) gave luciduline (3) in 87% yield and >90% purity, elution of a sample through neutralized silica gel gave a lower yield (56%) but afforded analytically pure material.
- (17).(a) Prusov E, Marier ME. Tetrahedron. 2007;63:10486–10469. [Google Scholar]; (b) Gracias V, Gasiecki AF, Morre JD, Akritopoulou-Zanze I, Djuric SW. Tetrahedron Lett. 2006;47:8977–8980. [Google Scholar]; (c) Wright DL, Schulte JP, II, Page MA. Org. Lett. 2000;2:1847–1850. doi: 10.1021/ol005903b. [DOI] [PubMed] [Google Scholar]
- (18).(a) Scholl M, Ding S, Woo Lee C, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; (b) Scholl M, Trnka TM, Morgan JP, Grubbs RH. Tetrahedron Lett. 1999;40:2247–2250. [Google Scholar]
- (19).For an overview of metathesis processes with nitrogen-containing substrates, see: Compain P. Adv. Synth. Catal. 2007;349:1829–1846..
- (20).Caine D, Procter K, Cassell RA. J. Org. Chem. 1984;49:2647–2648. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.