

Abstract
Sugars such as trehalose, sucrose, and glucose are effectively used by a variety of animals (e.g., brine shrimp, tardigrades, some frogs, and insects), as well as by bacteria, yeasts, and plant seeds to survive freezing and extreme drying. The objective of this study was to examine the potential application of sugars to mammalian oocyte cryopreservation. To this end, we used trehalose, a nonreducing disaccharide, and mouse metaphase II oocytes as models. Our experiments show that extracellular trehalose alone affords some protection at high subzero temperatures (e.g., −15°C), which diminishes with further cooling of the oocytes to −30°C and below. When present both intracellularly and extracellularly, trehalose dramatically improves the cryosurvival with increasing extracellular concentrations to 0.5 M, even after cooling to −196°C. Furthermore, the combination of intracellular and extracellular trehalose with small amounts of a conventional penetrating cryoprotectant (i.e., 0.5 M dimethylsulfoxide) provide high survival, fertilization, and embryonic development rates statistically similar to untreated controls. When transferred to foster mothers, cryopreserved oocytes give rise to healthy offspring showing the proof of principle. Our experiments with differential scanning calorimetry indicate that when cooled using the same cryopreservation protocol, the mixture of 0.5 M trehalose and cryopreservation medium undergoes glass transition at high subzero temperatures, which further substantiates the use of sugars as intracellular and extracellular cryoprotectants. Taken together, our results are in agreement with the survival schemes in nature and demonstrate the successful use of sugars in cryopreservation of mammalian oocytes.
Keywords: freezing, glass transition temperature, in vitro fertilization, microinjection, oocyte cryopreservation, sugar, trehalose
Combination of intracellular and extracellular trehalose with small amounts of a conventional cryoprotectant provides high cryosurvival, fertilization, and development rates.
INTRODUCTION
Oocyte cryopreservation offers future fertility for women expecting loss of their ovarian function as a result of cancer therapy. Current statistics suggest that 1 in 52 females younger than age 40 years is diagnosed with cancer [1]. Successful cryopreservation of the oocytes before treatment would not only provide future fertility, but could also alleviate the emotional consequences of cancer therapy for women afflicted with such devastating disease. Furthermore, oocyte cryopreservation also represents a good alternative to embryo freezing by avoiding many legal and ethical issues that are encountered in embryo freezing today. In addition, oocyte banking has wide-ranging research and commercial applications in livestock breeding, and may help conservation of genetic material of endangered species.
The first successful cryopreservation of mammalian oocytes was achieved in the 1970s [2, 3]. A decade later, the successful cryopreservation of human oocytes was reported [4]. It took another decade and the use of intracytoplasmic sperm injection to reproduce the initial success of human oocyte cryopreservation [5–8]. In recent years, encouraging results have been reported by several groups using both slow cooling [9–12] and vitrification [13–16] techniques. However, the overall success rate of human oocyte cryopreservation is still lower than that of its unfrozen counterparts, whereas vitrification techniques requiring direct contact between oocytes and liquid nitrogen pose a biosafety risk [17, 18]. Consequently, there is a need for more reliable protocols, as well as for alternative approaches. To this end, we have focused our efforts on sugar-based cryopreservation techniques inspired by the survival scheme in nature where a variety of organisms, from certain types of frogs and tardigrades to brine shrimp, bacteria, and yeast, cope with extreme conditions, such as freezing and almost complete drying [19, 20]. The ability to survive such extreme conditions is closely associated with the accumulation of large amounts of intracellular and extracellular sugars, such as trehalose, sucrose, and glucose [19]. The protective effect of sugars has also been shown in a number of model systems, including liposomes, membranes, and proteins during both air and freeze drying [21–25]. One of the major advantages of sugars is their high glass transition temperature compared with conventional cryoprotectants (CPAs), such as dimethylsulfoxide (DMSO), ethylene glycol (EG), and 1,2-propanediol (PROH) [26–29]. The high glass transition temperature would, in theory, allow long-term storage of cells at high subzero and even suprazero temperatures. In fact, many organisms in nature can remain in suspended animation for years, if not centuries, by undergoing a glassy state using sugars [19]. Therefore, sugars offer a unique prospect for high-temperature storage of mammalian cells. However, mammalian cell membranes are practically impermeable to sugars. Consequently, sugars have been used as extracellular additives [30, 31]. In recent years, several groups overcame the permeability barrier using different approaches, such as thermotropic lipid-phase transition [32], reversible poration by a genetically engineered protein [33], transfection [34], and ATP poration [35], and demonstrated the beneficial effect of intracellular sugars with somatic cells. To introduce sugars into mammalian oocytes, we developed a quantitative microinjection technique because the aforementioned approaches were not practical for the oocytes due to their small surface-to-volume ratios [36]. Our study with mouse pronuclear eggs revealed the nontoxicity of microinjected trehalose at its effective concentrations up to 0.15 M [36]. We were also able to obtain improved cryosurvival after microinjection of trehalose into failed-to-fertilize human oocytes [37].
In the present study, we microinjected mouse metaphase II (MII) oocytes with our model sugar trehalose to systematically address the cryoprotective effect of intracellular and extracellular trehalose as a sole CPA as well as in combination with small amounts of a conventional penetrating CPA, DMSO. In addition, we addressed the changes in glass transition temperature of freeze-concentrated trehalose/medium mixtures and showed that such mixtures undergo a glassy state at high subzero temperatures, which has implications for long-term storage of cells at high subzero temperatures. Furthermore, we also demonstrated the proof of principle by obtaining healthy pups from cryopreserved oocytes.
MATERIALS AND METHODS
Reagents and Media
All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise stated. A concentrated trehalose (Fluka) solution (0.8 M) was prepared for microinjection into oocytes by dissolving pure trehalose in 15 mM HEPES (pH 7.4) that was prepared in ultrapure water. Initially, HEPES-buffered Dulbecco Modified Eagle Medium (DMEM)/F-12 mixture (Gibco, Grand Island, NY) containing 4 mg/ml BSA and 50 mg/ml gentamycin was used for manipulation of oocytes and embryos under air. DMEM/F-12 was supplemented with various concentrations of trehalose and galactose for manipulations related to microinjection and cryopreservation. Microinjection was also initially performed in this medium after supplementation with 0.10–0.15 M trehalose. Later, DMEM/F-12 was replaced with HEPES-buffered Hypermedium [36] as indicated in specific experiments. Hypermedium consisted of 95 mM NaCl, 4.78 mM KCl, 0.38 mM KH2PO4, 0.2 mM MgSO4·7H2O, 2.0 mM CaCl2·2H2O, 20 mM Na-lactate, 0.33 mM Na-pyruvate, 2.78 mM glucose, 1 mM glutamine, 0.03 mM EDTA, 4 mg/ml BSA, 50 mg/ml gentamycin, 50× essential amino acids (Gibco), 100× nonessential amino acids (Gibco), and either 15 mM HEPES or 25 mM NaHCO3, depending on oocyte manipulations or culture, respectively. For in vitro fertilization (IVF) and subsequent culture of noninjected and microinjected oocytes, Hypermedium with an osmolality of 315–320 mOsm was used to partially compensate for the increased intracellular osmolality of trehalose-injected oocytes [36]. Our previous experiments revealed that the Hypermedium supports mouse embryonic development at least as well as other commonly used culture media [36]. Before use, drops of the Hypermedium were overlaid with embryo-tested mineral oil and equilibrated overnight under a humidified atmosphere of 5% CO2 in air at 37°C.
Oocyte Isolation
All animal experiments were approved by the Institutional Animal Care and Use Committee at the Massachusetts General Hospital and subsequently at the Medical College of Georgia. Metaphase II oocytes were obtained from 4- to 8-wk-old B6D2F1 (C57BL/6NCrl × DBA/2NCrl; Charles River Laboratories, Wilmington, MA) hybrid mice. Superovulation was induced by a combination of 5 IU equine chorionic and 2.5 IU human chorionic gonadotropins (PG 600; Intervet, Millsboro, DE), followed by 7.5 IU hCG alone 48–49 h later. Both hormone solutions were given intraperitoneally. To collect MII oocytes, the oviducts were excised from euthanized mice 13–14 h after hCG injection, and oocyte-cumulus masses were released from the ampulla. To remove cumulus cells, the oocyte-cumulus masses were exposed to 120 IU/ml bovine testis hyaluronidase (Type IV-S) at ambient temperature for 3–4 min. Next, the oocytes were washed in DMEM/F-12 or HEPES-buffered Hypermedium twice and then transferred to the Hypermedium for recovery before experimentation. For each experiment, MII oocytes were typically isolated from three or more female mice, pooled, and then randomly distributed among the experimental groups.
Microinjection of Trehalose
Quantitative microinjection of sugars was described in detail elsewhere [36]. Briefly, microinjection and holding pipettes were manufactured from 1-mm borosilicate thin-wall (B100-75-10; Sutter, Novato, CA) and thick-wall (B100-50-10; Sutter) glass capillaries, respectively. Both types of pipettes were pulled using a horizontal micropipette puller (Model P-97; Sutter). The holding pipettes were broken off at an external diameter of 70–80 μm and then fire polished using an MF-900 microforge (Narishige, East Meadow, NY). To obtain a sharp tip with an inside diameter of 0.5–0.7 μm, the injection pipettes were beveled at an angle of 40° on a modified Sutter micropipette beveler (BV-10; Sutter), and their tip diameters were determined by bubble pressure measurement in order to calibrate them. Microinjection was accomplished using two identical sets of MMN-1 coarse mechanical and MMO-202D fine hydraulic micromanipulators (Narishige) mounted on an inverted Diaphot 300 Microscope (Nikon, Melville, NY). To minimize vibration during microinjection, the micromanipulation system was set up on a vibration isolation table (TCM, Peabody, MA). A modified Stoelting piezo injector (PM-20; Stoelting, Chicago, IL) was used to facilitate puncture of the plasma membrane by a controlled sudden movement of the injection pipette. To deliver predetermined amounts of intracellular trehalose, a PLI-100 Pico-Injector (Medical Systems Co., Greenvale, NY) was used, and intracellular trehalose concentrations were confirmed by volumetric response of microinjected oocytes as described previously [36]. Oocytes that were degenerated after microinjection were easily identifiable using morphological criteria and were excluded from the experiments.
Cryosurvival Experiments
This set of experiments was designed to examine the effect of both intracellular and extracellular trehalose on the cryosurvival of mouse oocytes. The cryosurvival refers to the morphological integrity of the oocytes after thawing and culturing at 37°C at least for 1 h. The rate of the cryosurvival was calculated based on the number of frozen oocytes. The aim of these experiments also was to quickly identify effective concentrations of trehalose by evaluating the morphological survival of frozen-thawed oocytes and to establish a basis for subsequent experiments on fertilization and development. To this end, we first studied the survival of frozen-thawed oocytes as a function of different extracellular trehalose concentrations by adding 0, 0.15, 0.3, and 0.5 M trehalose to the freezing medium while the simultaneous presence (∼0.15 M) and absence of intracellular trehalose allowed us to uncover the effect of intracellular trehalose. The intracellular trehalose concentration was fixed around 0.15 M based on previous data showing that mouse zygotes injected with 0.15 M trehalose develop to the blastocyst stage at a rate similar to that of controls [36]. Cryosurvival was assessed after cooling trehalose-injected (intracellular trehalose group) and noninjected (no-trehalose and extracellular trehalose groups) oocytes to glass transition temperature (Tg′) of maximally freeze-concentrated trehalose solutions (i.e., approximately −30°C). Before cooling, oocytes were transferred to respective freezing media containing different amounts of trehalose, and then aspirated into 0.25-cc plastic straws (TS Scientific, Perkasie, PA). Next, straws containing oocytes were placed in a programmable freezer (Cryomed 1010; Forma Scientific, Marietta, OH) at 0°C and cooled to −6°C at a rate of 2°C/min. After seeding of extracellular ice and holding at −6°C for 10 min, the straws were cooled to −30°C at 1°C/min, where they were held for 2 min before thawing. Thawing was done by holding the straws in air. Subsequently, the contents of the straws were released into DMEM/F-12 containing 0.2–0.5 M galactose at an ambient temperature depending on the extracellular trehalose concentration. Further dilution of extracellular trehalose and galactose was carried out by transferring oocytes first to 0.2 M galactose in DMEM/F-12, and then to 0.15 M galactose in the Hypermedium with 10-min intervals. The latter also was used for overnight culture. Survival of cryopreserved oocytes was assessed after overnight culture by morphological criteria that included translucent appearance of cytoplasm, integrity of the plasma membrane and the zona pellucida, and the size of the perivitelline space. The thawing rate and dilution steps of extracellular trehalose were modified in the final series of experiments, as described later.
In the next set of cryosurvival experiments, we further examined the cryoprotective effect of trehalose as a function of final freezing temperature by cooling trehalose-injected and noninjected oocytes to slightly below the average intracellular ice formation (IIF) temperature (−15°C), Tg′ (−30°C), and well below Tg′ of trehalose (i.e., −60°C). Based on the results in the initial experiments, intracellular and extracellular trehalose concentrations were set to 0.15 M and 0.50 M, respectively. In addition to intracellular and extracellular trehalose groups, this set of cryosurvival experiments also included a control group in which noninjected oocytes were cooled to −15°C, −30°C, and −60°C in plain DMEM/F-12 mixture without extracellular trehalose. Cooling from −30°C to −60°C was carried out at 1°C/min. Otherwise, the cooling and thawing protocol was identical.
Fertilization and Development of Cryopreserved Oocytes
These experiments were designed to examine the fertilization and developmental competence of cryopreserved oocytes by comparison to controls. To test our hypothesis that the combination of intracellular and extracellular trehalose with low concentrations of conventional CPAs can further improve the cryoprotection while minimizing CPA toxicity [38–42], one third (0.5 M) of the typical DMSO concentration (1.5 M) employed in slow-freezing protocols was also used in these series of experiments.
In the initial set of embryonic developmental experiments, we studied the fertilization and embryonic development of MII oocytes after cooling to −60°C using the same protocol described above with two exceptions: 1) HEPES-buffered Hypermedium containing 10% fetal bovine serum was used as a cryopreservation medium instead of DMEM/F-12 and 2) intracellular trehalose concentration was reduced to approximately 0.08 M based on excellent survival rates achieved in the presence of 0.5 M DMSO. The lower intracellular trehalose concentrations facilitate embryo culture at osmolalities closer to physiological values around 290 mOsm and simplify the dilution of extracellular trehalose. The extracellular trehalose concentration was 0.5 M, as before. Experimental groups included 1) combination of only extracellular trehalose with 0.5 M DMSO, 2) intracellular and extracellular trehalose, and 3) combination of intracellular and extracellular trehalose with 0.5 M DMSO. In our preliminary studies, 0.5 M DMSO alone yielded poor survival (39%) after cooling to −60°C, and therefore it was not tested further. Untreated oocytes served as controls. In vitro fertilization and culture of inseminated oocytes were carried out in Hypermedium at 37°C under a humidified atmosphere of 5% CO2 in air. For IVF, sperm were obtained from the cauda epididymides of a mature (4–6 mo old) BDF1 males (C57BL/6NCrl × DBA/2NCrl; Charles River Laboratories). The cauda epididymides were dissected and placed in a large drop (0.4 ml) of pre-equilibrated, BSA-free Hypermedium. Sperm were released into the medium by gently puncturing the epididymides (five to seven times) with a hypodermic needle and allowed to disperse for 15 min at 37°C. After dispersion, sperm concentration was determined using a hemocytometer. To give a final concentration of 1 × 106 to 2 × 106 sperm/ml, an appropriate volume of the sperm suspension was added to each insemination drop containing Hypermedium with BSA supplementation. The insemination drops were then incubated for 1–2 h in order to capacitate the sperm before introducing control and microinjected oocytes. After 5–6 h of incubation with sperm, all oocytes were washed twice in the Hypermedium and then cultured in fresh drops of the same medium. Cleavage to the two-cell stage was examined after overnight culture, whereas development to the blastocyst stage was evaluated after 5 days of culture. Fertilization and blastocyst rates were calculated based on the number of survived and fertilized oocytes, respectively.
The aim of the next and final set of experiments was to evaluate the survival, fertilization, and development of cryopreserved oocytes after cooling to −196°C. To this end, mouse oocytes were microinjected with 0.05–0.08 M trehalose and cooled to −60°C at 1°C/min in the presence of 0.5 M extracellular trehalose and DMSO, and then plunged into liquid nitrogen. After storage in liquid nitrogen for several days, cryopreserved oocytes were slowly thawed and then inseminated along with both untreated and trehalose-injected but unfrozen controls. The cryopreservation protocol in this final set of experiments was further modified by slowing the thawing rate and simplifying the dilution steps to reduce osmotic stresses, as explained later in Discussion. The slower thawing rates (∼76°C/min from −196°C to −40°C and ∼9°C/min from −40°C to 0°C) were achieved by holding the straws within the neck region of a liquid nitrogen tank for 5 min. After thawing, the oocytes were released into HEPES-buffered Hypermedium containing 0.1 M galactose, where they were held for 7 min. Next, the oocytes were transferred to a fresh drop of 0.1 M galactose for 7–10 min before being transferred to the culture medium for a recovery period of 1 h. Following the recovery period, both cryopreserved and control oocytes were inseminated as descibed above to evaluate fertilization and embryonic development. The control groups included untreated controls and injection controls that were microinjected with the same amount of trehalose but not frozen. To demonstrate the proof-of-principle that mouse oocytes cryopreserved using small amounts of trehalose and DMSO can develop to term, we also carried out embryo transfer, as described next.
Embryo Transfer
Nontreated control and cryopreserved oocytes were inseminated and cultured in the Hypermedium (∼320 mOsm) overnight. On Day 1.5, the resulting two-cell embryos were transferred into the oviducts of Day 0.5 pseudopregnant CD1 females that were obtained by mating them to vasectomized CD1 males. In cases of blastocyst transfer, embryos were transferred into uterine horns of Day 3.5 pseudopregnant CD1 females. The pseudopregnant CD1 females were anesthetized by an intraperitoneal injection of avertin (0.3–0.4 mg/g body weight), and 10–13 embryos from either the control or the treatment group were transferred to each female. The recipients were individually housed after embryo transfer. Embryo transfer into oviducts and uterine horns was carried out as described by Hogan et al. [43].
Determination of Glass Transition Temperature
To measure the glass transition temperature of freeze-concentrated cryopreservation media by differential scanning calorimetry (DSC), 20 μl of 0.5 M trehalose in DMEM/F-12 was pipetted into a DSC aluminum pan and hermetically sealed. The sample pan was run against a similarly sealed empty reference pan. A modified Mettler DSC-FP90 (Mettler Instrument Corp., Highttown, NJ) with FP99 system software was used to control the sample temperature and scan the heat flow. First, the sample and reference pan were brought to −6°C, and then ice was seeded in the sample. After being held at −6°C for 3 min, the sample and reference pan were next cooled to −60°C at 1°C /min and equilibrated at −60°C for 5 min. Subsequently, the pans were heated to 20°C at 10°C/min to produce a thermogram. A typical change in heat flow indicated glass transition of the sample (Fig. 1). The midpoint between the onset and endpoint of the transition was taken as the glass transition temperature. To probe the effect of cooling rates, some sample pans were first immersed into LN2 and then quickly transferred to the DSC cell at −60°C. Next, the sample was equilibrated with the start tempertature (i.e., −60°C) and heated to 20°C at 10°C/min to determine the glass transition temperature. Measurements were repeated three times. Plain DMEM/F-12 without any disaccharide was used as a control solution. In addition, DSC measurements were repeated with the mixture of 0.5 M trehalose and 0.5 M DMSO, as well as with 0.5 M sucrose in DMEM/F-12 for comparison.
FIG. 1.
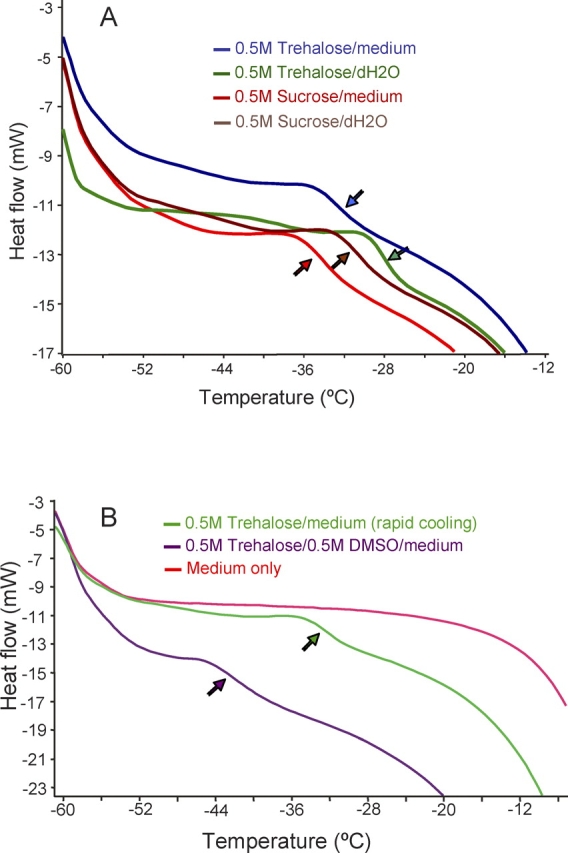
Glass transition temperatures of freeze-concentrated (A) disaccharide/medium mixtures compared with their binary solutions and (B) medium alone, trehalose/DMSO/medium, and trehalose/medium mixtures. All samples were cooled slowly after inducing ice formation, except the trehalose medium mixture in B, which was cooled rapidly. Arrows show the midpoint of glass transition.
Statistical Analysis
The data were analyzed by chi-square test, Fisher exact test, or ANOVA as appropriate, and using GraphPad Prism (GraphPad Software Inc., San Diego, CA). Before ANOVA, arcsine transformation was performed on proportional data. Differences between the groups were considered statistically significant when the P value was less than 0.05. Experiments in each series were repeated at least three times. Data reported are means of survival, fertilization, and development rates, with error bars representing standard error of mean.
RESULTS
Glass Transition Temperature of Disaccharide/Medium Mixtures
The glass transition temperature (Tg′) of maximally freeze-concentrated binary disaccharide solutions (i.e., disaccharide/water) has already been published [26, 44, 45]. However, it was unclear how the components of a cryopreservation medium (e.g., salts, proteins, and other carbohydrates such as glucose), the presence of 0.5 M DMSO, and the cooling rate used in the present study (1°C/min) affected Tg′. This information is relevant to designing cooling protocols and determining possible storage temperatures other than in liquid nitrogen. To clarify this issue, we performed DSC measurements with 0.5 M trehalose and sucrose prepared in DMEM/F-12, and we used plain DMEM/F-12 without disaccharide supplementation as a control. In addition, we measured the Tg′ of our cryopreservation medium containing 0.5 M trehalose and 0.5 M DMSO. The results of these measurements are shown in Figure 1. Upon freezing and warming, the mixtures of trehalose and sucrose with DMEM/F-12 showed a clear glass transition around at −33°C and −35°C, respectively. These Tg′ values for the trehalose/medium and sucrose/medium mixtures were slightly lower than those (approximately −28°C and −32°C, respectively) of their respective binary solutions in distilled water (Fig. 1A). Unlike the disaccharide/medium mixture, DMEM/F-12 alone showed no typical change in heat flow indicative for glass transition. The thermogram of plain DMEM/F-12 displayed only ice melting within the scanned temperature range (−60°C to 20°C), as expected (Fig. 1B). To address the effect of the cooling rate, we directly immersed the sample DSC pan into LN2, equilibrated it with the start temperature (−60°C), and then measured Tg′ as usual. Interestingly, the fast cooling rate (i.e., directly immersing the DSC pan into LN2 vs. slow cooling to −60°C) did not significantly alter the Tg′ of the 0.5 M trehalose/medium mixture, both of which were around −33°C (Fig. 1). In contrast, the presence of 0.5 M DMSO in 0.5 M trehalose/medium mixture depressed the Tg′ to −42°C. Nevertheless, even this depressed Tg′ is much higher than those of conventional penetrating CPAs and would in theory allow the cryostorage at a −80°C freezer or transport on dry ice if the intracellular Tg′ also occurs at such high subzero temperatures. Because macromolecules increase the Tg′ and the oocyte cytoplasm largely consists of macromolecules and the sugar/CPA mixture after freezing-induced dehydration, it is likely that the intracellular Tg′ occurs at a similar high subzero temperature.
Survival after microinjection.
The microinjection procedure itself induced minimal damage when performed properly. On average, 96% of the microinjected oocytes survived the procedure when assesed by morphological criteria after 30–40 min of incubation at 37°C.
Cryosurvival.
To evaluate the protective effect of intracellular and extracellular trehalose against cryopreservation-associated stresses, both trehalose-injected and noninjected mouse oocytes were subjected to freezing and thawing. First, we examined the cryosurvival of mouse oocytes as a function of different extracellular trehalose concentrations by adding 0, 0.15, 0.3, and 0.5 M trehalose to the freezing medium while the intracellular trehalose concentration was kept at 0.15 M. Oocytes that were frozen in the absence of intracellular and extracellular trehalose served as controls. The results of these experiments involving a total of 459 mouse oocytes are summarized in Figure 2. In the absence of intracellular and extracellular trehalose, no oocytes survived freezing to −30°C. Adding 0.15 or 0.3 M trehalose to the freezing medium resulted in minimal cryosurvival of 4% to 5% without a significant difference. Further increase of the extracellular trehalose concentration to 0.5 M raised the cryosurvival to 17%, which was statistically significant. However, more dramatic improvement in the cryosurvival was observed when increasing extracellular trehalose concentrations were combined with intracellular trehalose. Specifically, the cryosurvival rates in the presence of 0.15 M intracellular trehalose were 18%, 55%, and 84% for 0.15, 0.3, and 0.5 M extracellular trehalose, respectively. This almost linear increase in the survival rate as a function of increasing extracellular trehalose concentrations was highly significant, indicating the critical role of both intracellular and extracellular sugars in cryoprotection.
FIG. 2.

Cryosurvival of trehalose-injected and noninjected mouse oocytes as a function of extracellular trehalose concentrations. The final cooling temperature for these experiments was −30°C. The survival rates represent mean ± SEM. Different lowercase letters for each category indicate significant differences between the groups (P < 0.05).
To further probe the protective effect of trehalose, we next compared the survival of trehalose-injected and noninjected oocytes as a function of final freezing temperature by cooling them to slightly below the average IIF temperature (−15°C), Tg′ (−30°C), and well below the Tg′ of trehalose (i.e., −60°C). A total of 488 oocytes were subjected to the freezing in three groups: 1) control group without intracellular and extracellular trehalose; 2) extracellular trehalose group with 0.5 M trehalose in the freezing medium; and 3) intracellular and extracellular trehalose group with 0.15 M intracellular and 0.5 M extracellular trehalose. A fourth group with only 0.15 M intracellular trehalose was omitted because it requires increasing the osmolarity of the freezing medium. It is also important to note that the cryoprotective effect of 0.15 M intracellular and 0.5 M extracellular trehalose was examined after cooling to −30°C in the first series of experiments (Fig. 2). Therefore, we have not repeated the same experiments for these series. Instead, we combined those two data points with the results of this series of experiments to present a complete picture of the cryosurvival as a function of final cooling temperature. The results of these experiments are shown in Figure 3. In the absence of intracellular and extracellular trehalose, only 9% of control oocytes survived cooling to −15°C, whereas all control oocytes were degenerated with further cooling to −30°C or −60°C. Compared with the control (no-trehalose) group, the presence of 0.5 M extracellular trehalose alone significantly improved the survival to 60% after cooling to −15°C (P < 0.001). However, survival of the oocytes in the extracellular trehalose group steeply declined to 17% and then 14% as the final cooling temperature lowered to −30°C and −60°C, respectively. In contrast, 90% of mouse oocytes in the intracellular and extracellular trehalose group survived cooling to −15°C, whereas 83% and 81% of them were able to maintain their viability as the temperature was lowered to −30°C and −60°C, respectively. These survival rates that were significantly higher than those in the control and extracellular trehalose groups suggest that sugars should be present both inside and outside the cell for optimal cryoprotection.
FIG. 3.

Cryosurvival of trehalose-injected and noninjected mouse oocytes as function of final cooling temperature. The survival rates represent mean ± SEM. Different lowercase letters for a given temperature indicate significant differences between the groups, whereas different capital letters denote significant differences within each group with respect to final cooling temperature (P < 0.05).
Fertilization and Development of Cryopreserved Oocytes
In the initial set of embryonic developmental experiments, we investigated the fertilization and development of cryopreserved oocytes after cooling to well below the Tg′ of trehalose (i.e., −60°C). As explained in Materials and Methods, we modified the freezing protocol in this set of the developmental experiments to test our hypothesis that the combination of sugars with low concentrations of conventional CPAs would further improve the cryoprotection. These modifications included 1) adding small amounts (i.e., 0.5 M) of DMSO to the cryopreservation medium, 2) reducing intracellular trehalose concentration to around 0.08 M, and 3) using HEPES-buffered Hypermedium instead of DMEM/F-12. A total of 358 MII oocytes were used in four groups that were 1) control, 2) extracellular trehalose + DMSO, 3) intracellular and extracellular trehalose, and 4) intracellular and extracellular trehalose + DMSO groups. Our preliminary studies showed that 0.5 M DMSO alone was insufficient to provide adequate protection. Therefore, we have not included that group (i.e., 0.5 M DMSO alone) in this series of experiments.The MII oocytes in the three freezing groups were cooled to −60°C at 1°C/min, held at this temperature for 10 min, thawed in air, and then inseminated along with unfrozen controls by conventional IVF. The results of these experiments are summarized in Figure 4. In the absence of intracellular trehalose, the combination of extracellular trehalose with DMSO provided a reasonable cryosurvival rate of 77% after cooling to −60°C. The cryosurvival rate was significantly improved even further when we combined intracellular and extracellular trehalose (92%) or intracellular and extracellular trehalose with DMSO (96%). Similarly, the fertilization rates in the latter two groups (74% and 83%, respectively) were comparable to that of untreated controls (83%), whereas the combination of extracellular trehalose with DMSO yielded an acceptable but significantly lower fertilization rate (63%) than untreated controls. However, only a small portion of the fertilized eggs in the intracellular and extracellular trehalose group (21%) developed to the blastocyst stage, similar to the ones in the extracellular trehalose + DMSO group (21%). In contrast, the majority of the fertilized eggs in the intracellular and extracellular trehalose + DMSO group (66%) developed to the blastocyst stage, although it was still significantly lower than untreated controls (98%). Nevertheless, our results indicate that intracellular and extracellular trehalose in combination with small amounts of a conventional penetrating CPA can greatly improve the survival, fertilization, and embryonic development of frozen-thawed mouse oocytes.
FIG. 4.

Cryosurvival, fertilization, and embryonic development of mouse oocytes after cooling to −60°C. The rates represent mean ± SEM. Different lowercase letters for each category indicate significant differences between the groups (P < 0.05).
In the next set of embryonic development experiments, we studied fertilization and development of cryopreserved oocytes after cooling them to −196°C and further modified our cryopreservation protocol to improve the results by using slower warming rates and a simplified dilution procedure. The intracellular trehalose concentration was kept between 0.05 and 0.08 M. A total of 355 MII oocytes were used in three groups, which included 1) untreated controls, 2) trehalose-injected but unfrozen injection controls, and 3) trehalose-injected oocytes cryopreserved in the presence of extracellular trehalose and DMSO. The results of these experiments are summarized in Figure 5. The vast majority (90%) of cryopreserved oocytes survived the freezing and thawing procedure and had a fertilization rate (87%) similar to that of untreated (94%) and injected unfrozen (94%) controls. Likewise, a high percentage of fertilized oocytes developed to the blastocyst stage in the cryopreservation group (65%), which was not significantly lower than the blastocyst rates of untreated (80%) and injection controls (79%), showing the positive effect of our modifications. Taken together, these results suggest that intracellular and extracellular trehalose with small amounts of DMSO affords an adequate cryoprotection for mouse oocytes during freezing to liquid nitrogen temperature and subsequent thawing.
FIG. 5.

Cryosurvival, fertilization, and embryonic development of mouse oocytes after cooling to −196°C. The rates represent mean ± SEM. There was no significant difference between the groups (P > 0.05). N/A indicates that control oocytes were not cryopreserved, and thus the cryosurvival is not applicable.
To demonstrate the proof-of-principle that MII oocytes cryopreserved using small amounts of trehalose and DMSO can develop to term, we froze and thawed the oocytes as described above, inseminated the surviving oocytes, and then transferred the resulting embryos into the oviducts or uterine horns of pseudopregnant CD1 females. The transfer of two-cell embryos from the control (22) and cryopreservation (21) groups into the oviducts of pseudopregnant females resulted in the birth of five and four pups with delivery rates of 23% and 19%, respectively. We also transferred 25 control blastocysts and 22 blastocysts that originated from cryopreserved oocytes into the uterine horns of pseudopregnant CD1 females and obtained seven (28%) and three (14%) pups, respectively. All offspring in both control and cryopreservation groups were healthy, grew, and bred normally, giving birth to a second generation of healthy pups.
DISCUSSION
In the present study we used trehalose as a model sugar to test our two hypotheses that 1) sugars such as trehalose can protect mammalian oocytes against freezing-associated stresses when present both intracellularly and extracellularly and 2) the combination of sugars with low concentrations of conventional CPAs such as DMSO can further improve cryoprotection while minimizing CPA toxicity. Our results showing that 1) in the presence of intracellular trehalose, increasing extracellular trehalose concentrations provide excellent cryosurvival rates and 2) the combination of intracellular and extracellular trehalose with small amounts of DMSO results in high cryosurvival, fertilization, and development rates support both hypotheses. Furthermore, we demonstrated the proof-of-principle by obtaining healthy offspring from cryopreserved oocytes.
The results of this study are consistent with the survival scheme in nature. It is well known that the accumulation of intracellular and extracellular sugars such as trehalose and sucrose plays a major role in the survival of a diversity of animals (e.g., certain frogs, nematodes, tardigrades, insects, brine shrimp), as well as bacteria, yeasts, and plant seeds under extreme drying and freezing [19, 20]. Findings of several studies suggest that sugars afford their protection 1) by stabilizing lipid membranes and proteins as a result of direct interactions with polar residues through hydrogen bonding (so-called water replacement hypothesis) and 2) by their excellent glass-forming properties [21–23]. In the present study the Tg′ of binary solutions of trehalose (approximately −28°C) and the Tg′ of sucrose (approximately −32°C) were similar to those in previously published results [26, 27, 45], whereas the components of the cryopreservation medium in the absence of DMSO slightly depressed the Tg′ of trehalose/medium (−33°C) and sucrose/medium (−35°C) mixtures. These Tg′ values are significantly higher than those of conventional penetrating CPAs, which are typically below −85°C [27–29]. Therefore, sugars like trehalose are more effective glass formers than the conventional CPAs. Because the objective of any cryopreservation procedure is to safely bring the cells into a glassy state, the low Tg′ of conventional CPAs requires dehydration of the cells through the homogenous ice nucleation temperature. This makes the cells susceptible to IIF, particularly in the case of insufficient dehydration due to dramatically reduced or stopped water transport at such low subzero temperatures. In contrast, the high Tg′ and relatively flat melting curve of sugars such as trehalose allow cell dehydration at high subzero temperatures without crossing homogenous ice nucleation temperature while the water permeability across the cell membrane is still high. A high Tg′ also means dramatically increased viscosity at relatively high subzero temperatures around Tg′, which may protect cells during the freeze-thaw process by preventing nucleation and growth of lethal intracellular ice crystals. Furthermore, a high Tg′ would potentially permit long-term storage and transportation of cells at higher subzero temperatures (e.g., −80°C freezer and on dry ice, respectively) than −196°C. This would also reduce thermal stresses that occur, particularly during the plunging of samples from an intermediate temperature to the LN2 temperature and subsequently during rapid warming [46–48]. Our results further indicate that trehaloselike sugars can be combined with small amounts of conventional CPAs without much lowering the Tg′ while maximizing the cryoprotection. The Tg′ of 0.5 M trehalose and DMSO (i.e., approximately −42°C ) would still allow the cryostorage at a −80°C freezer or transport on dry ice if intracellular glass transition occurs at a similar subzero temperature. In the present study we have not documented the glassy state of oocyte cytoplasm. It is unclear how intracellular compartmentalization and cytoplasmic contents of the oocytes affect the glass transition temperature. Further research is needed to address these issues.
The cryosurvival, fertilization, and embryonic developmental rates obtained in this study are comparable to the best results in the literature achieved using much higher CPA concentrations [49–54]. This indicates the efficiency of trehaloselike sugars in cryopreserving mammalian oocytes. The effectiveness of sugars has also been shown using several other cell types [32, 33, 55]. In the present study trehalose as a sole cryoprotectant provided excellent cryosurvival and fertilization (cleavage to the two-cell stage) rates. However, further development of cryopreserved oocytes to the blastocyst stage was impaired. The reason for this is unclear. Insufficient cryoprotection of cell organelles by cytoplasmic trehalose might have contributed to poor blastocyst rates. As shown in cryosurvival experiments and earlier studies [33, 37], optimal cryoprotection requires the presence of trehalose at both sides of membranes. Although we injected trehalose into the cytoplasm, it might have not entered into organelles, such as mitochondria and endoplasmic reticulum, that are vital for proper cell functionality. Consequently, insufficient protection of vital cell organelles might have been manifested after activation of the embryonic genome at the two-cell stage, until which point cytoplasmic stores accumulated during oocyte growth and maturation are used. Support for this explanation comes from the experiments that also included 0.5 M DMSO as a penetrating CPA. Indeed, the addition of small amounts (0.5 M) of DMSO that also enters into cell organelles was sufficient to greatly improve the development rates to the blastocyst stage. This finding also suggests that the development of cell-permeable sugars may dramatically improve the cryoprotection afforded by sugars and opens the door for cell/tissue storage at ambient temperatures.
Earlier studies on human oocyte cryopreservation typically used 0.1 M sucrose as extracellular additive [56–59]. When we used higher concentrations of extracellular sugars with mouse and failed-to-fertilize human oocytes, we obtained improved cryopreservation outcome [37, 60, 61]. In 2001, Fabbri et al. [62] published a comprehensive study with human oocytes showing that increasing the concentration of extracellular sucrose from 0.1 to 0.3 M in slow-freezing protocols significantly improves the cryosurvival of human oocytes. This study stimulated further studies, which also demonstrated improved spindle morphology, cryosurvival, and fertilization rates after cryopreservation of human oocytes using 0.3 M extracellular sucrose and 1.5 M 1,2-propanediol [63, 64], although the improvement in pregnancy and implantation rates is still debated [12, 65, 66]. Since the current slow-freezing protocols of human oocytes use moderate concentrations (1.5 M) of penetrating CPAs, such as 1,2-propanediol, the beneficial effect of higher concentrations of extracellular sucrose may not be obvious in such protocols. However, the favorable effect of an even higher extracellular sugar concentration (i.e., 0.5 M) became apparent when we used low intracellular CPA concentrations in the present study. This might be a result of temporary increase in the intracellular CPA concentration and initial suppression of IIF by cell dehydration in the presence of 0.5 M extracellular trehalose, as well as better stabilization of the extracellular milieu in a glassy state.
During the course of this study we made several modifications in our protocol. Based on our earlier studies, we initially injected mouse oocytes with 0.15 M trehalose. Over the course of this study, we found that even lower concentrations of intracellular trehalose were very effective, particularly when combined with small amounts of DMSO (0.5 M). Consequently, we lowered the intracellular trehalose concentration with the progress of the study. The lower intracellular trehalose concentrations facilitate embryo culture in terms of overall osmolality of the culture medium, allow faster elimination of intracellular trehalose after freezing and thawing, and also simplify the dilution procedure. Another modification was adjustment of the warming rate. Initially, we thawed the samples in air, which resulted in a warming rate of approximately 220°C/min. However, after thawing in air some oocytes seemed to experience osmotic stresses due to swelling. Consequently, we attempted to slow down the warming rate by holding the straws within the neck region of our cryotank because our current controlled-rate freezer (Cryomed 1010) does not allow programming of warming rates. Indeed, we were able to slow down the warming rates (∼76°C/min to −40°C and ∼9°C/min to 0°C), which improved the results. Based on our glass transition measurements, we can predict that a faster warming rate to the glass transition temperature (i.e., around −30°C) or slightly above it, and then a slower warming rate to 0°C would give better results by reducing osmotic shock. In the future, we plan to revisit this issue using a controlled-rate freezer with programmable warming rates.
Typically, slow cooling and vitrification protocols use high concentrations (from 1.5 M up to 6.0 M) of penetrating conventional CPAs compared with 0.5 M DMSO in the present study. Two early studies showing that mouse embryos can be frozen using lower concentrations of CPAs (i.e., 1.0 M DMSO and 1.2 M EG) with good success rates represent an exception to this [67, 68]. However, it is well known that mammalian embryos are more resistant to cryopreservation-associated stresses than oocytes and have been successfully cryopreserved routinely for many decades, in contrast to mammalian oocytes. Another exception is the study of Schroeder et al. [54], who were able to successfully cryopreserve mouse oocytes using a slow-cooling protocol and 1.0 M DMSO, although their DMSO concentration was 2-fold higher than that in the present study. The use of lower concentrations of penetrating CPAs are important to avoid/reduce the potential toxicity of such CPAs [38–42].
Any intervention causing even temporary change in the equilibrium of the physiological state could potentially be toxic to cells. This applies to microinjection of sugars, as well as loading conventional CPAs. Nevertheless, human intracytoplasmic sperm injection, which introduces small amounts of PVP, a synthetic polymer, into oocytes, proved to be an extremely useful manipulation. To address the safety issue, we performed a series of experiments. Our studies with trehalose-injected mouse oocytes revealed that microinjected trehalose is rapidly eliminated during embryonic development [69]. When we cultured mouse pronuclear eggs injected with 0.10–0.15 M trehalose, we obtained embryonic development rates similar to those in untreated controls [36]. Embryo transfer of trehalose-injected eggs also resulted in implantation and delivery rates similar to those in controls [36]. Furthermore, studies involving intracellular loading of higher concentrations of sugars into different mammalian cells, such as embryonic stem cells and fibroblasts, showed that these cells function and grow normally [33, 70]. Taken together, a large body of experimental evidence indicates nontoxicity of the sugars that are used by many organisms as osmolytes against osmotic stresses [71, 72], as well as against chemical [73] and hypoxic [74] stresses. Our current results showing that healthy pups can be obtained from trehalose-injected and cryopreserved mouse oocytes further confirm the safety of our cryopreservation technique, although the delivery rates for untreated control and cryopreserved oocytes were at the lower range of published data due to suboptimal embryo transfer setup (e.g., transportation of embryos to another building for embryo transfer, exposure to nonphysiologic temperature and pH, and poor microscopy setting for embryo transfer).
In conclusion, the results of this study showing that mouse oocytes can be cryopreserved successfully by low concentrations of trehalose and DMSO support potential use of sugars, such as trehalose, as intracellular and extracellular cryoprotectants in cryopreservation of mammalian oocytes and open the door for convenient long-term storage of the oocytes at high subzero temperatures, as well as for transportation on dry ice.
Acknowledgments
The authors would like to thank Edyta Szurek and Marissa Markyna for their technical assistance.
Footnotes
1Supported by grant R01HD049537 from the National Institute Of Child Health and Human Development to A.E.
REFERENCES
- Jemal A, Thomas A, Murray T, Thun M.Cancer statistics, 2002. CA Cancer J Clin 2002; 52: 23–47. [DOI] [PubMed] [Google Scholar]
- Parkening TA, Tsunoda Y, Chang MC.Effects of various low temperatures, cryoprotective agents and cooling rates on the survival, fertilizability and development of frozen-thawed mouse eggs. J Exp Zool 1976; 197: 369–374. [DOI] [PubMed] [Google Scholar]
- Whittingham DG.Fertilization in vitro and development to term of unfertilized mouse oocytes previously stored at −196 degrees C. J Reprod Fertil 1977; 49: 89–94. [DOI] [PubMed] [Google Scholar]
- Chen C.Pregnancy after human oocyte cryopreservation. Lancet 1986; 1: 884–886. [DOI] [PubMed] [Google Scholar]
- Porcu E, Fabbri R, Seracchioli R, Ciotti PM, Magrini O, Flamigni C.Birth of a healthy female after intracytoplasmic sperm injection of cryopreserved human oocytes. Fertil Steril 1997; 68: 724–726. [DOI] [PubMed] [Google Scholar]
- Tucker MJ, Wright G, Morton PC, Massey JB.Birth after cryopreservation of immature oocytes with subsequent in vitro maturation. Fertil Steril 1998; 70: 578–579. [DOI] [PubMed] [Google Scholar]
- Kuleshova L, Gianaroli L, Magli C, Ferraretti A, Trounson A.Birth following vitrification of a small number of human oocytes: case report. Hum Reprod 1999; 14: 3077–3079. [DOI] [PubMed] [Google Scholar]
- Yoon TK, Chung HM, Lim JM, Han SY, Ko JJ, Cha KY.Pregnancy and delivery of healthy infants developed from vitrified oocytes in a stimulated in vitro fertilization-embryo transfer program [letter]. Fertil Steril 2000; 74: 180–181. [DOI] [PubMed] [Google Scholar]
- Porcu E, Fabbri R, Damiano G, Giunchi S, Fratto R, Ciotti PM, Venturoli S, Flamigni C.Clinical experience and applications of oocyte cryopreservation. Mol Cell Endocrinol 2000; 169: 33–37. [DOI] [PubMed] [Google Scholar]
- Borini A, Lagalla C, Bonu MA, Bianchi V, Flamigni C, Coticchio G.Cumulative pregnancy rates resulting from the use of fresh and frozen oocytes: 7 years' experience. Reprod Biomed Online 2006; 12: 481–486. [DOI] [PubMed] [Google Scholar]
- Boldt J, Tidswell N, Sayers A, Kilani R, Cline D.Human oocyte cryopreservation: 5-year experience with a sodium-depleted slow freezing method. Reprod Biomed Online 2006; 13: 96–100. [DOI] [PubMed] [Google Scholar]
- Chen SU, Lien YR, Chen HF, Chang LJ, Tsai YY, Yang YS.Observational clinical follow-up of oocyte cryopreservation using a slow-freezing method with 1,2-propanediol plus sucrose followed by ICSI. Hum Reprod 2005; 20: 1975–1980. [DOI] [PubMed] [Google Scholar]
- Yoon TK, Kim TJ, Park SE, Hong SW, Ko JJ, Chung HM, Cha KY.Live births after vitrification of oocytes in a stimulated in vitro fertilization-embryo transfer program. Fertil Steril 2003; 79: 1323–1326. [DOI] [PubMed] [Google Scholar]
- Kuwayama M, Vajta G, Kato O, Leibo SP.Highly efficient vitrification method for cryopreservation of human oocytes. Reprod Biomed Online 2005; 11: 300–308. [DOI] [PubMed] [Google Scholar]
- Lucena E, Bernal DP, Lucena C, Rojas A, Moran A, Lucena A.Successful ongoing pregnancies after vitrification of oocytes. Fertil Steril 2006; 85: 108–111. [DOI] [PubMed] [Google Scholar]
- Antinori M, Licata E, Dani G, Cerusico F, Versaci C, Antinori S.Cryotop vitrification of human oocytes results in high survival rate and healthy deliveries. Reprod BioMed Online 2007; 14: 73–79. [DOI] [PubMed] [Google Scholar]
- Tedder RS, Zuckerman MA, Goldstone AH, Hawkins AE, Fielding A, Briggs EM, Irwin D, Blair S, Gorman AM, Patterson KG, Linch DC, Heptonstall J, Brink NS.Hepatitis B transmission from contaminated cryopreservation tank. Lancet 1995; 346: 137–140. [DOI] [PubMed] [Google Scholar]
- Bielanski A, Nadin-Davis S, Sapp T, Lutze-Wallace C.Viral contamination of embryos cryopreserved in liquid nitrogen. Cryobiology 2000; 40: 110–116. [DOI] [PubMed] [Google Scholar]
- Crowe JH, Hoekstra FA, Crowe LM.Anhydrobiosis. Annu Rev Physiol 1992; 54: 579–599. [DOI] [PubMed] [Google Scholar]
- Potts M.Desiccation tolerance of prokaryotes. Microbiol Rev 1994; 58: 755–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe JH, Crowe LM, Carpenter JF.Preserving dry biomaterials: the water replacement hypothesis, Part I. BioPharm 1993; 28: 31 [Google Scholar]
- Crowe JH, Crowe LM, Carpenter JF.Preserving dry biomaterials: the water replacement hypothesis, Part II. BioPharm 1993; 28: 40–44. [Google Scholar]
- Crowe JH, Leslie SB, Crowe LM.Is vitrification sufficient to preserve liposomes during freeze-drying? Cryobiology 1994; 31: 355–366. [DOI] [PubMed] [Google Scholar]
- Crowe JH, Crowe LM, Carpenter JF, Aurell Wistrom C.Stabilization of dry phospholipid bilayers and proteins by sugars. Biochem J 1987; 242: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmele RL, Jr, Stushnoff C, Carpenter JF.Real-time in situ monitoring of lysozyme during lyophilization using infrared spectroscopy: dehydration stress in the presence of sucrose. Pharm Res 1997; 14: 1548–1555. [DOI] [PubMed] [Google Scholar]
- Chen T, Fowler A, Toner M.Literature review: supplemented phase diagram of the trehalose-water binary mixture. Cryobiology 2000; 40: 277–282. [DOI] [PubMed] [Google Scholar]
- Levine H, Slade L.Thermomechanical properties of small-carbohydrate water glasses and rubbers - kinetically metastable systems at sub-zero temperatures. J Chem Soc Faraday Transactions 1988; 84: 2619–2633. [Google Scholar]
- Murthy SS.Some insight into the physical basis of the cryoprotective action of dimethyl sulfoxide and ethylene glycol. Cryobiology 1998; 36: 84–96. [DOI] [PubMed] [Google Scholar]
- Jabrane S, Letoffe JM, Claudy P.Vitrification and crystallization in the R(-)1,2-propanediol-S(+)1,2-propanediol system. Thermochim Acta 1995; 258: 33–47. [Google Scholar]
- Mazur P, Farrant J, Leibo SP, Chu EH.Survival of hamster tissue culture cells after freezing and thawing. Interactions between protective solutes and cooling and warming rates. Cryobiology 1969; 6: 1–9. [DOI] [PubMed] [Google Scholar]
- Leibo SP, Farrant J, Mazur P, Hanna MG, Jr, Smith LH.Effects of freezing on marrow stem cell suspensions: interactions of cooling and warming rates in the presence of PVP, sucrose, or glycerol. Cryobiology 1970; 6: 315–332. [DOI] [PubMed] [Google Scholar]
- Beattie GM, Crowe JH, Lopez AD, Cirulli V, Ricordi C, Hayek A.Trehalose: a cryoprotectant that enhances recovery and preserves function of human pancreatic islets after long-term storage. Diabetes 1997; 46: 519–523. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Russo MJ, Bieganski R, Fowler A, Cheley S, Bayley H, Toner M.Intracellular trehalose improves the survival of cryopreserved mammalian cells. Nat Biotechnol 2000; 18: 163–167. [DOI] [PubMed] [Google Scholar]
- Guo N, Puhlev I, Brown DR, Mansbridge J, Levine F.Trehalose expression confers desiccation tolerance on human cells. Nat Biotechnol 2000; 18: 168–171. [DOI] [PubMed] [Google Scholar]
- Elliott GD, Liu XH, Cusick JL, Menze M, Vincent J, Witt T, Hand S, Toner M.Trehalose uptake through P2X7 purinergic channels provides dehydration protection. Cryobiology 2006; 52: 114–127. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Lawitts JA, Toner M, Toth TL.Quantitative microinjection of trehalose into mouse oocytes and zygotes, and its effect on development. Cryobiology 2003; 46: 121–134. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Toner M, Toth TL.Beneficial effect of microinjected trehalose on the cryosurvival of human oocytes. Fertil Steril 2002; 77: 152–158. [DOI] [PubMed] [Google Scholar]
- Johnson MH, Pickering SJ.The effect of dimethylsulphoxide on the microtubular system of the mouse oocyte. Development 1987; 100: 313–324. [DOI] [PubMed] [Google Scholar]
- Vanderelst J, Vandenabbeel E, Nerinckx S, Vansteirteghem A.Parthenogenetic activation pattern and microtubular organization of the mouse oocyte after exposure to 1,2-propanediol. Cryobiology 1992; 29: 549–562. [DOI] [PubMed] [Google Scholar]
- Huang JYJ, Chen HY, Tan SL, Chian RC.Effects of osmotic stress and cryoprotectant toxicity on mouse oocyte fertilization and subsequent embryonic development in vitro. Cell Preserv Technol 2006; 4: 149–160. [Google Scholar]
- Larman MG, Katz-Jaffe MG, Sheehan CB, Gardner DK.1,2-propanediol and the type of cryopreservation procedure adversely affect mouse oocyte physiology. Hum Reprod 2007; 22: 250–259. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Szurek E.Effect of trehalose and dimethylsulfoxide on oocyte cytoskeleton. Cryobiology 2007; 55: 341 [Google Scholar]
- Hogan B, Beddington B, Constantini F, Lacy E. Manipulating the Mouse Embryo. Woodbury, NY:: Cold Spring Harbor Laboratory Press;; 1994. [Google Scholar]
- Chang LQL, Milton N, Rigsbee D, Mishra DS, Tang XLC, Thomas LC, Pikal MJ.Using modulated DSC to investigate the origin of multiple thermal transitions in frozen 10% sucrose solutions. Thermochim Acta 2006; 444: 141–147. [Google Scholar]
- Blond G, Simatos D, Catte M, Dussap CG, Gros JB.Modeling of the water-sucrose state diagram below 0 degrees C. Carbohyd Res 1997; 298: 139–145. [Google Scholar]
- Lehn-Jensen H, Rall WF.Cryomicroscopic observations of cattle embryos during freezing and thawing. Theriogenology 1983; 19: 263–277. [DOI] [PubMed] [Google Scholar]
- Todorow SJ, Siebzehnrubl ER, Spitzer M, Koch R, Wildt L, Lang N.Comparative results on survival of human and animal eggs using different cryoprotectants and freeze-thawing regimens. II. Human. Hum Reprod 1989; 4: 812–816. [DOI] [PubMed] [Google Scholar]
- Rall WF, Meyer TK.Zona fracture damage and its avoidance during the cryopreservation of mammalian embryos. Theriogenology 1989; 31: 683–692. [DOI] [PubMed] [Google Scholar]
- Carroll J, Wood MJ, Whittingham DG.Normal fertilization and development of frozen-thawed mouse oocytes: protective action of certain macromolecules. Biol Reprod 1993; 48: 606–612. [DOI] [PubMed] [Google Scholar]
- Chen SU, Lien YR, Cheng YY, Chen HF, Ho HN, Yang YS.Vitrification of mouse oocytes using closed pulled straws (CPS) achieves a high survival and preserves good patterns of meiotic spindles, compared with conventional straws, open pulled straws (OPS) and grids. Hum Reprod 2001; 16: 2350–2356. [DOI] [PubMed] [Google Scholar]
- O'Neil L, Paynter SJ, Fuller BJ.Vitrification of mature mouse oocytes: improved results following addition of polyethylene glycol to a dimethyl sulfoxide solution. Cryobiology 1997; 34: 295–301. [DOI] [PubMed] [Google Scholar]
- Stachecki JJ, Willadsen SM.Cryopreservation of mouse oocytes using a medium with low sodium content: effect of plunge temperature. Cryobiology 2000; 40: 4–12. [DOI] [PubMed] [Google Scholar]
- Lane M, Gardner DK.Vitrification of mouse oocytes using a nylon loop. Mol Reprod Dev 2001; 58: 342–347. [DOI] [PubMed] [Google Scholar]
- Schroeder AC, Champlin AK, Mobraaten LE, Eppig JJ.Developmental capacity of mouse oocytes cryopreserved before and after maturation in vitro. J Reprod Fertil 1990; 89: 43–50. [DOI] [PubMed] [Google Scholar]
- Buchanan SS, Gross SA, Acker JP, Toner M, Carpenter JF, Pyatt DW.Cryopreservation of stem cells using trehalose: evaluation of the method using a human hematopoietic cell line. Stem Cells Dev 2004; 13: 295–305. [DOI] [PubMed] [Google Scholar]
- Mandelbaum J, Junca AM, Tibi C, Plachot M, Alnot MO, Rim H, Salat-Baroux J, Cohen J.Cryopreservation of immature and mature hamster and human oocytes. Ann N Y Acad Sci 1988; 541: 550–561. [DOI] [PubMed] [Google Scholar]
- Toth TL, Baka SG, Veeck LL, Jones HW, Jr, Muasher S, Lanzendorf SE.Fertilization and in vitro development of cryopreserved human prophase I oocytes. Fertil Steril 1994; 61: 891–894. [DOI] [PubMed] [Google Scholar]
- Tucker M, Wright G, Morton P, Shanguo L, Massey J, Kort H.Preliminary experience with human oocyte cryopreservation using 1,2-propanediol and sucrose. Hum Reprod 1996; 11: 1513–1515. [DOI] [PubMed] [Google Scholar]
- Gook DA, Osborn SM, Johnston WI.Cryopreservation of mouse and human oocytes using 1,2-propanediol and the configuration of the meiotic spindle. Hum Reprod 1993; 8: 1101–1109. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Toth TL, Toner M.Alterations of the cytoskeleton and polyploidy induced by cryopreservation of metaphase II mouse oocytes. Fertil Steril 1998; 69: 944–957. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Toth TL, Toner M.Cryoprotection of mouse and human oocytes by intracellular trehalose. Cryobiology 2001; 43: 320 [Google Scholar]
- Fabbri R, Porcu E, Marsella T, Rocchetta G, Venturoli S, Flamigni C.Human oocyte cryopreservation: new perspectives regarding oocyte survival. Hum Reprod 2001; 16: 411–416. [DOI] [PubMed] [Google Scholar]
- Chen SU, Lien YR, Tsai YY, Chang LJ, Ho HN, Yang YS.Successful pregnancy occurred from slowly freezing human oocytes using the regime of 1.5 mol/l 1,2-propanediol with 0.3 mol/l sucrose. Hum Reprod 2002; 17: 1412; author reply 1412–1413. [DOI] [PubMed] [Google Scholar]
- Coticchio G, De Santis L, Rossi G, Borini A, Albertini D, Scaravelli G, Alecci C, Bianchi V, Nottola S, Cecconi S.Sucrose concentration influences the rate of human oocytes with normal spindle and chromosome configurations after slow-cooling cryopreservation. Hum Reprod 2006; 21: 1771–1776. [DOI] [PubMed] [Google Scholar]
- Borini A, Sciajno R, Bianchi V, Sereni E, Flamigni C, Coticchio G.Clinical outcome of oocyte cryopreservation after slow cooling with a protocol utilizing a high sucrose concentration. Hum Reprod 2006; 21: 512–517. [DOI] [PubMed] [Google Scholar]
- De Santis L, Cino I, Rabellotti E, Papaleo E, Calzi F, Fusi FM, Brigante C, Ferrari A.Oocyte cryopreservation: clinical outcome of slow-cooling protocols differing in sucrose concentration. Reprod BioMed Online 2007; 14: 57–63. [DOI] [PubMed] [Google Scholar]
- Whittingham DG, Leibo SP, Mazur P.Survival of mouse embryos frozen to −196 degrees and −269 degrees C. Science 1972; 178: 411–414. [PubMed] [Google Scholar]
- Miyamoto H, Ishibashi T.The protective action of glycols against freezing damage of mouse and rat embryos. J Reprod Fertil 1978; 54: 427–432. [DOI] [PubMed] [Google Scholar]
- Eroglu A, Elliott G, Wright DL, Toner M, Toth TL.Progressive elimination of microinjected trehalose during mouse embryonic development. Reprod Biomed Online 2005; 10: 503–510. [DOI] [PubMed] [Google Scholar]
- Potter H.Protocols for using electroporation to stably or transiently transfect mammalian cells. Chang DC, Chassy BM, Saunders JA, Showers AE.Guide to Electroporation and Electrofusion San Diego, CA:Academic Press;1992: 457–463. [Google Scholar]
- Singer MA, Lindquist S.Thermotolerance in Saccharomyces cerevisiae: the Yin and Yang of trehalose. Trends Biotechnol 1998; 16: 460–468. [DOI] [PubMed] [Google Scholar]
- Somero GN.Protons, osmolytes, and fitness of internal milieu for protein function. Am J Physiol 1986; 251: R197–R213. [DOI] [PubMed] [Google Scholar]
- Sola-Penna M, Ferreira-Pereira A, Lemos AP, Meyer-Fernandes JR.Carbohydrate protection of enzyme structure and function against guanidinium chloride treatment depends on the nature of carbohydrate and enzyme. Eur J Biochem 1997; 248: 24–29. [DOI] [PubMed] [Google Scholar]
- Chen Q, Haddad GG.Role of trehalose phosphate synthase and trehalose during hypoxia: from flies to mammals. J Exp Biol 2004; 207: 3125–3129. [DOI] [PubMed] [Google Scholar]