

Abstract
The glutamate receptor 6 (GluR6 or GRIK2, one of the kainate receptors) gene resides in a genetic linkage region (6q21) associated with bipolar disorder (BPD), but its function in affective regulation is unknown. Compared with wild-type (WT) and GluR5 knockout (KO) mice, GluR6 KO mice were more active in multiple tests and super responsive to amphetamine. In a battery of specific tests, GluR6 KO mice also exhibited less anxious or more risk-taking type behavior and less despair-type manifestations, and they also had more aggressive displays. Chronic treatment with lithium, a classic antimanic mood stabilizer, reduced hyperactivity, aggressive displays and some risk-taking type behavior in GluR6 KO mice. Hippocampal and prefrontal cortical membrane levels of GluR5 and KA-2 receptors were decreased in GluR6 KO mice, and chronic lithium treatment did not affect these decreases. The membrane levels of other glutamatergic receptors were not significantly altered by GluR6 ablation or chronic lithium treatment. Together, these biochemical and behavioral results suggest a unique role for GluR6 in controlling abnormalities related to the behavioral symptoms of mania, such as hyperactivity or psychomotor agitation, aggressiveness, driven or increased goal-directed pursuits, risk taking and supersensitivity to psychostimulants. Whether GluR6 perturbation is involved in the mood elevation or thought disturbance of mania and the cyclicity of BPD are unknown. The molecular mechanism underlying the behavioral effects of lithium in GluR6 KO mice remains to be elucidated.
Keywords: glutamate receptor 6 (GluR6), kainate receptors (KARs), mood-related behaviors, bipolar disorder (BPD), knockout mice (KO), lithium
Introduction
Bipolar disorder (BPD) (also known as manic-depressive illness) is one of the most severely debilitating medical illnesses, affecting the lives and functioning of millions worldwide.1 Emerging data suggest that BPD arises from the complex inheritance of multiple susceptibility genes.2–4 Phenotypically, it is a very complex disease in which patients alternate between episodes of mania and depression. Manic episodes are characterized by euphoric or irritable mood, thought disturbances, such as racing thoughts and grandiosity, and behavioral symptoms, including hyperactivity, poor judgment, recklessness, aggressive behavior, increased goal-directed behavior, increased pursuit of pleasurable activities with potentially painful consequences and substance abuse. Episodes of depression are characterized by depressed or irritable mood accompanied by markedly diminished interest or pleasure in everyday activities, psychomotor agitation or retardation, changes in body weight or appetite, poor memory and concentration, fatigue and recurrent thoughts of death or suicide.1
There is a growing appreciation that BPD can be best conceptualized as a genetically influenced disorder of synapses and circuits. It is thus noteworthy that recent human genetic studies have identified GRIK2 (which encodes for GluR6, a kainite receptor implicated in synaptic plasticity) as a potential BPD susceptibility gene.5 Genetic linkage of BPD to chromosome 6q21 has been demonstrated in several studies, and genome-wide significant linkage was recently established by meta-analysis.6–9 Buervenich and colleagues investigated the broad linkage peak on 6q and found that a specific haplotype, located on the regulatory region of the human GluR6 gene, was associated with BPD. In addition, very recent post-mortem studies demonstrate a reduction in GluR6 mRNA levels in the brains of BPD patients,10 thus pointing to GluR6 as a potential candidate gene in the pathophysiology of BPD.
A completely independent genetic study has also recently implicated the GRIK2 gene in a behavioral phenotype that may be associated with BPD.11 Intriguingly, the very same gene has been found to confer sensitivity to treatment-emergent suicidal ideation in a large collaborative genetic study of individuals with mood disorders (Sequenced Treatment Alternatives to Relieve Depression (STAR*D)).11 Individuals with variations in genes encoding GluR6 and GluR3 (an AMPA receptor) were 15 times more likely to experience treatment-induced suicidal ideation. Although the neurobiologic basis of this very rare (but serious) side effect is unknown, it is noteworthy that individuals with BPD—and potentially those with a bipolar diathesis—are susceptible to antidepressant-induced dysphoric activation/agitation and potential suicidality.12,13
Among the ionotropic glutamate receptors are the kainate receptor (KAR) family comprising five subunits: GluR 5–7 and KA-1-2 (also named glutamate receptor ionotropic kainate (GRIK) 1–5, respectively). KARs are expressed in the areas of neuronal circuits involved in mood regulation.14,15 They have been shown to bidirectionally regulate the release of glutamate at the mossy fiber to CA3 synapses,16 and notably, they are also involved in long-term potentiation induction at the hippocampus and the amygdala.17 Moreover, it is now widely accepted that presynaptic KARs control the release of GABA (γ-aminobutyric acid) in the hippocampus and the amygdala.16,18 Furthermore, a growing body of evidence demonstrates that KARs have metabotropic actions in addition to their ionotropic activity that modulates both hippocampal inhibitory19 and excitatory postsynaptic currents20 as well as pyramidal cell excitability.21,22 The metabotropic actions include interaction with G proteins, induction of second messengers and activation of PKA (protein kinase A), PKC (protein kinase C) and MAP (mitogen-activated protein) kinases.19–22
In view of the GluR6 receptor as a putative BPD susceptibility gene, we undertook a series of animal studies to elucidate the role of this receptor in modulating behavioral patterns related to facets of the complex symptoms of BPD. Furthermore, to determine potential specificity of this receptor to mood-related behaviors, we also investigated the role of GluR5 receptors. We used a battery of behavioral studies tailored to monitor diverse symptoms of either mania23 or depression.24 The chronic effects of lithium—the classic antimanic mood stabilizer—on behavioral alterations of mutant mice were also investigated. Finally, to assess any potential biological changes associated with GluR6 ablation or a possible biochemical mechanism mediating aberrant behaviors in GluR6 KO mice, a series of biochemical studies were also conducted.
Materials and methods
Animals
KAR KO mice were originated from 129/Sv and C57/ Bl6 background, and then backcrossed with 129Sv/Ev mice for at least 10 generations to provide an isogenic 129Sv/Ev strain.25 GluR6 KO, GluR5 KO and WT male mice, aged 8 weeks old, were single housed in an animal room at a constant temperature (22±1 °C) and a 12-h light/dark cycle with free access to food and water. Female mice were not used in the studies to avoid the potential confounding effects of the menstrual cycle on behavioral and neurochemical measures. Mice were subjected to multiple tests in the following sequence designed from less to more intrusive as recommended previously:26 open-field, social interaction, elevated plus maze, forced swim, resident–intruder, saline–response and psychostimulant–response tests. The tests were performed at least 24 h apart. Additional cohorts of mice were used for other behavioral evaluations, such as passive avoidance test, and for experiments with lithium treatment. Mice to be compared were tested concurrently in the same room under dim illumination (30 lux) by the same testers between 10:00 and 18:00 hours. All experimental procedures were approved by the Animal Use Committee of the National Institute of Mental Health (NIMH) and were conducted according to the National Institutes of Health guidelines.
Passive avoidance test
The test was conducted as described previously by El-Ghundi,27 with the modification of using GEMINI Avoidance System driven by in-house software. In brief, all mice were given one habituation trial to explore both chambers for 120 s, followed by two training trials separated by 5 min. During the training trials, individual mice were placed in the bright chamber for 30 s, after which a guillotine door was opened and the latency to enter the dark chamber was measured for 300 s. On the second trial, on entrance into the dark compartment (all four paws and tail inside), the guillotine door was closed and the mouse was confined in the dark compartment for 10 s, followed by two consecutive 3-s (inescapable) foot shocks (0.6 mA), left in the compartment for an additional 10 s, and then returned to their home cage and left there undisturbed until the next acquisition trial. Acquisition (learning) and retention (memory) of passive avoidance response were assessed 5 min, 24 h, 48 h and 14 days, respectively, after the training. During assessment, each individual mouse was placed in the light chamber, and the latency to enter the dark chamber was recorded for up to 300 s. Passive avoidance response was assessed by comparing the step-through latencies during training and assessment trials.
Open-field test
The experiment was carried out as described previously.28 In brief, a 50 ×50 cm arena was used for the open-field test. To study spontaneous locomotion, mice were placed in one corner of the arena and their behavior was recorded for 60 min with the Ethovision video tracking system (Noldus, Leesburg, VA, USA) and videotaped. The experiment was repeated for 3 consecutive days and again 14 and 15 days after the first exposure to the arena to test persistency of the activity pattern. To study saline injection response as a control for amphetamine injection, mice were first placed in one corner of the arena and their behavior was recorded for 30 min; they then received saline injection (2 ml kg−1, i.p.), were placed back to the same arena and their behavior was recorded for an additional 30 min. To test amphetamine response, the same procedure was carried out, substituting amphetamine (2 mgkg−1, Sigma, St Louis, MO) for saline. Measures of locomotor activity, total time spent in the center and frequency measures for center visits (30 × 30 cm) were collected. The open field was wiped between trials with a 10% alcohol solution.
Home cage activity
Mice were individually housed in the new home cage at the beginning of the testing period. After 1 day, mice were transported in the same cage and placed in the testing room from 08:00 and 10:00 hours every day for 2 consecutive days to avoid the potential effects of transportation and testing room on behavior. Activity of the mouse was then videotaped with a digital camera for 30 min between 08:00 and 10:00 hours every day for the following 2 consecutive days. Digital video files were analyzed using CleverSystems' HomeCageScan Suite (CleverSys. Inc., Reston, VA, USA). To validate the software, behavioral events identified by HomeCageScan were compared with the findings of observers; between the software and the observers, consistency of each reported behavior exceeded 90%. For the purpose of this study, two types of home cage activities were scored: general behavior (turning, walking, digging, grooming, drinking and eating) and exploratory behavior (rearing, sniffing and foraging).
Social interaction test
Pairs of mice of the same genotype were placed in opposite corners of a 50×50 cm open-field arena. Their activities were videotaped for 5 min, and the frequency of social behaviors was scored for olfactory investigations (sniffing), side-by-side behavior, tail rattling and attack biting. Each arena was wiped clean between trials with a 10% alcohol solution.
Elevated plus-maze test
The procedure was carried out as reported previously.28,29 A Plexiglas plus-shaped maze containing two dark and enclosed arms and two open and lit arms, elevated 50 cm above ground, was used to examine anxiety-related behaviors. The arms were 30×5 cm with a 5×5 cm center area, and the walls of the closed arms were 40 cm high. Individual mice were placed in the center of the maze, tracked for 5 min with a video camera, and then returned to their home cage. The plus maze was wiped clean between trials with a 10% alcohol solution. Time spent in the maze and frequency of visits to the different zones of the maze were scored using Ethovision (Noldus) or the TopScan Suite of CleverSystems (CleverSys Inc.).
Forced swim test
The test was conducted as described previously.28,30 Briefly, transparent Plexiglas cylinders, 50 cm high with a diameter of 20 cm, were filled with tap water at 22–24 °C to approximately 25 cm in a way that mice were not able to touch the floor or escape. Individual mice were placed in the water for a 6-min session, and their behavior was videotaped for later analysis. At the end of each session, mice were dried with a paper towel and returned to their home cage. Water was replaced after each trial. The time of immobility, defined as a lack of activity except movements needed to keep their nose above water, was scored during the last 4 min of the session.
Resident intruder test
A 129Sv/Ev naive male mouse was introduced into the home cage of each mouse tested. The interaction between the two mice was videotaped for 5 min, and the frequency of attacks on the intruder was counted in each trial. Each naive mouse was used no more than once as an intruder.
Lithium treatment
The treatment was conducted using a previously tested regimen that maintains the trunk blood level of lithium within the human therapeutic range (0.4–1.2 mM).1 WT and GluR6 KO mice were fed with lithium carbonate chow (2.4 gkg−1, Bio-Serv, French-town, NJ, USA) or control chow identical to lithium carbonate chow with the exception of lithium salt for 4–6 days (short-term treatment) and 4 weeks (chronic treatment). Brain lithium levels after chronic treatment were 0.55±0.01 mM (mean±s.e.m.). Owing to the well-known lithium side effects of polyuria and polydipsia, cage bedding was changed twice a week and animals were supplied with 0.9% NaCl (saline) solution in addition to tap water to prevent possible electrolyte imbalance.
Immunoblotting
Cytosolic and membrane fractions were prepared using a previously described method.31 In brief, the frontal cortex (FCX) and hippocampus samples were homogenized in a hypotonic protein extraction buffer containing a protease inhibitor cocktail (Sigma) and a phosphatase inhibitor cocktail I and II (Sigma) by passing through a 19-gauge needle 10 times, followed by a 22-gauge needle 10 times. The homogenates were then centrifuged at 1200 r.p.m. for 12 min at 4 °C to remove non-dissolved debris and nuclear components. The particulate (representing largely cell membrane) and soluble (representing largely cytoplasmic) fractions were then separated by centrifugation at 14 000 r.p.m. for 30 min at 4 °C. To make it soluble, the membrane pellet was suspended in the protein extraction buffer added with 1% Triton X-100. Protein concentrations were determined using BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL). The linearity of the protein concentration for immunoblotting was ascertained by resolution of selected concentrations of protein.
Protein immunoblotting was conducted using previously described methods.32 In brief, 5–10 μg protein samples were separated by 10% sodium dodecyl sulfate-gradient gel electrophoresis (Invitrogen, Carlsbad, CA) and immunoblotted with anti-GluR5 (1:500; Millipore, Billerica, MA), anti-KA-1 (1:1000; Abcam, Cambridge, MA), anti-KA-2 (1:1000; Upstate), anti-GluR1 (1:1000; Millipore), anti-GluR2 (1:1000; Zymed Laboratories, San Francisco, CA), anti-GluR3 (1:1000; Chemicon), anti-NR1 (1:500; Chemicon) or anti-NR2A (1:500; Upstate) antibodies. Anti-rabbit or anti-mouse IgG, horseradish peroxidase-linked antibodies (Cell Signaling Technology, Danvers, MA) were used as secondary antibodies. The immunoreactive bands were visualized by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) and exposed to Kodak BioMax or Biolight film (Eastman Kodak, Rochester, NY). The signal intensities were quantified using Kodak Imaging System, based on standard curves of each protein.
Statistical analysis
Two- and one-way analysis of variance (ANOVA), post hoc Fisher LSD (least significant difference) and Student's t-tests were conducted using the software STATISTICA 7 (Statsoft, Tulsa, OK, USA) and Prism 4 (GraphPad Software Inc., San Diego, CA, USA).
Results
Previous studies have shown that GluR6 KO mice have no gross neuroanatomical abnormalities or sensorimotor deficits.33,34 GluR6 KO mice frequently engage in fighting within their home cages; therefore, mice of all genotypes were individually housed throughout these experiments. As previously shown by others,33 GluR6 KO mice weighed less (24.2± 0.52 g (mean±s.e.m., n = 13)) than GluR5 KO (25.86± 0.52 g (n = 13)) and WT mice (27.47±0.54 g (n = 12)) (ANOVA, F2,3 = 9.57, P < 0.001). Consistent with a lack of spatial learning and memory impairment,33,34 no significant effects of genotype were detected on the outcome measures of the passive avoidance test (Figure 1), a test that assesses fear-associated learning and memory.
Figure 1.
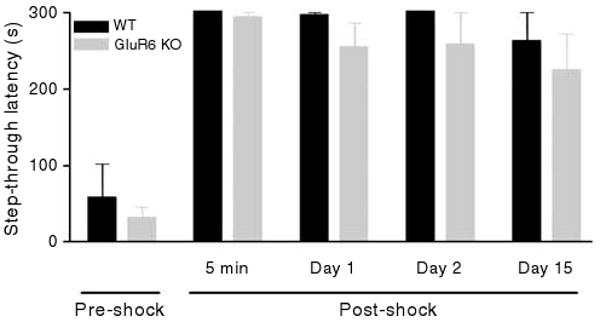
GluR6 KO mice have no significant impairment in the passive avoidance test. Acquisition (learning) and retention (memory) of the passive avoidance response did not differ in GluR6 KO and WT mice. During the training trial, all mice promptly entered the dark compartment, whereas after receiving foot shocks, all mice quickly learned to avoid entering the dark chamber (previously paired with foot shock), as indicated by increased latencies during the post-shock test trials. No statistical difference was found between the two groups in the avoidance latencies in the pre-shock trial and during retention trials conducted 5 min, 1 day, 2 days and 15 days post-shock (two-way ANOVA: genotype effect, F1,50 = 2.75, P > 0.1 (NS)). Results are means±s.e.m.
GluR6 KO mice exhibited more spontaneous activity
The spontaneous activities of mice were examined in the open-field test and in their home cage. In the open-field test, all three groups displayed habituation to the novel environment, as indicated by gradual movement reduction in the arena (Figure 2a). Compared with GluR5 KO and WT mice, GluR6 KO mice displayed increased spontaneous locomotor activity (two-way ANOVA, genotype effect, F2,35 = 87.55, P < 0.0001) in a pattern consistent with enhanced exploratory behavior rather than locomotor unrest. That is, if the GluR6 KO mice were simply hyperlocomoting, they would be expected to maintain similar levels of activity throughout the trial. When introducing the animals to the same arena in repeated tests, GluR5 KO and WT mice showed reduced activity; in contrast, the GluR6 KO mice continued to display hyperactivity in the same arena for at least 15 days (Figure 2b) (two-way ANOVA: genotype F2,174 = 148.3, P < 0.0001; day F4,174 = 4.9, P < 0.001; genotype × day F8,174 = 1.2, P =NS (nonsignificant) and one-way ANOVA (day): for GluR6 KO mice, F4,60 = 1.0, P = NS; for GluR5 KO mice, F4,60 = 4.6, P < 0.01; for WT mice, F4,54 = 3.6, P = 0.01).
Figure 2.
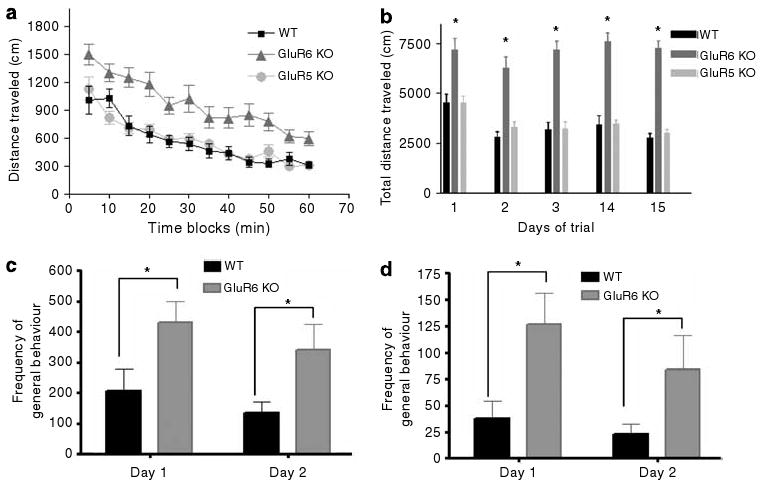
GluR6 KO mice exhibit increased spontaneous activity in the open field and home cage. (a) To study spontaneous locomotion in the open field, mice were placed in one corner of an open-field arena and their behavior was recorded for 60 min. GluR6 KO mice displayed increased spontaneous locomotor activity during 1 h of the first day of the open-field test compared with GluR5 KO and WT mice (two-way ANOVA, genotype effect, F2,35 = 87.55, P < 0.0001). (b) GluR6 KO mice displayed persistent hyperactivity in the open-field arena at each trial conducted over 3 consecutive days and also at days 14 and 15 from the first exposure (two-way ANOVA: genotype F2,174 = 148.3, P < 0.0001; day F4,174 = 4.9, P < 0.001; genotype × day F8,174 = 1.2, P = NS and one-way ANOVA (day): GluR6 KO F4,60 = 1.0, P = NS; GluR5 KO F4,60 = 4.6, P < 0.01; WT F4,54 = 3.6, P = 0.01). To study the home cage activity, WT and GluR6 KO mice were monitored in their home cages using a digital camcorder for 1 h between 0800 and 1000 hours for 2 consecutive days. Digital video files were analyzed using CleverSystems' HomeCageScan Suite for general activity (including, turning, walking, digging, grooming, drinking and eating) and exploratory activity. (c) GluR6 KO mice displayed significantly more home cage general activity, which was consistent over 2 days of testing (two-way ANOVA: genotype F1,14 = 8.194, P = 0.0125; time F1,14 = 1.926, P = 0.1869; genotype × time F1,14 = 0.02712, P = 0.8715). (d) GluR6 KO mice also displayed significantly more home cage exploratory activity, which was consistent over 2 days of testing (two-way ANOVA: genotype F1,14 = 10.99, P = 0.0051; time F1,14 = 1.279, P = 0.2771; genotype × time F1,14 = 0.3059, P = 0.5890). Results are means±s.e.m. *P ≤ 0.05.
Home cage activity monitoring showed that GluR6 KO mice displayed increased home cage general activity (Figure 2c) (two-way ANOVA: genotype F1,14 = 8.194, P = 0.0125; time F1,14 = 1.926, P = 0.1869; genotype × time F1,14 = 0.02712, P = 0.8715) as well as increased exploratory activity (Figure 2d) (two-way ANOVA: genotype F1,14 = 10.99, P = 0.0051; time F1,14 =1.279, P = 0.2771; genotype × time F1,14 = 0.3059, P = 0.5890).
GluR6 KO mice were more responsive to amphetamine
Saline injection alone did not change the activity levels in any of the three groups of mice (Figure 3a). For mice of all three genotypes, amphetamine injection increased locomotor activity above the activity level before the injection (Figure 3b). The amphetamine-induced increases in locomotion were significantly higher in GluR6 KO mice (Figure 3c) (ANOVA for genotype, F2,32 = 5.529, P = 0.0087; Tukey's multiple comparison test, for WT vs GluR6 KO mice, P < 0.05; for GluR5 KO vs GluR6 KO mice, P < 0.05).
Figure 3.
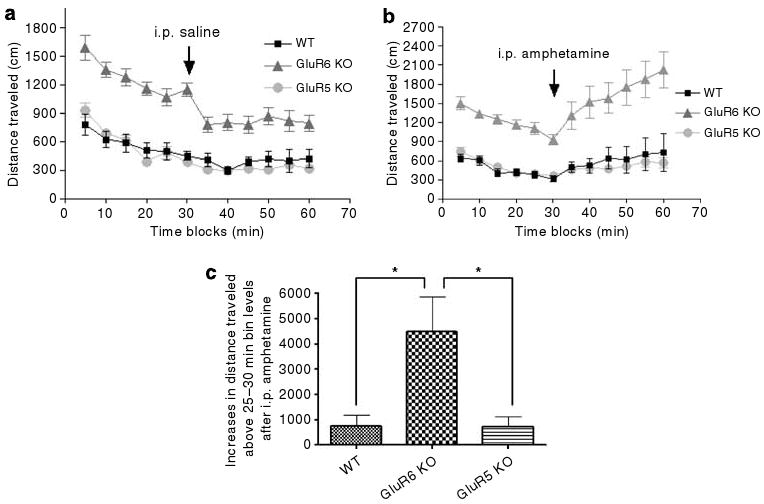
GluR6 KO mice exhibit increased amphetamine-induced response. (a) To study injection-induced irritability, mice were first placed in one corner of the arena and their behavior was recorded for 30 min as a control for amphetamine injection; immediately afterwards they were intraperitoneally injected with saline and placed back in the same arena for an additional 30 min. Saline injection did not affect locomotor activity for any of the three groups. (b) To study amphetamine-induced hyperactivity, the same procedure as in panel (a) was carried out, except that instead of saline, 2 mg kg−1 amphetamine was administered. Amphetamine increased locomotor activity above 25–30 min activity levels in mice of all three genotypes. (c) GluR6 KO mice displayed significantly more increases above the 25–30-min activity level after amphetamine injection compared with WT and GluR5 KO mice (ANOVA for genotype, F2,32 = 5.529, P = 0.0087; Tukey's multiple comparison test, WT vs GluR6 KO mice, P < 0.05; GluR5 KO vs GluR6 KO mice, P < 0.05). Results are means±s.e.m. *P≤0.05.
GluR6 KO mice were robustly aggressive
As mentioned before, there were more home cage fights among GluR6 KO mice. To further assess social behavior and aggressiveness in these mice, the social interaction and resident–intruder tests were conducted. In the social interaction test (Figure 4a), the number of side-by-side events did not differ between the groups. Compared with GluR5 KO and WT mice, GluR6 KO mice exhibited a significantly higher frequency of sniffing, which is an index of investigational activity (ANOVA, F2,19 = 3.44, P = 0.05; post hoc Fisher LSD test: GluR6 KO vs WT mice, P < 0.03). GluR6 KO mice displayed significantly more attack bites (ANOVA, F2,19 = 3.9, P < 0.04) and events of tail rattling (ANOVA, F2,19 = 11.4, P < 0.001); these are thought to reflect a state of high arousal and characteristic of dominant mice.35 In the resident–intruder test (Figure 4b), GluR6 KO mice attacked the intruder significantly more frequently than WT mice (Student's t-test: t23 =−2.12, P < 0.05).
Figure 4.
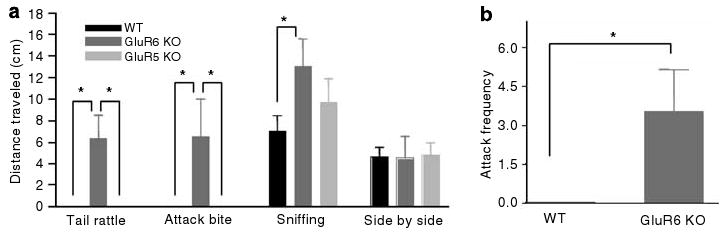
GluR6 KO mice display robustly aggressive behavior. (a) In the social interaction test, pairs of mice within the same strain were placed in opposite corners of an open-field arena. The frequency of social behaviors was counted over 5 min. GluR6 KO mice displayed a high frequency of destructive (aggressive) behaviors compared with GluR5 KO and WT mice (one-way ANOVA for ‘tail rattling’: F2,19 = 11.4, P < 0.001; ‘attack bite’: F2,19 = 3.9, P < 0.04). GluR6 KO mice displayed a high frequency of exploratory behavior compared with WT mice (one-way ANOVA for ‘sniffing’: F2,19 = 3.44, P = 0.05; post hoc Fisher LSD test for ‘sniffing’: GluR6 KO vs WT mice, P < 0.03). GluR6 KO mice displayed the same side-by-side behavior as GluR5 KO and WT mice (one-way ANOVA for ‘side by side’ F2,19 = 0.02, P = NS). (b) In the resident–intruder test, a 129Sv/Ev naive male mouse was introduced to the home cage of each mouse tested. The frequency of attacks on the intruder was counted over 5 min. GluR6 KO mice exhibited a high frequency of attack events toward their intruder compared with WT mice, who exhibited no attacks toward their intruder (Student's t-test: t23 =−2.12, P < 0.05). Results are means±s.e.m. *P≤0.05.
GluR6 KO mice exhibited less anxious and more risk-taking behavior
To measure anxiety- or risk-taking-related behavior in the mice, center activity in the open-field test (Figure 5) and open- or closed-arm activity in the elevated plus-maze test (Figure 6) were monitored. In these tests, the amount of time spent in the anxiety-provoking spaces (center of the open field and open arms of the maze) is thought to be driven by curiosity and prohibited by fear and self-protection, and are sensitive to treatment with anxiolytic drugs.36,37 GluR6 KO mice exhibited significantly increased entries to (Figure 5a) (one-way ANOVA: F2,35 = 33.6, P < 0.0001) and time spent in (Figure 5b) (one-way ANOVA: F2,35 = 13.4, P < 0.0001) the center of the open field compared with GluR5 KO and WT mice. In the elevated plus-maze test, GluR6 KO mice exhibited proportional (open arm vs all arms) increases in open-arm duration (Figure 6a) (ANOVA: F2,16 = 5.783, P = 0.0129; WT vs GluR6 KO mice, P < 0.05; WT vs GluR6 KO mice, P < 0.05), entry frequency (Figure 6b) (ANOVA: F2,16 = 6.256, P = 0.0098; WT vs GluR6 KO mice, P < 0.05; WT vs GluR6 KO mice, P < 0.05) and distance traveled (Figure 6c) (ANOVA: F2,16 = 7.264, P = 0.0057; WT vs GluR6 KO mice, P < 0.05; WT vs GluR6 KO mice, P < 0.05).
Figure 5.
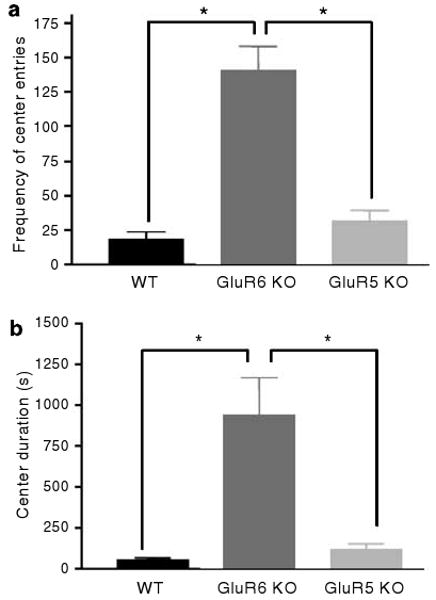
GluR6 KO mice show increased activity in anxiety-provoking subregions of the open-field test. To further study the activity in an anxiety-provoking environment, mice were placed in one corner of an open-field arena; the frequency of entering events to the center of the arena and the total time spent in it was recorded over 60 min. (a) GluR6 KO mice displayed a higher frequency of entries to the center of the open-field arena compared with GluR5 KO or WT mice (one-way ANOVA: F2,35 = 33.6, P < 0.0001). (b) GluR6 KO mice increased their time spent in the center of the open-field arena compared with GluR5 KO or WT mice (one-way ANOVA: F2,35 = 13.4, P < 0.0001). Results are means±s.e.m. *P < 0.0001.
Figure 6.
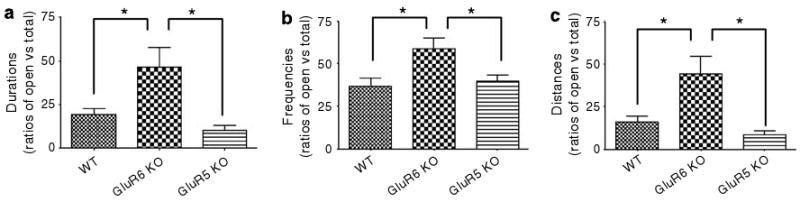
GluR6 KO mice exhibited increased activity in anxiety-provoking regions of the elevated plus maze test. A plus-shaped maze elevated above ground containing two dark closed arms and two open and lit arms without walls was used to examine activity in anxiety-provoking regions. Each mouse was placed in the center of the maze; time and frequency of visits to the different zones of the maze, as well as locomotion measures, were collected for 5-min sessions. (a) Ratios of open-arm vs total (open and closed arms) duration of time spent. GluR6 KO mice exhibited increases in duration of time spent in the open arms of the maze (ANOVA: F2,16 = 5.783, P = 0.0129; WT vs GluR6 KO mice, P < 0.05; WT vs GluR6 KO mice, P < 0.05). (b) Ratios of open-arm vs total arm entries. GluR6 KO mice exhibited increased open-arm entries (ANOVA: F2,16 = 6.256, P = 0.0098; WT vs GluR6 KO mice, P < 0.05; WT vs GluR6 KO mice, P < 0.05). (c) Ratios of open-arm vs total arm distance traveled. GluR6 KO mice exhibited increased distance traveled in the open arms (ANOVA: F2,16 = 7.264, P = 0.0057; WT vs GluR6 KO mice, P < 0.05; WT vs GluR6 KO mice, P < 0.05). Results are means±s.e.m. *P < 0.05.
GluR6 KO mice displayed less immobility in the forced swim test
The forced swim test, a test for the behavioral effects of antidepressants, is thought to monitor behavioral despair in a goal-directed task (for example, escaping).38 In this test, GluR6 KO mice displayed reduced immobility time compared to the mice of other strains (Figure 7) (one-way ANOVA: F2,35 = 4.5762, P < 0.05). Whether this reduced immobility in GluR6 KO mice reflects hyperactivity, increased goal-directed behavior or both, cannot be fully distinguished by this test alone.
Figure 7.
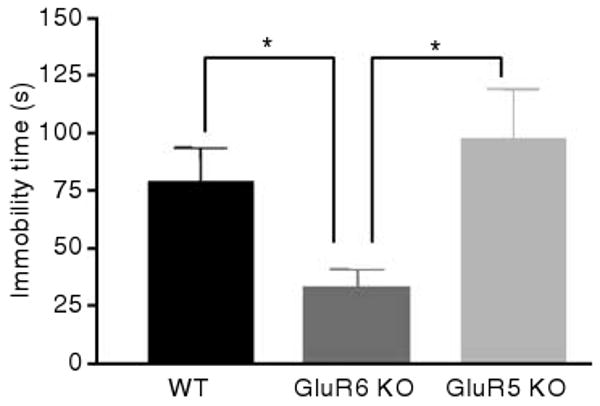
GluR6 KO mice displayed decreased immobility time in the forced swim test. Mice were placed in cylinders filled with water in a way that they were not able to touch the floor or escape for 6 min. Immobility time, defined as a lack of activity aside from small movements needed to keep the body floating, was measured throughout the last 4 min of the session. Immobility time was reduced for GluR6 KO mice compared with GluR5 KO or WT mice (one-way ANOVA: F2,35 = 4.5762, P < 0.05). Results are means±s.e.m. *P < 0.05.
Behavioral effects of chronic lithium treatment on GluR6 KO and WT mice
Chronic lithium treatment relieves symptoms in manic patients, but does not alter mood state in healthy subjects.1 Lithium, alone or as an adjunct to antidepressant therapy, is also used to treat depression.1 To determine if the behavioral excitement and aggression observed in the GluR6 mice were responsive to lithium, WT and GluR6 KO mice were treated with lithium using a clinically relevant regimen.28 The effects of short-term lithium treatment (4–6 days) were investigated. In WT mice, no significant behavioral effects of chronic lithium treatment were detected in the open-field test (Figure 8a), social interaction test (Figure 9a), resident–intruder test (Figure 9b) and center activity in the open-field test (Figure 10a). Chronic lithium treatment did not significantly alter open-arm proportional measures of the elevated plus-maze test in WT mice (duration: t10 = 0.9000, P = 0.3893; frequency: t10 = 0.435, P = 0.6728; distanced traveled: t10 = 1.390, P = 0.1947).
Figure 8.
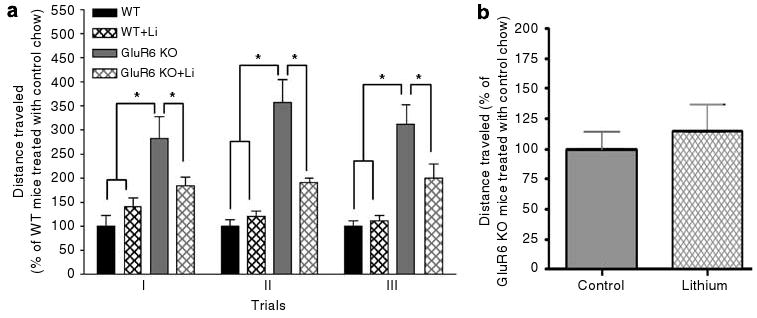
Chronic lithium treatment reduced spontaneous locomotor activity in GluR6 KO mice. (a) Four weeks of lithium treatment attenuated the spontaneous locomotor activity of GluR6 KO mice studied over 3 consecutive days of open-field test (two-way ANOVA: genotype F3,57 = 647.7, P < 0.0001; day F2,57 = 1512, P = NS; genotype×day F6,57 = 0.8, P = NS; one-way ANOVA: day 1: F3,19 = 7.7, P < 0.01; day 2: F3,19 = 25.7, P < 0.001; day 3: F3,19 = 14.2, P < 0.001; post hoc Fisher LSD test: P < 0.05, P < 0.001). Results are mean percentage of WT total activity of the same day. (b) Effects of 4-day lithium treatment on spontaneous locomotor activity of GluR6 KO mice. Short-term lithium treatment did not significantly alter spontaneous locomotor activity of GluR6 KO mice. Data are percent of mean value of GluR6 KO mice treated with control food. Results are means±s.e.m. *P≤0.05.
Figure 9.
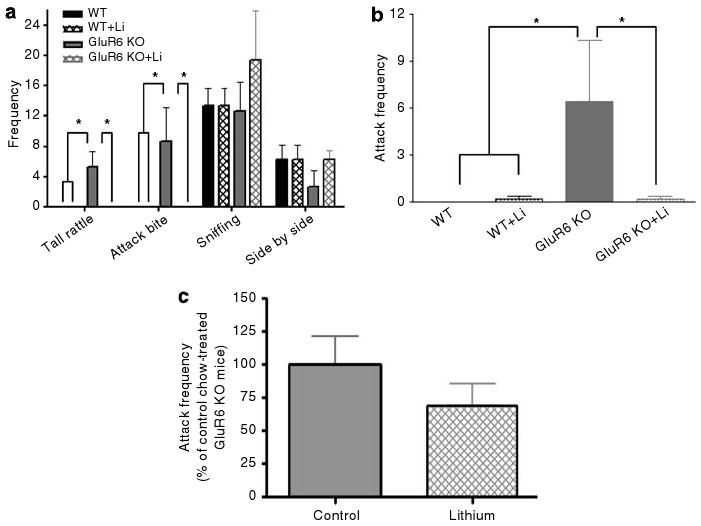
Chronic lithium dramatically decreased aggressive behavior in GluR6 KO mice. (a) Social interaction test: 4 weeks of lithium treatment caused a complete loss of tail rattling and attack bite events displayed by GluR6 KO mice toward their opponents from the same group (one-way ANOVA: tail rattling: F3,8 = 6.9, P < 0.05; attack bite F3,8 = 3.9, P = 0.05; sniffing: F3,8 = 1.3, P = 0.3 (NS); side by side: F3,8 = 0.63, P = 0.6 (NS)). (b) Resident–intruder test: 4 weeks of lithium treatment caused a robust reduction in attacks displayed by GluR6 KO mice toward a naive male intruder of the same strain (one-way ANOVA: F3,19 = 3.2, P < 0.05). Results are mean of percentage relative to WT±s.e.m. Li, lithium (chronic treatment). (c) Effects of 6 days of lithium treatment on measures of the resident–intruder test in GluR6 KO mice. Short-term lithium treatment did not significantly reduce attack frequencies of GluR6 KO mice (t14 = 1.171, P = 0.2611). Results are means±s.e.m. *P < 0.05.
Figure 10.
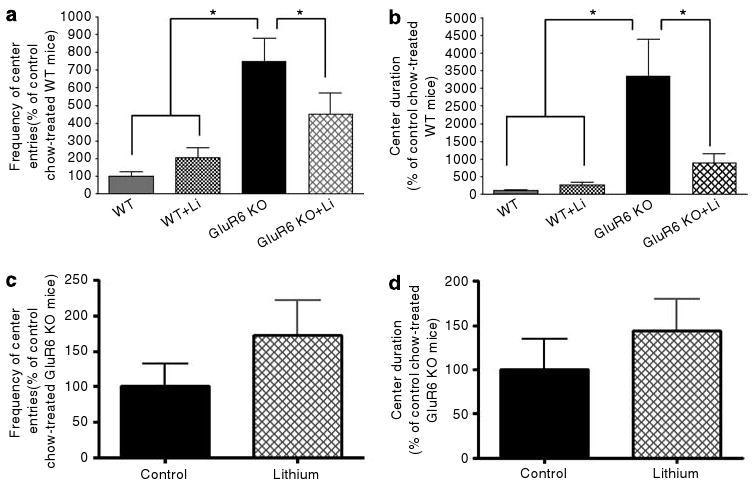
Chronic lithium treatment attenuated increased activity in anxiety-provoking regions. (a) Frequency of entries to the center of the arena during the first day of the open-field test. Four weeks of lithium treatment reduced the frequency of entries to the center of the open-field arena in GluR6 KO mice (one-way ANOVA: F3,19 = 9.8, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li, WT+Li or WT mice, P < 0.05; GluR6 KO+Li vs WT+Li or WT mice, P = 0.01). (b) Duration of time spent in the center of the arena during the first day of open-field test. Four weeks of lithium treatment reduced the amount of time spent in the center of the open-field arena in GluR6 KO mice (one-way ANOVA: F3,19 = 9.2, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li, WT+Li or WT mice, P < 0.05; GluR6KO+Li vs WT mice, P = 0.01). Results are means of percentage relative to WT±s.e.m. Li, lithium (chronic treatment). The effects of 5 days of lithium treatment on the increased activity in anxiety-provoking regions of the open-field test in GLuR6 KO mice were also studied. (c) The frequency of center entries of GluR6 KO mice was not reduced by short-term lithium treatment. (d) The time that GluR6 KO mice spent in the center of the arena was also not reduced by short-term lithium treatment. Results are means±s.e.m. *P≤0.05.
As noted previously, GluR6 KO mice were more active on the locomotion measures (Figure 8a). Chronic (Figure 8a), but not short-term (Figure 8b), lithium treatment significantly lowered locomotor activity in GluR6 KO mice (two-way ANOVA: genotype F3,57 = 647.7, P < 0.0001; day F2,57 = 1512, P = NS; genotype×day F6,57 = 0.8, P = NS; one-way ANOVA: day 1: F3,19 = 7.7, P < 0.01; day 2: F3,19 = 25.7, P < 0.001; day 3: F3,19 = 14.2, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs WT and WT+Li mice, P < 0.001; GluR6 KO vs GluR6 KO+Li mice, P < 0.05).
As described above, GluR6 KO mice were aggressive (Figures 9a and b). Chronic (Figures 9a and b), but not short-term (Figure 9c), lithium treatment significantly lowered tail rattle frequency (ANOVA, F3,8 = 6.9, P < 0.05) and attack bite frequency in the social interaction test (ANOVA, F3,8 = 3.9, P < 0.05), and attack frequency in the resident–intruder test (one-way ANOVA: F3,19 = 3.2, P < 0.05) in GluR6 KO mice.
In this cohort of animals, GluR6 KO mice also displayed more activity in the center of the open-field arena (Figures 10a and b). Chronic (Figures 10a and b), but not short-term (Figures 10c and d), lithium treatment significantly attenuated center entries (one-way ANOVA: F3,19 = 9.8, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li, WT+Li or WT mice, P < 0.05; GluR6 KO+Li vs WT+Li or WT mice, P = 0.01) and time spent in the center of the open field (one-way ANOVA: F3,19 = 9.2, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li, WT+Li or WT mice, P < 0.05; GluR6KO+Li vs WT mice, P = 0.01).
Consistent with the findings described above, the GluR6 KO mice showed more proportional open-arm duration (t9 = 6.257, P = 0.0001), frequency (t9 = 4.681, P = 0.0009) and traveled distance (t9 = 4.458, P = 0.0016). Chronic or short-term lithium treatment did not significantly alter measures of this test (Figure 11).
Figure 11.
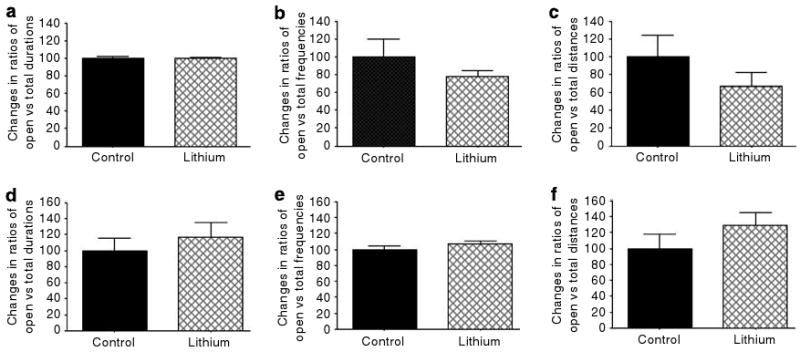
Effects of chronic and short-term lithium treatment on measures of the elevated plus maze test. Chronic (4 weeks, (a–c)) and short-term (4 days, (d–f)) lithium treatment did not significantly alter duration of time spent in the open arms of the maze (a and d), frequency of open-arm entries (b and e) and distance traveled in the open arms (c and f) in GluR6 KO mice. Results are means±s.e.m.
In the forced swim test, the GluR6 KO mice exhibited less immobility compared to the WT mice (Figure 12). Chronic lithium treatment reduced the immobility of WT mice. Interestingly, lithium's effects on the GluR6 KO mice did not represent a generalized attenuation of activity, because lithium treatment further reduced immobility in the forced swim test (Figure 12) (two-way ANOVA: genotype F1,19 = 6.6, P < 0.02; treatment F1,19 = 13.5, P < 0.01 with no interaction).
Figure 12.
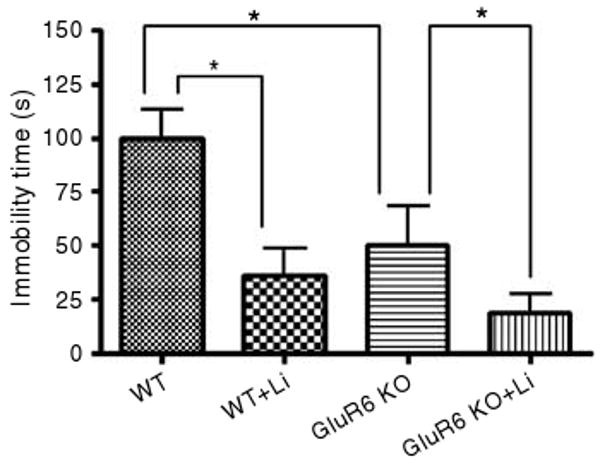
Chronic lithium treatment further reduced GluR6 KO and WT mice immobility time in the forced swim test. Both GluR6 KO and WT mice reduced their immobility time in the forced swim test by more than 60% after chronic lithium treatment (two-way ANOVA: genotype F1,19 = 6.6, P < 0.02; treatment F1,19 = 13.5, P < 0.01 with no interaction). Results are means of percentage relative to WT±s.e.m. Li, lithium (chronic treatment). *P < 0.01.
Effects of ablation of GluR6 and chronic lithium treatment on cell membrane levels of ionotropic glutamatergic receptors
To understand the potential involvement of other ionotropic glutamatergic receptors in the behavioral alterations of GluR6 KO mice, and the mechanisms underlying the behavioral effects of lithium in GluR6 KO mice, the cell membrane levels of ionotropic glutamatergic receptors were monitored in WT and GluR6 KO mice with or without chronic lithium treatment. The membrane levels of GluR5 were significantly reduced in GluR6 KO mice, and the reductions were not significantly reversed by chronic lithium treatment in the hippocampus (one-way ANOVA: F2,22 = 51.1, P < 0.001; post hoc Fisher LSD test: GluR6 KO and GluR6 KO+Li vs WT mice, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li mice, P = 0.80 (NS)) and in the FCX (one-way ANOVA: F2,22 = 18.159, P < 0.001; post hoc Fisher LSD test: GluR6 KO and GluR6 KO+Li vs WT mice, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li mice, P = 0.92 (NS)). The cytosol levels of GluR5 were not significantly altered by GluR6 ablation and lithium treatment in the hippocampus and pre-FCX (Figures 13c and d).
Figure 13.
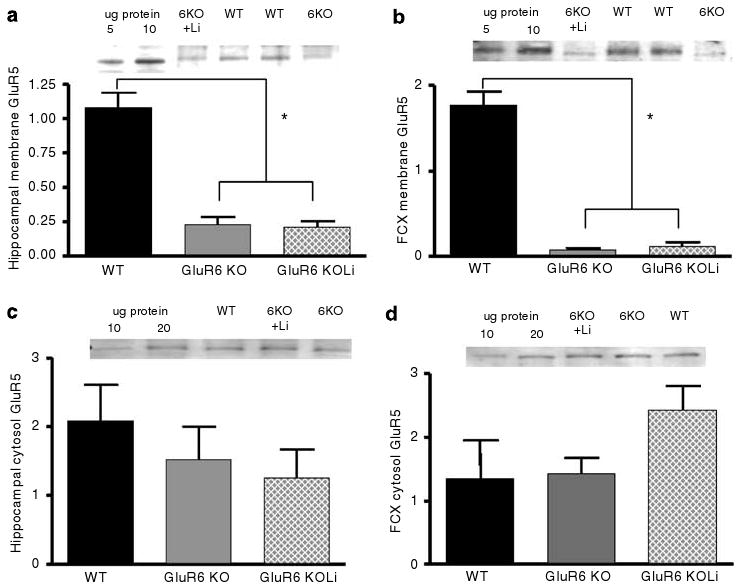
The GluR5 subunit membrane expression levels were reduced in the hippocampus and FCX of GluR6 KO mice. (a) The GluR5 protein levels were reduced in the hippocampal membrane fraction of the GluR6 KO mice (one-way ANOVA: F2,22 = 51.1, P < 0.001; post hoc Fisher LSD test: GluR6 KO and GluR6 KO+Li vs WT mice, P < 0.001). Four weeks of chronic lithium treatment did not affect GluR5 protein level reduction in the hippocampal membrane fraction of the GluR6 KO mice (post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li mice, P = 0.80 (NS)). (b) The GluR5 protein levels were reduced in the FCX membrane fraction of the GluR6 KO mice (one-way ANOVA: F2,22 =18.159, P < 0.001; post hoc Fisher LSD test: GluR6 KO and GluR6KO+Li vs WT mice, P < 0.001). Four weeks of chronic lithium treatment did not affect GluR5 protein level reduction in the FCX membrane fraction of the GluR6 KO mice (post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li mice, P = 0.92 (NS)). (c) The GluR5 protein levels were not reduced in the hippocampal cytosol fraction of GluR6 KO mice. Four weeks of chronic lithium treatment did not affect GluR5 levels in the hippocampal cytosolic fraction of GluR6 KO mice (one-way ANOVA: F2,21 = 0.61, P = 0.55). (d) The GluR5 protein levels were not reduced in the FCX cytosol fraction of GluR6 KO mice. Four weeks of chronic lithium treatment did not affect the GluR5 levels in the FCX cytosolic fraction of GluR6 KO mice (one-way ANOVA: F2,18 = 2.63, P = 0.10 (NS)). FCX, frontal cortex; Li, lithium (chronic treatment). Results are mean±s.e.m. in arbitrary units (AU). *P < 0.001.
The membrane levels of KA-2 receptors were significantly reduced in GluR6 KO mice, but the reductions were not significantly reversed by chronic lithium treatment in the hippocampus (Figures 14a and b) (one-way ANOVA: F2,22 = 168.34, P < 0.001; post hoc Fisher LSD test: GluR6 KO and GluR6 KO+Li vs WT mice, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li mice, P = 0.98 (NS)) and in the FCX (one-way ANOVA: F2,22 = 84.14, P < 0.001; post hoc Fisher LSD test: GluR6 KO and GluR6 KO+Li vs WT mice, P < 0.001; post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li mice, P = 0.89 (NS)). The cytosol levels of KA-2 receptors were not significantly altered by GluR6 ablation and lithium treatment in the hippocampus and pre-FCX (Figures 14c and d).
Figure 14.
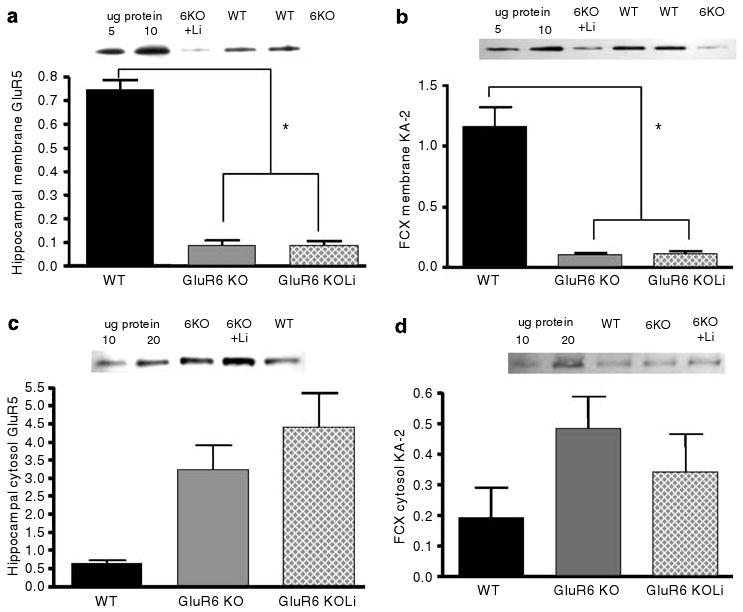
KA-2 subunit membrane expression levels were reduced in the hippocampus and FCX of GluR6 KO mice. (a) The KA-2 protein levels were reduced in the hippocampal membrane fraction of GluR6 KO mice (one-way ANOVA: F2,22 = 168.34, P < 0.001; post hoc Fisher LSD test: GluR6 KO and GluR6 KO+Li vs WT mice, P < 0.001). Four weeks of chronic lithium treatment did not affect KA-2 protein level reduction in the hippocampal membrane fraction of GluR6 KO mice (post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li mice, P = 0.98 (NS)). (b) The KA-2 protein levels were reduced in the FCX membrane fraction of GluR6 KO mice (one-way ANOVA: F2,22 = 84.14, P < 0.001; post hoc Fisher LSD test: GluR6 KO and GluR6 KO+Li vs WT mice, P < 0.001). Four weeks of chronic lithium treatment did not affect the KA-2 protein level reduction in the FCX membrane fraction of GluR6 KO mice (post hoc Fisher LSD test: GluR6 KO vs GluR6 KO+Li mice, P = 0.89 (NS)). (c) A trend toward increased KA-2 protein levels in the hippocampal cytosol fraction of GluR6 KO mice was observed; this trend was not affected by lithium treatment (one-way ANOVA: F2,21 = 3.38, P = 0.053 (NS)). (d) A trend toward increased KA-2 protein levels in the FCX cytosol fraction of GluR6 KO mice was observed; this trend was not affected by lithium treatment (one-way ANOVA: F2,18 = 1.37, P = 0.28 (NS)). FCX, frontal cortex; Li, lithium (chronic treatment). Results are mean±s.e.m. in arbitrary units (AU). *P < 0.001.
KARs subunit KA-1, the N-methyl-D-aspartate (NMDA) receptor subunits NR1 and NR2A, and the α-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA) receptor subunits GluR1, GluR2 and GluR3 were not significantly changed in the membrane fractions of the hippocampus or FCX of the GluR6 KO mice, nor were the membrane fraction levels of these receptors affected by lithium treatment (Table 1).
Table 1.
Ionotropic glutamate receptor subunits membrane expression levels are not altered in the hippocampus or FCX of GluR6 KO mice, and are not affected by lithium treatment
| Hippocampus | FCX | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Receptor type | Subunit | WT | GluR6 KO | GluR6 KO+Li | ANOVA F2,22 | WT | GluR6 KO | GluR6 KO+Li | ANOVA F2,22 |
| Kainate | KA-1 | 0.28 ± 0.18 | 0.31 ± 0.09 | 0.36 ± 0.08 | 0.18; P = 0.83 | 2.09 ± 0.46 | 1.68 ± 0.34 | 2.36 ± 0.31 | 1.10; P = 0.35 |
| NMDA | NR1 | 0.17 ± 0.04 | 0.19 ± 0.03 | 0.18 ± 0.03 | 0.07; P = 0.93 | 0.36 ± 0.10 | 0.34 ± 0.08 | 0.52 ± 0.07 | 1.74; P = 0.19 |
| NR2A | 1.61 ± 0.22 | 1.12 ± 0.16 | 1.33 ± 0.15 | 1.55; P = 0.23 | 1.25 ± 0.88 | 1.02 ± 0.66 | 2.09 ± 0.59 | 0.79; P = 0.46 | |
| AMPA | GluR1 | 0.76 ± 0.21 | 1.06 ± 0.16 | 0.80 ± 0.14 | 1.06; P = 0.36 | 0.98 ± 0.12 | 0.77 ± 0.08 | 0.75 ± 0.08 | 1.41; P = 0.26 |
| GluR2 | 0.76 ± 0.14 | 0.62 ± 0.11 | 0.59 ± 0.09 | 0.49; P = 0.61 | 0.88 ± 0.19 | 0.92 ± 0.15 | 0.87 ± 0.13 | 0.44; P = 0.96 | |
| GluR3 | 0.53 ± 0.07 | 0.51 ± 0.05 | 0.57 ± 0.05 | 0.27; P = 0.76 | 0.77 ± 0.24 | 0.93 ± 0.18 | 0.91 ± 0.16 | 0.27; P = 0.86 | |
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole; ANOVA, analysis of variance; AU, arbitrary units; FCX, frontal cortex; KO, knockout; Li, lithium; NMDA, N-methyl-d-aspartate; WT, wild type.
Li (chronic treatment).
Results are mean of protein levels in AU±s.e.m.
Discussion
In this study, we were able to demonstrate, for the first time, that GluR6, a KAR subunit, plays a critical role in the general control of diverse behavioral features, including hyperactivity, drive, aggressiveness, risk taking (or less anxious behavior) and sensitivity to psychostimulants. These features were demonstrated repeatedly in multiple tests (Figures 2–12). GluR6 KO mice displayed these features without any impairment of consciousness, sensory-motor function, learning or memory (Figure 1 and Mulle et al.33,34). Furthermore, GluR6 ablation was associated with reduced membrane GluR5 or KA-2 receptors (Figures 13, 14). Notably, this behavioral phenotype was highly selective for the GluR6 receptor, because GluR5 ablation (another ionotropic KA receptor with many similarities to GluR6) was completely without effect in any of these behavioral paradigms (Figures 2–8). Membrane levels of other ionotropic glutamatergic receptors tested remained unchanged in the hippocampus and FCX of GluR6 KO mice (Table 1). Thus, GluR6 appears to be uniquely able to control a subset of behavioral manifestations.
KARs are known to function in the neuronal circuitry of mood regulation. For instance, GluR5 is highly expressed in the amygdala, and GluR6 and KA-1-2 are prominently expressed in the dentate gyrus and the CA3 region of the hippocampus.14,15 KARs regulate glutamate release at the mossy fiber to CA3 synapses16 and, notably, are involved in long-term potentiation induction at the hippocampus and the amygdala.17 Presynaptic KARs control the release of GABA in the hippocampus and the amygdala.16,18 Evidence also suggests that KARs produce metabotropic actions, such as interaction with G proteins, induction of second messengers and the activation of PKA, PKC and MAP kinase. Through these actions, KARs are involved in the modulation of hippocampal inhibitory19 and excitatory postsynaptic currents,20 as well as pyramidal cell excitability.21,22
As mentioned in the introduction, the GluR6 gene is located on chromosome 6q21, a region reported by several groups to be linked with BPD.6–9 Moreover, it has recently been shown that cortical expression levels of GluR6 are lower in the cortex of BPD patients.10 Mania traditionally manifests as severe mood elevation (either euphoria or irritability) and is accompanied by behavioral excitement, including hyperactivity and psychomotor agitation, an increase in goal-directed activity and recklessness.39,40 Psychostimulants typically worsen manic symptoms,39,40 and lithium is the classic agent for alleviating those symptoms.39,40 Consistent with the genetic and postmortem brain findings on GluR6, GluR6 KO mice displayed behavioral alterations (Figures 2–12) that appear to be phenocopies of the behavioral symptoms of mania, although the mood and thought states of these mice are obviously unknown.
Hyperactivity, increased risk-taking behavior and aggressiveness per se can be observed in several neuropsychiatric illnesses, such as ADHD, personality disorders, organic brain disorders and brain trauma. Therefore, the pathophysiological mechanisms underlying each of these three symptoms are very diverse and might not be directly linked to those for manic symptoms. This is consistent with both animal studies and online data (http://www.informatics.jax.org) reporting that a single-related behavior can result from the mutation of a variety of genes. However, comorbidity of the three symptoms is more common in manic patients, and all three symptoms respond to lithium treatment in manic patients. There are no sufficient or convincing clinical data to support the notion that lithium therapy is effective in the treatment of ADHD or aggression. GluR6 KO mice co-displayed hyperactivity, risk-taking behavior and aggression (Figures 2–12). Chronic lithium treatment, at least, partially alleviated these behavioral alterations in GluR6 KO mice (Figure 8–10). Therefore, from a phenomenal similarity point of view, the current data collectively support the notion that GluR6 contributes to control mechanisms related to facets of manic symptoms.
We have chosen to use the term phenocopy, as this emphasizes apparent phenomenological similarity, which can result from the same or different underlying mechanism(s). In our opinion, the term ‘model’ is far more stringent, requiring additional criteria, including phenomenological similarity, treatment response similarity, induction mechanistic similarity and pathophysiological similarity.24,41,42 Despite intensive research and intriguing findings, such as changes in GluR6 levels reported in the post-mortem cerebral cortex of bipolar patients, the induction mechanism and the symptom pathophysiology of mania are still largely unknown. Although it is plausible that GluR6 dysfunction is involved in the pathophysiology of manic symptoms, further clinical investigations are needed to establish this link.
Bipolar disorder has a unique cycling feature. Although this study was initiated, in part, by the genetic findings that linked GluR6 polymorphism to the increased risk of BPD, the data presented here do not directly address whether GluR6 functional alterations change the risk for BPD. Furthermore, the behavioral patterns of GluR6 KO mice as well as GluR6 ablation-associated behavioral displays cannot be viewed as a model of BPD because the model would lack the spontaneous cycling feature. Rather, the current data support the notion that GluR6 might play a critical role in the control mechanism of some facet of manic symptoms.
The cell surface expression levels of the different KARs subunits have been shown to be regulated by alternative splicing, the process that determines the subunit trafficking properties from the ER (endoplasmic reticulum) to the membrane.43 The present biochemical studies revealed a significant reduction of both GluR5 and KA-2 subunits cell surface levels in the hippocampus and the FCX of the GluR6 KO mice. Low levels of KA-2 subunit at the cell membrane in the absence of the GluR6 subunit have been shown in vitro by several other groups (reviewed in Jaskolski et al.43). KA-2 subunits and the alternative splice variant GluR5c are known to fail to reach the plasma membrane because of the presence of a retention motif in their sequences that prevents them from exiting the ER unless they are assembled in heteromeric KARs.44–46 GluR5-a and -b splice variants are targeted to the plasma membrane but at low levels in homomeric forms. GluR6a splice variant contains a specific forwarding trafficking domain, making it a key subunit that, upon assembling in heteromeric receptors, promotes the cell surface expression of all other ER-retained KARs subunits.44 Thus, consistent with findings from in vitro studies, we found a tremendous decrease in KARs from the cell surface in the hippocampus and the FCX of the GluR6 KO mice. These decreases are likely due to GluR6 ablation-associated trafficking impairments of GluR5 and KA-2 receptors.
The question of whether GluR6 ablation-associated changes of GluR5 and KA-2 contribute to the behavioral displays of GluR6 KO mice remains unanswered. One of the limitations of the present study is that the role of KA-2 in behavioral regulation was not directly investigated. On the other hand, GluR5 KO mice did not display similar behavioral alterations to those observed in GluR6 KO mice (Figures 2–8). Chronic lithium treatment produced significant behavioral effects in GluR6 KO mice; however, it did not reverse reductions of GluR5 and KA-2 in the hippocampal and frontal cortical membrane fractions. The effects of chronic lithium treatment on membrane levels of GluR5 and KA-2 in other brain regions need to be investigated.
It is interesting to note that recent clinical studies show that ketamine, a non-competitive NMDA receptor antagonist, produces rapid-onset antidepressant effects in depressed patients.47,48 An animal study further showed that ketamine produces antidepressant-like effects in animal models and that the effect of ketamine can be blocked by NBQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide), an AMPA and KA receptor antagonist.30 Chronic lithium treatment decreased the levels of GluR1 in membrane fraction, whereas treatment with imipramine, lamotrigine and riluzole had the opposite effect. These data suggest that NMDA and AMPA receptors are crucially involved in regulating mood-related behaviors. Interestingly, the hippocampal and prefrontal-cortical membrane levels of GluR1-3, NR1 and NR2A were not altered by GluR6 ablation and were not affected by chronic lithium treatment in GluR6 KO mice. The plausible explanation is that the synaptic machinery responsible for behavioral excitement abnormalities differs from that used by antidepressant agents to produce desired behavioral outcomes; furthermore, ionotropic glutamatergic receptors appear to play a role in regulating behavior in a brain regional-specific manner.
These findings place GluR6 into a small group of molecules that are known to significantly contribute to the behavioral control mechanisms related to different facets of manic symptoms.10,49 These molecules include GSK-3b (glycogen synthase kinase 3β),50 CLOCK,51 ERK1 (also known as MAPK3)26 and mutant mtDNA polymerase.52 Mice with genetic alterations of these molecules display some common phenotypes, including hyperactivity in multiple tests,26,50,51,53,54 sensitivity to psychostimulants and rewards,26,51,53,55,56 less anxiety or more risk taking in anxiety-related tests,26,51 less behavioral despair or more drive in immobility tests26,50,51 and circadian alterations.51,52 The additional and unique feature of GluR6 KO mice is their aggression which, notably, can be reversed by lithium treatment. The role of GluR6 in circadian regulation is unknown. Pharmacological and KO mice studies both suggest that GluR6 is a crucial subunit for the metabotropic action of KARs,22,57 which has recently been shown to involve activation of the MAP kinase cascade.58 Interactions between GluR6 and implicated molecules are less clear. Whether the molecules discussed here act alone or in concert to control behaviors related to manic symptoms remains to be elucidated.
Nevertheless, the current coherent data from these behavioral and biochemical experiments imply a novel role for GluR6 in the control of behaviors related to the symptoms of mania, including hyperactivity, aggravated aggression, risk taking and sensitivity to psychostimulants. Further investigation is clearly needed to address whether GluR6 is only involved in the control mechanisms related to facets of manic symptoms or whether it is also involved in the pathophysiology of the manic state and susceptibility to BPD. Although further investigation is needed, targeting GluR6 appears to be a potential avenue for the development of novel mood-stabilizing agents.
Acknowledgments
We thank Ioline Henter for providing outstanding editorial assistance. This work was supported by the Intramural Program of the NIMH.
References
- 1.Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression. Oxford University Press; New York: 2007. [Google Scholar]
- 2.Baum A, Akula N, Cabenero M, Cardona I, Corona W, Klemens B, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2007;13:197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kato T. Molecular genetics of bipolar disorder and depression. Psychiatry Clin Neurosci. 2007;61:3–19. doi: 10.1111/j.1440-1819.2007.01604.x. [DOI] [PubMed] [Google Scholar]
- 4.The Wellcome Trust Case Control Consortium. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buervenich S, Detera-Wadleigh SD, Akula N, Thomas CJM, Kassem L, Rezvani A, et al. Fine mapping on chromosome 6q in the NIMH Genetics Initiative bipolar pedigrees (abstract 1882). Annual Meeting of The American Society of Human Genetics; Los Angeles, CA. 2003. http://www.genetics.faseb.org/genetics/ashg03s/index.shtml. [Google Scholar]
- 6.Dick DM, Foroud T, Flury L, Bowman ES, Miller MJ, Rau NL, et al. Genomewide linkage analyses of bipolar disorder: a new sample of 250 pedigrees from the National Institute of Mental Health Genetics Initiative. Am J Hum Genet. 2003;73:107–114. doi: 10.1086/376562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McQueen MB, Devlin B, Faraone SV, Nimgaonkar VL, Sklar P, Smoller JW, et al. Combined analysis from eleven linkage studies of bipolar disorder provides strong evidence of susceptibility loci on chromosomes 6q and 8q. Am J Hum Genet. 2005;77:582–595. doi: 10.1086/491603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulze TG, Buervenich S, Badner JA, Steele CJ, Detera-Wadleigh SD, Dick DM, et al. Loci on chromosomes 6q and 6p interact to increase susceptibility to bipolar affective disorder in the national institute of mental health genetics initiative pedigrees. Biol Psychiatry. 2004;56:18–23. doi: 10.1016/j.biopsych.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Schumacher J, Kaneva R, Jamra RA, Diaz GO, Ohlraun S, Milanova V, et al. Genomewide scan and fine-mapping linkage studies in four European samples with bipolar affective disorder suggest a new susceptibility locus on chromosome 1p35–p36 and provides further evidence of loci on chromosome 4q31 and 6q24. Am J Hum Genet. 2005;77:1102–1111. doi: 10.1086/498619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;14:14. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- 11.Laje G, Buervenich S, Manji HK, Rush AJ, Wilson A, Charney DS, et al. Genetic markers of suicidal ideation emerging during citalopram treatment of major depression. Am J Psychiatry. 2007;164:1530–1538. doi: 10.1176/appi.ajp.2007.06122018. [DOI] [PubMed] [Google Scholar]
- 12.McElroy SL, Kotwal R, Kaneria R, Keck PEJ. Antidepressants and suicidal behavior in bipolar disorder. Bipolar Disord. 2006;8:596–617. doi: 10.1111/j.1399-5618.2006.00348.x. [DOI] [PubMed] [Google Scholar]
- 13.Yerevanian BI, Koek RJ, Mintz J, Akiskal HS. Bipolar pharmacotherapy and suicidal behavior Part 2. The impact of antidepressants. J Affect Disord. 2007;103:5–11. doi: 10.1016/j.jad.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Chen A, Xing G, Wei ML, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci. 2001;4:612–620. doi: 10.1038/88432. [DOI] [PubMed] [Google Scholar]
- 15.Wisden W, Seeburg PH. A complex mosaic of high-affinity kainate receptors in rat brain. J Neurosci. 1993;13:3582–3598. doi: 10.1523/JNEUROSCI.13-08-03582.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lerma J. Kainate receptor physiology. Curr Opin Pharmacol. 2006;6:89–97. doi: 10.1016/j.coph.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Bortolotto ZA, Nistico R, More JC, Jane DE, Collingridge GL. Kainate receptors and mossy fiber LTP. Neurotoxicology. 2005;26:769–777. doi: 10.1016/j.neuro.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Braga MF, Aroniadou-Anderjaska V, Li H. The physiological role of kainate receptors in the amygdala. Mol Neurobiol. 2004;30:127–141. doi: 10.1385/MN:30:2:127. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Moreno A, Herreras O, Lerma J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron. 1997;19:893–901. doi: 10.1016/s0896-6273(00)80970-8. [DOI] [PubMed] [Google Scholar]
- 20.Frerking M, Schmitz D, Zhou Q, Johansen J, Nicoll RA. Kainate receptors depress excitatory synaptic transmission at CA3→CA1 synapses in the hippocampus via a direct presynaptic action. J Neurosci. 2001;21:2958–2966. doi: 10.1523/JNEUROSCI.21-09-02958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melyan Z, Lancaster B, Wheal HV. Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J Neurosci. 2004;24:4530–4534. doi: 10.1523/JNEUROSCI.5356-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melyan Z, Wheal HV, Lancaster B. Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron. 2002;34:107–114. doi: 10.1016/s0896-6273(02)00624-4. [DOI] [PubMed] [Google Scholar]
- 23.Einat H. Modeling facets of mania—new directions related to the notion of endophenotypes. J Psychopharmacol. 2006;20:714–722. doi: 10.1177/0269881106060241. [DOI] [PubMed] [Google Scholar]
- 24.Cryan JF, Holmes A. The ascent of mouse: advances in modelling human depression and anxiety. Nat Rev Drug Discov. 2005;4:775–790. doi: 10.1038/nrd1825. [DOI] [PubMed] [Google Scholar]
- 25.Fisahn A, Contractor A, Traub RD, Buhl EH, Heinemann SF, McBain CJ. Distinct roles for the kainate receptor subunits GluR5 and GluR6 in kainate-induced hippocampal gamma oscillations. J Neurosci. 2004;24:9658–9668. doi: 10.1523/JNEUROSCI.2973-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crawley JN. What's Wrong with My Mouse: Behavioral Phenotyping of Transgenic and Knockout Mice. 2nd. Wiley; New York: 2007. [Google Scholar]
- 27.El-Ghundi M, O'Dowd BF, George SR. Prolonged fear responses in mice lacking dopamine D1 receptor. Brain Res. 2001;892:86–93. doi: 10.1016/s0006-8993(00)03234-0. [DOI] [PubMed] [Google Scholar]
- 28.Engel SR, Creson TK, Hao Y, Shen Y, Maeng S, Nekrasova T, et al. The extracellular signal-regulated kinase pathway contributes to the control of behavioral excitement. Mol Psychiatry. 2008 January 29; doi: 10.1038/sj.mp.4002135. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Einat H, Yuan P, Manji HK. Increased anxiety-like behaviors and mitochondrial dysfunction in mice with targeted mutation of the Bcl-2 gene: further support for the involvement of mitochondrial function in anxiety disorders. Behav Brain Res. 2005;165:172–180. doi: 10.1016/j.bbr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Maeng S, Zarate CA, Jr, Du J, Schloesser R, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of AMPA receptors. Biol Psychiatry. 2007;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 31.Gould TD, Chen G, Manji HK. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology. 2004;29:32–38. doi: 10.1038/sj.npp.1300283. [DOI] [PubMed] [Google Scholar]
- 32.Du J, Gray NA, Falke CA, Chen W, Yuan P, Szabo ST, et al. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci. 2004;24:6578–6589. doi: 10.1523/JNEUROSCI.1258-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mulle C, Sailer A, Perez-Otano I, Dickinson-Anson H, Castillo PE, Bureau I, et al. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- 34.Mulle C, Sailer A, Swanson GT, Brana C, O'Gorman S, Bettler B, et al. Subunit composition of kainate receptors in hippocampal interneurons. Neuron. 2000;28:475–484. doi: 10.1016/s0896-6273(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 35.Miczek KA, Maxson SC, Fish EW, Faccidomo S. Aggressive behavioral phenotypes in mice. Behav Brain Res. 2001;125:167–181. doi: 10.1016/s0166-4328(01)00298-4. [DOI] [PubMed] [Google Scholar]
- 36.Mi XJ, Chen SW, Wang WJ, Wang R, Zhang YJ, Li WJ, et al. Anxiolytic-like effect of paeonol in mice. Pharmacol Biochem Behav. 2005;81:683–687. doi: 10.1016/j.pbb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 37.Suaudeau C, Rinaldi D, Lepicard E, Venault P, Crusio WE, Costentin J, et al. Divergent levels of anxiety in mice selected for differences in sensitivity to a convulsant agent. Physiol Behav. 2000;71:517–523. doi: 10.1016/s0031-9384(00)00383-8. [DOI] [PubMed] [Google Scholar]
- 38.Cryan JF, Mombereau C. In search of a depressed mouse: utility of models for studying depression-related behavior in genetically modified mice. Mol Psychiatry. 2004;9:326–357. doi: 10.1038/sj.mp.4001457. [DOI] [PubMed] [Google Scholar]
- 39.Belmaker RH. Bipolar disorder. N Engl J Med. 2004;351:476–486. doi: 10.1056/NEJMra035354. [DOI] [PubMed] [Google Scholar]
- 40.Goodwin FK, Jamison KR. Manic-Depressive Illness. Oxford University Press; New York: 1990. [Google Scholar]
- 41.McKinney R, Bunney WE. Animal models of depression. I. Review of evidence: implications for research. Arch Gen Psychiatry. 1969;21:240–248. doi: 10.1001/archpsyc.1969.01740200112015. [DOI] [PubMed] [Google Scholar]
- 42.Willner P. The validity of animal models for depression. Psychopharmacology (Berl) 1984;83:1–16. doi: 10.1007/BF00427414. [DOI] [PubMed] [Google Scholar]
- 43.Jaskolski F, Coussen F, Mulle C. Subcellular localization and trafficking of kainate receptors. Trends Pharmacol Sci. 2005;26:20–26. doi: 10.1016/j.tips.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 44.Jaskolski F, Coussen F, Nagarajan N, Normand E, Rosenmund C, Mulle C. Subunit composition and alternative splicing regulate membrane delivery of kainate receptors. J Neurosci. 2004;24:2506–2515. doi: 10.1523/JNEUROSCI.5116-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren Z, Riley NJ, Garcia EP, Sanders JM, Swanson GT, Marshall J. Multiple trafficking signals regulate kainate receptor KA2 subunit surface expression. J Neurosci. 2003;23:6608–6616. doi: 10.1523/JNEUROSCI.23-16-06608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren Z, Riley NJ, Needleman LA, Sanders JM, Swanson GT, Marshall J. Cell surface expression of GluR5 kainate receptors is regulated by an endoplasmic reticulum retention signal. J Biol Chem. 2003;278:52700–52709. doi: 10.1074/jbc.M309585200. [DOI] [PubMed] [Google Scholar]
- 47.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 48.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 49.Kato T, Kubota M, Kasahara T. Animal models of bipolar disorder. Neurosci Biobehav Rev. 2007;31:832–842. doi: 10.1016/j.neubiorev.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 50.Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I, et al. Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci. 2006;26:9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA. 2007;104:6406–6411. doi: 10.1073/pnas.0609625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kasahara T, Kubota M, Miyauchi T, Noda Y, Mouri A, Nabeshima T, et al. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol Psychiatry. 2006;11:577–593. doi: 10.1038/sj.mp.4001824. [DOI] [PubMed] [Google Scholar]
- 53.Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 54.Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferguson SM, Fasano S, Yang P, Brambilla R, Robinson TE. Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology. 2006;31:2660–2668. doi: 10.1038/sj.npp.1301014. [DOI] [PubMed] [Google Scholar]
- 56.McClung CA, Sidiropoulou K, Vitaterna MH, Takahashi JS, White FJ, Cooper DC, et al. Regulation of dopaminergic transmission and cocaine reward by the clock gene. Proc Natl Acad Sci USA. 2005;102:9377–9381. doi: 10.1073/pnas.0503584102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fisahn A, Heinemann SF, McBain CJ. The kainate receptor subunit GluR6 mediates metabotropic regulation of the slow and medium AHP currents in mouse hippocampal neurons. J Physiol. 2005;562:199–203. doi: 10.1113/jphysiol.2004.077412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grabauskas G, Lancaster B, O'Connor V, Wheal H. Protein kinase signaling requirements for metabotropic action of kainate receptors in rat CA1 pyramidal neurons. J Physiol. 2006;7:7. doi: 10.1113/jphysiol.2006.122051. [DOI] [PMC free article] [PubMed] [Google Scholar]