

Abstract
Autocrine ligands are important regulators of many normal tissues and have been implicated in a number of disease states, including cancer. However, because by definition autocrine ligands are synthesized, secreted, and bound to cell receptors within an intrinsically self-contained “loop,” standard pharmacological approaches cannot be used to investigate relationships between ligand/receptor binding and consequent cellular responses. We demonstrate here a new approach for measurement of autocrine ligand binding to cells, using a microphysiometer assay originally developed for investigating cell responses to exogenous ligands. This technique permits quantitative measurements of autocrine responses on the time scale of receptor binding and internalization, thus allowing investigation of the role of receptor trafficking and dynamics in cellular responses. We used this technique to investigate autocrine signaling through the epidermal growth factor receptor by transforming growth factor alpha (TGFα) and found that anti-receptor antibodies are far more effective than anti-ligand antibodies in inhibiting autocrine signaling. This result indicates that autocrine-based signals can operate in a spatially restricted, local manner and thus provide cells with information on their local microenvironment.
Awareness of the crucial role that autocrine ligands play in tissue physiology and pathology is increasing across a wide spectrum of fields including embryonic and tissue development, immunology, cancer, angiogenesis, dermatology, neuroscience, and biotechnology (1–8). The concept of autocrine ligand/receptor cell signaling was introduced almost two decades ago (9), and a range of physiological and pathological situations are now known to be regulated by self-secreted factors (10). Unfortunately, understanding how autocrine systems work is severely limited by an inability to construct something as simple and fundamental to receptor biology as a dose–response curve for ligand/receptor binding. This is due to the recursive nature of autocrine signaling and the difficulty of selectively labeling autocrine ligands. Without being able to quantify relationships between ligand production and receptor binding, interpreting cell behavioral changes after a molecular intervention remains ambiguous. Current methodologies for analyzing autocrine signaling are indirect, but whether they are measuring end-point cell functions such as migration, proliferation, or differentiation (e.g., see refs. 11–13) or short-term receptor activation events such as phosphorylation (e.g., see refs. 14, 15), they are laborious and time-consuming—therefore not “real-time”—and are only poorly quantitative at best.
The Cytosensor microphysiometer (Molecular Devices) (16, 17) uses a light-addressable potentiometric sensor to measure rapid (<30 sec) and small (<0.1 unit) changes in solution pH in the cellular microenvironment in an ≈1 μl chamber above the sensor. These pH changes [termed “extracellular acidification rate” (ECAR)] can arise from both metabolic and regulatory events and have been shown to be quantitatively related to specific activation of many types of cell receptors, including tyrosine kinase receptors, G protein receptors, and ion channel receptors (16) with EC50 values similar to those derived from direct-labeled ligand binding (17). Hence, for exogenous ligands the microphysiometer can be used to obtain real-time, kinetic measurements of receptor binding once a calibration curve has been generated relating ECAR data to labeled ligand/receptor-binding data. This has proven of special value for high-throughput screening of pharmacological compounds.
Because the quantitative relationship between ECAR and ligand/receptor binding should be identical regardless of whether the particular ligand is added exogenously or is self-produced in an autocrine fashion, we reasoned that we could adapt the microphysiometer to permit real-time quantitative determination of autocrine ligand binding in an analogous manner. We demonstrate here this new methodology by calibrating ligand/receptor binding to ECAR and establishing key dose–response relationships. As an example of the utility of this approach, we test a theoretical prediction of the comparative effectiveness of anti-receptor (“blocker”) versus anti-ligand (“decoy”) antibodies in interrupting autocrine signaling.
MATERIALS AND METHODS
Materials.
Parental B82 mouse fibroblasts lacking epidermal growth factor receptor (EGFR) and EGFR-expressing B82 cells were a gift from Gordon Gill (University of California, San Diego). Use of the tetracycline-controlled two-plasmid system (20) to create the transforming growth factor alpha (TGFα) autocrine cell system has been described by Oehrtman et al. (18). The constructed cell lines relevant to the present work are denoted as R+/L− (B82 cells with EGFR) and R+/L+ (B82 cells with EGFR and TGFα). The R+/L+ cells can express TGFα at a range of levels depending on the medium concentration of the suppresser tetracycline (18).
Dialyzed bovine calf serum (10,000 Mr-cutoff dialyzed against 0.15 M NaCl), DMEM, methotrexate, geneticin sulfate (G418), histidinol, BSA fraction IV (RIA grade), glutamine, penicillin, and streptomycin were purchased from Sigma. Hybridoma producing mAb 225 were obtained from the American Type Culture Collection and the antibodies produced as described by Opresko et al. (34). Goat anti-TGFα antibody was obtained from R & D Systems. Rabbit IgG was obtained from Sigma. Both anti-EGFR blocking antibody 225 and anti-TGFα antibody bind to EGFR and TGFα, respectively, with affinity comparable with TGFα/EGFR binding (Kd = 2 nM) (27).
R+/L− cells were grown in DMEM with 1 mM glutamine, 100 units/ml penicillin, and 2.5 mg/ml streptomycin along with 10% dialyzed bovine calf serum and 1 mM methotrexate to maintain selection on the EGFR expression plasmid pXER (19). R+/L+ cells were grown in DMEM with 10% dialyzed bovine calf serum, 1 mM glutamine, 100 units/ml penicillin, and 2.5 mg/ml streptomycin along with maintaining plasmid selection with 600 μg/ml G418 (pUHD15.1), 2,400 mM histidinol (pUHD10.3 and pR8), and 1 mM methotrexate (pXER).
Microphysiometer Assay.
Cells (250,000) were seeded into each Cytosensor transwell (Corning Transwell #3402; 3 μm pore size, 12 mm diameter) in the appropriate growth medium. Twenty-four hours later, cell-transwell assemblies were prepared for placement on the Cytosensor by inserting a spacer (50 mm high, 6 mm inner diameter; #R7026B, Molecular Devices) into the transwell medium directly over the cells followed by a transwell insert (3 μm pore size; #R7025, Molecular Devices), fitting flush inside the original transwell and the addition of 1 ml of running buffer to each insert cup to seat the insert into the transwell. The cell-transwell assemblies were then placed in Cytosensor silicon sensor chambers and equilibrated for 2–3 hr in DV/cyto medium (DMEM with 2.59 g/liter NaCl and 0.1 mg/ml BSA, no bicarbonate) at a 100 μl/min flow rate. Pump cycles for all experiments were 30 sec on and 30 sec off. ECAR was measured during the pump-off period for 20 sec, starting 8 sec into this period. A value of 100% denotes a steady–state baseline established before changing conditions. Upon challenge of cells with exogenous growth factor, maximal ECAR was achieved at 10 min, after which buffer flow was reestablished and cells were allowed to acquire a new steady-state baseline.
In experiments with antibodies, cells were allowed to reestablish baseline ECAR before exposing them to a 30-min challenge with competing antibodies. To test effects of autocrine ligand production rates, uninduced R+/L+ cells were seeded at 250,000 cells/transwell in normal growth medium containing 1 μg/ml tetracycline. Twenty-four hours later, cells were equilibrated on the Cytosensor in DV/cyto running buffer with 1 μg/ml tetracycline; on reaching steady-state ECAR, tetracycline concentration was altered to induce TGFα production under partially induced or fully induced conditions for 7 hr before measurement of ECAR or labeled ligand binding. In experiments with exogenous ligand, cells were seeded at 250,000 cells/transwell in normal growth medium. Induced R+/L+ cells were plated in tetracycline-free growth medium for overnight expression. Cells were equilibrated to steady–state ECAR on the Cytosensor in DV/cyto buffer and then exposed to a series of EGF concentrations—0.1, 0.3, 0.6, 1.2, 2.5, 5, 10, 25, and 50 ng/ml, according to the following distribution across the multiple Cytosensor lanes: in lane A, 0.1, 1.2, and 10 ng/ml EGF; in lane B, 0.3, 2.5, and 25 ng/ml EGF; in lane C, 0.6, 5, and 50 ng/ml EGF; in lane D, 0 EGF. After each EGF addition, cells were allowed to reequilibrate ECAR to baseline before next ligand addition. EGF was removed immediately when cells reached maximal ECAR (≈10 min).
Ligand-Binding Assay.
Cells were seeded at approximately 100,000 cells/35-mm culture dish in normal growth medium. When induced R+/L+ cells were used, induction was achieved by replacing tetracycline-containing with tetracycline-free medium on day 2. On day 3, the medium was switched to D/H/B medium (DMEM with 25 mM Hepes and 1 mg/ml BSA) for 3 hr and 125I-EGF in D/H/B solution was added to the cells in a 37°C water bath incubator. After 10 min, cells were placed on ice, the 125I-EGF medium was removed, and the cells were washed three times with 1× WHIPS saline (1 mg/ml PVP/130 mM NaCl/5 mM KCl/0.5 mM MgCl2/1 mM CaCl2/20 mM Hepes). Bound ligand was determined by lysing cells with 1 M NaOH for 10 min and counting in a gamma spectrophotometer. Free ligand concentration was determined by counting an aliquot of the binding medium.
RESULTS AND DISCUSSION
For our experimental autocrine cell system, we used a B82 mouse fibroblast cell line in which we have created an artificial autocrine loop consisting of TGFα and EGFR (18). These cells do not normally express EGFR or its ligands (TGFα or EGF). After expression of EGFR in B82 cells by transfection, there is no significant activation of the receptor in the absence of exogenous EGF ligands (19). TGFα expression is under control of the “tetracycline-off” two-plasmid system (20) permitting quantitative manipulation of the level of autocrine ligand production. We have previously shown that in these cells, TGFα is expressed in its normal manner as a transmembrane precursor, and is efficiently processed at the plasma membrane to yield almost exclusively the 5.5-kDa soluble form (18). Clones transfected with both EGFR and TGFα (therefore, autocrine clones) were designated R+/L+. They were fully induced for ligand production in the absence of tetracycline and uninduced in the presence of 1 μg/ml tetracycline. Control clonal lines transfected with EGFR only were designated R+/L−. The TGFα production rates for the R+/L+ autocrine clones were determined to be approximately 6 × 103 and 1 × 102 no./cell-min under fully induced and uninduced conditions, respectively. This was measured by ELISA in the presence of high concentrations of anti-EGFR antibodies that block ligand capture and permit full escape into the extracellular bulk medium (18). The EGFR production rate for all clones is approximately 4 × 103 no./cell-min, resulting in a steady-state number of ≈1.2 × 105 no./cell with a constitutive turnover rate constant of ≈0.03 min−1 (21).
To directly determine whether our methodology can detect ECAR responses to autocrine ligand binding, we measured ECAR for uninduced and fully induced R+/L+ cells in the absence and presence of saturating levels of anti-receptor antibody after 7 hr of induction. We have previously found that this is the time period needed for approach to the full ligand synthesis rate (22). The ECAR responses averaged over several runs are shown in Fig. 1. ECAR for the uninduced R+/L+ cells in the presence of anti-receptor antibody is indistinguishable from background and so is set at zero. All other cases were normalized pair-wise to this control case. ECAR response was highest for the fully induced R+/L+ cells in the absence of anti-receptor antibody, at a value of ≈16 ± 3, as expected because this situation should correspond to the greatest autocrine signaling. ECAR was reduced to essentially background level by the presence of anti-receptor antibody, due to the ability of the antibody to inhibit ligand binding. Consistent with this finding, ECAR response of the uninduced R+/L+ cells was significantly higher in the absence of the anti-receptor antibody, 7 ± 3, compared with background level in its presence. We conclude that the microphysiometer can indeed detect substantial differences in ECAR in the presence of active autocrine signaling.
Figure 1.

EGFR-mediated ECAR response varies with production rate of autocrine TGFα. B82 cells (R+/L+) were induced to produce TGFα by withdrawal of tetracycline in either the presence or absence of 1 μg/ml of anti-EGFR mAb 225 for 7 hr. Shown are the average ECAR values at the 7-hr time point from three experiments normalized to uninduced autocrine cells with blocking antibody. Error bars represent SEM. Notice that the uninduced cells secrete TGFα constitutively at a background level, giving rise to an ECAR signal when blocking antibody is absent.
To quantitatively relate microphysiometer assay data to ligand/receptor binding levels, we calibrated ECAR to number of occupied receptors under identical experimental conditions (a 10-min stimulation with exogenous ligand) by using R+/L−, uninduced R+/L+, and fully induced R+/L+ cells and 125I-labeled EGF as the exogenous ligand. EGF and TGFα bind to EGFR on B82 cells with essentially identical kinetics, affinity constants (23), and microphysiometer responses (data not shown). Shown in Fig. 2 (Upper) are the amounts of 125I-labeled EGF bound to the cells following a 10-min incubation for the R+/L−, uninduced R+/L+, and induced R+/L+ cells. After converting labeled binding data from radioactive counts (cpm) to ligand/receptor complexes (no./cell) and free ligand (nM), the data were fit to a quasi-equilibrium-binding equation to determine the quasi-equilibrium dissociation constant, K′d, and total receptor number, RT, from the number of labeled complexes, C, as a function of exogenous, labeled ligand concentration, Lexog, and effective autocrine ligand concentration, Laut:
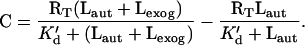 |
1 |
This equation assumes that the lower levels of labeled ligand/receptor complexes observed in the autocrine cells is due to the presence of unlabeled autocrine ligand binding. [It has been previously established that for the EGF/EGFR system on B82 cells ligand/receptor binding can be considered at quasi-equilibrium within this 10-min time period, and although endocytic internalization has begun within this time period, the vast majority of the complexes is still on the cell surface (21)]. We obtained RT = 1.2 × 105 no./cell, K′d = 1.7 nM, and Laut = 0.21 nM and 0.72 nM for the uninduced and induced cells, respectively. That is, the binding data for the autocrine cells are simply shifted up the binding curve by an effective amount of ligand secreted and captured by the producing cells. The numbers of “background” complexes generated by autocrine ligand binding can therefore be calculated by using the values of RT and K′d to be 0.12 × 105 no./cell and 0.26 × 105 no./cell for the uninduced and induced autocrine cells, respectively.
Figure 2.

EGFR-mediated binding and ECAR responses stimulated by exogenous EGF are reduced by autocrine TGFα. (Left, Center, and Right) Results from control cells (R+/L−), uninduced autocrine cells (R+/L+), and fully induced autocrine cells (R+/L+), respectively. (Upper) Cells were exposed to the indicated concentrations of 125I-EGF for 10 min and the amount of specifically bound ligand was measured as described in Material and Methods. (Lower) The response of cells to a 10-min exposure to the indicated concentrations of EGF was determined on a Cytosensor as described in Material and Methods. The data are the average of four experiments ± SD. The lines through the data are nonlinear regression lines calculated as described in the text.
Fig. 2 (Lower) shows corresponding ECAR values recorded at each exogenous ligand concentration, showing the stimulation above the background values generated by autocrine signaling. These data were fit to an equation analogous to that for labeled ligand binding but replacing RT with ECARmax and K′d with the ligand concentration necessary for EC50:
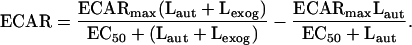 |
2 |
This equation assumes that the reduction in ECAR response in autocrine cells is due a “baseline” ECAR value in response to autocrine ligand binding. From our data, we calculate ECARmax = 67%, EC50 = 1.1 nM, and an Laut = 0.11 nM and 1.3 nM for the uninduced and induced cells respectively. The lower value of EC50 compared with K′d suggests that the ECAR signal transduction pathway approaches saturation at lower EGF/EGFR complex levels than does binding. The values of Laut that were independently estimated from the binding studies and ECAR responses studies were similar (within a factor of 2). As expected, the estimated value of Laut for the induced cells was substantially greater than for the uninduced cells.
Fig. 3 shows the resulting calibration curve between ECAR and ligand/receptor complex number, defined by calculating total (i.e., including autocrine plus exogenous ligand) complex numbers and ECAR across the range of exogenous ligand concentrations by using the values of RT, K′d, ECARmax, and EC50 determined above. The asymptotically plateauing nature of ECAR as a function of complex number is consistent with the relatively lower values of K′d versus EC50 noted above. Superimposed on this curve are all the measured data points from the three cell cases for each concentration of exogenous ligand, corrected for the binding generated by autocrine ligand. It is evident that the curve satisfactorily accounts for all three sets of data, strongly supporting the validity of the calibration.
Figure 3.

ECAR correlates directly with the total number of ligand/receptor complexes due to the combination of exogenous EGF and autocrine TGFα. Total ECAR for the three sets of data shown in the Bottom of Fig. 2 are plotted against the total number of ligand–receptor complexes calculated from the equations outlined in the text.
To investigate the utility of this methodology, we used the microphysiometer assay to test the hypothesis that anti-receptor antibodies should be more effective in disrupting autocrine signaling than anti-ligand antibodies (24). Although anti-receptor antibodies can inhibit cell responses by directly blocking the receptor from binding ligand, inhibition by anti-ligand antibodies is more complicated. Receptors are confined to the cell surface whereas anti-ligand antibodies exist primarily in the bulk medium. The relative ability of anti-ligand antibodies to compete with receptors for ligand binding, therefore, strongly depends on the extent to which the autocrine ligand diffuses into the medium (25).
To test this hypothesis, ECAR was first measured continuously for fully induced, partially induced, and uninduced R+/L+ cells after addition of anti-EGFR antibody. As shown in Fig. 4, addition of anti-receptor antibody rapidly reduced ECAR in the autocrine cells. The extent of the reduction was proportional to the levels of autocrine ligand expression, with ECAR of fully induced, partially induced (using 5 ng/ml tetracycline, yielding a ligand production rate of ≈2,000–3,000 no./cell-min), and uninduced cells decreasing approximately 15%, 10%, and 5% compared with the level maintained for uninduced cells without antibody. The half-time of ECAR reduction (≈3–4 min) is similar to the half-time of occupied receptors on the cell surface (26), suggesting that internalization and dissociation of previously formed receptor/ligand complexes is rate-limiting in the termination of the ECAR response. These data show that we can measure the kinetics of interruption of autocrine loops on the time scale of receptor/ligand processes.
Figure 4.

Effects of varying ligand induction on autocrine cell ECAR. Autocrine cells were plated onto Cytosensor transwells and were either fully induced with no tetracycline (—▴—) or partially induced with 5 ng/ml of tetracycline (—•—). Noninduced cells (1 ng/ml tetracycline) were used as controls (—■—, —♦—). Steady–state ECAR was set to 100%, and the cells received the indicated concentration of anti-EGFR antibody except for control cells receiving no additions (—■—).
Next, ECAR was measured for fully induced R+/L+ cells as a function of anti-EGFR and anti-TGFα antibodies. Fig. 5 compares the dose-dependence of inhibition of the ECAR response in autocrine cells by anti-EGFR and anti-TGFα antibodies. As a control for specificity, addition of a nonspecific rabbit IgG at 700 nM was found to not affect ECAR of the autocrine cells. Significant inhibition by the anti-EGFR antibodies was observed at 1 nM, and essentially complete inhibition was achieved by 10 nM. In contrast, no inhibition by the anti-TGFα was observed at concentrations lower than 1–10 μM. Thus, concentrations approximately 103-fold higher of anti-ligand antibody than of anti-receptor antibody were needed to yield similar levels of inhibition. It is not clear what concentration of anti-ligand antibodies might be required to obtain essentially complete inhibition.
Figure 5.

Anti-EGFR antibodies inhibit autocrine TGFα binding much more effectively than do anti-TGFα antibodies. Equilibrated, fully induced autocrine cells were challenged by the addition of the indicated concentrations of anti-EGFR 225 mAb (□), anti-TGFα antibody (•), and nonspecific rabbit IgG (×). ECAR was measured at a steady–state after antibody addition in each case, and this measurement was converted to ligand/receptor complex number by using the calibration curve illustrated in Fig. 3.
The difference in the ability of anti-EGFR and anti-TGFα antibodies to block autocrine signaling is unlikely due to differences in their binding affinity, for each antibody exhibits a KD value for its target antigen of approximately 1 nM (27). Instead, this difference is likely due to autocrine TGFα not entering the bulk medium before capture by cell receptors. To confirm that the anti-TGFα antibody could effectively block receptor binding if the ligand was supplied from the bulk medium, we measured the ECAR of R+/L− cells in response to exogenous TGFα in the absence and presence of 300 nM anti-ligand antibody. Exogenous TGFα effectively stimulated an increase in ECAR in the absence of anti-ligand antibody but not in its presence (Fig. 6). Therefore, the anti-ligand antibody can effectively block receptor signaling if the ligand is supplied from the bulk medium. This result suggests that a significant amount of autocrine TGFα does not enter the bulk medium before binding to the EGFR. We also can rule out the alternative explanation that the anti-ligand antibody is depleted from the medium by membrane-associated, noncleaved TGFα. The observed 103-fold lower effectiveness of inhibition by anti-ligand antibody compared with by anti-receptor antibody would require that 99.9% of the TGFα remains membrane-associated. We have in fact found that most of the TGFα is cleaved and secreted (18). Yet another alternative explanation is conceivable, that the difference in inhibition capabilities between the anti-ligand and anti-receptor antibodies arises from a strong disparity in association and dissociation kinetics despite the very similar equilibrium affinities. This would require that the rate constants be different by more than 103-fold, however, because the dependence of inhibition effectiveness on the values of these rate constants is milder than linear (25).
Figure 6.

Anti-TGFa antibodies neutralize the ligand when added together in the medium. Autocrine cells were equilibrated on the Cytosensor. Cells were either exposed to 2 nM TGFα alone (A) or together with 50 mg/ml anti-TGFα antibodies for 17-min time interval indicated by the double-arrow (B). The relative ECAR was normalized to zero time.
We therefore conclude that, for our cell system at least, EGFR possesses a high capture efficiency for autocrine TGFα and thus the ligand does not travel far between its release and its subsequent capture. This is a conservative conclusion because of at least two reasons. First, the microphysiometer apparatus offers the greatest possible opportunity for secreted ligand to travel far distances before binding to cell receptors. Because cells are present as a monolayer on a transwell filter adjacent to a fluid chamber, secreted ligand can easily be transported through the bulk chamber medium by diffusion and convective mixing to distant cells. Hence, if secreted ligand escaped local capture to a substantial extent, the anti-ligand antibodies should have been easily able to bind them in the bulk chamber medium. Second, the response of the cells to autocrine ligand is substantially greater than would be the case if the ligand mixed with the bulk medium. Quantitatively, for approximately 6 × 104 fully induced cells in contact with the Cytosensor chamber medium, secreting ≈2 × 103 TGFα molecules/min, up to 1.2 × 108 TGFα molecules could accumulate in the chamber medium within the 30-sec interflow period giving an average concentration of ≈0.15 nM TGFα in the 1.4 μl of bulk medium assuming complete ligand mixing within the bulk. According to Fig. 2, this concentration should be barely sufficient to generate an ECAR response. The actual measured ECAR response was much higher than this prediction, indicating that the ligand concentration is much higher near the cell surface than what would be predicted from complete ligand mixing.
The number of antibody molecules present in the chamber medium at a concentration of 10 nM (sufficient for inhibition by anti-receptor antibodies but not by anti-ligand antibodies) is approximately 1010 in excess of what should be needed from stoichiometric considerations. The relative inability of anti-ligand antibodies to inhibit autocrine ligand binding to cell receptors is most likely due to the predominance of binding proximal to the site of release. Again, this strongly suggests that autocrine TGFα does not enter the bulk medium. In some physiological tissue situations, the bulk extracellular medium can be as closely available to the producing cells as in the microphysiometer, permitting direct correspondence of this conclusion. In other tissue situations, the cells will be present at an even higher density so that long-distance travel of secreted ligand through the extracellular medium would be even less likely, again making this conclusion conservative.
In summary, then, our findings are twofold. First, we have demonstrated a methodology for measuring dynamics of autocrine signaling. Second, we have shown that anti-receptor antibodies may in general be more effective inhibitors of autocrine signaling than anti-ligand antibodies, implying that autocrine signals may be highly localized spatially.
If a ligand is captured locally instead of first entering the bulk medium, what does that imply with respect to the function of autocrine signaling? In the simplest view, it implies that autocrine loops can provide spatial information regarding the local cell microenvironment. If the distance traveled by the ligand before capture is significantly less that the diameter of a cell, which seems likely based on theoretical considerations (28), then autocrine signaling can discern features of the environment on the same spatial scale. Several EGFR ligands, such as amphiregulin and heparin-binding EGF, bind to glycosaminoglycans and thus could provide cells with information regarding the composition of the extracellular matrix (29, 30). Interruption of autocrine loops also could be restricted to a subdomain of the cell surface. Localized disruption of autocrine signaling could be important in directing spatial processes such as cell migration and tissue organization. The observation that disruption of signaling through the EGFR strongly affects these processes may therefore be highly significant (31–33).
Acknowledgments
We thank Lee Opresko for critically reading the manuscript. We also gratefully acknowledge financial support from a grant from the National Science Foundation Biotechnology Program, Division of Bioengineering and Environmental Systems.
ABBREVIATIONS
- EGFR
epidermal growth factor receptor
- ECAR
extracellular acidification rate
References
- 1.Guo C S, Wherle-Haller B, Rossi J, Ciment G. Dev Biol. 1997;184:61–69. doi: 10.1006/dbio.1997.8520. [DOI] [PubMed] [Google Scholar]
- 2.Anastasi S, Giordano S, Sthandier O, Gambarotta G, Maione R, Comoglio P, Amati P. J Cell Biol. 1997;137:1057–1068. doi: 10.1083/jcb.137.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen S B A, Parry S L, Feldmann M, Foxwell B. J Immunol. 1997;158:5596. [PubMed] [Google Scholar]
- 4.Okamoto M, Lee C, Oyasu R. Endocrinology. 1997;138:5071. doi: 10.1210/endo.138.11.5653. [DOI] [PubMed] [Google Scholar]
- 5.Seghezzi G, Patel S, Ren C J, Pintucci G, Fernandez H, Kallenbach K, Shapiro R L, Colvin S B, Rifkin D B, Mignatti P, et al. Surg Forum. 1997;48:836. [Google Scholar]
- 6.Pincelli C, Haake A R, Benassi L, Grassilli E, Magnoni C, Ottani D, Polakowska R, Franceschi C, Giannetti A. J Invest Dermatol. 1997;109:757–764. doi: 10.1111/1523-1747.ep12340768. [DOI] [PubMed] [Google Scholar]
- 7.Pesheva P, Gloor S, Schachner M, Probstmeier R. J Neurosci. 1997;17:4642–4651. doi: 10.1523/JNEUROSCI.17-12-04642.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pak S C O, Hunt S M N, Bridges M W, Sleigh M J, Gray P P. Cytotechnology. 1996;22:139–146. doi: 10.1007/BF00353933. [DOI] [PubMed] [Google Scholar]
- 9.Sporn M B, Todaro G J. N Engl J Med. 1980;303:878–880. doi: 10.1056/NEJM198010093031511. [DOI] [PubMed] [Google Scholar]
- 10.Sporn M B, Roberts A B. Ann Intern Med. 1992;117:408–414. doi: 10.7326/0003-4819-117-5-408. [DOI] [PubMed] [Google Scholar]
- 11.Liotta L A, Mandler R, Murano G, Katz D A, Gordon R K, Chang P K, Schiffman E. Proc Natl Acad Sci USA. 1986;83:3302–3306. doi: 10.1073/pnas.83.10.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunbar C E, Browder T M, Abrams J S, Nienhuis A W. Science. 1989;245:1493–1496. doi: 10.1126/science.2789432. [DOI] [PubMed] [Google Scholar]
- 13.Bejcek B E, Li D Y, Deuel T F. Science. 1989;243:1496–1499. doi: 10.1126/science.2551043. [DOI] [PubMed] [Google Scholar]
- 14.Keating M T, Williams L T. Science. 1988;239:914–916. doi: 10.1126/science.2829358. [DOI] [PubMed] [Google Scholar]
- 15.Van de Vijver M J, Kumar R, Mendelsohn J. J Biol Chem. 1991;266:7503–7508. [PubMed] [Google Scholar]
- 16.McConnell H M, Owicki J C, Parce J W, Miller D L, Baxter G T, Wada H G, Pitchford S. Science. 1992;257:1906–1912. doi: 10.1126/science.1329199. [DOI] [PubMed] [Google Scholar]
- 17.Hirst M A, Pitchford S. J NIH Res. 1993;5:69. [Google Scholar]
- 18.Oehrtman G T, Wiley H S, Lauffenburger D A. Biotech Bioeng. 1998;57:571–582. [PubMed] [Google Scholar]
- 19.Chen W S, Lazar C S, Lund K A, Welsh J B, Chang C P, Walton G M, Der C J, Wiley H S, Gill G N, Rosenfeld M G. Cell. 1989;59:33–43. doi: 10.1016/0092-8674(89)90867-2. [DOI] [PubMed] [Google Scholar]
- 20.Gossen M, Bujard H. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiley H S, Herbst J J, Walsh B J, Lauffenburger D A, Rosenfeld M G, Gill G N. J Biol Chem. 1991;266:11083–11094. [PubMed] [Google Scholar]
- 22.Will B H, Lauffenburger D A, Wiley H S. Tissue Eng. 1995;1:83–96. doi: 10.1089/ten.1995.1.81. [DOI] [PubMed] [Google Scholar]
- 23.French A R, Tadaki D K, Niyogi S K, Lauffenburger D A. J Biol Chem. 1995;270:4334–4340. doi: 10.1074/jbc.270.9.4334. [DOI] [PubMed] [Google Scholar]
- 24.Forsten K E, Lauffenburger D A. Biophys J. 1994;63:857–861. doi: 10.1016/S0006-3495(92)81661-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forsten K E, Lauffenburger D A. Biophys J. 1992;61:518–529. doi: 10.1016/S0006-3495(92)81856-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lund K A, Opresko L K, Starbuck C, Walsh B J, Wiley H S. J Biol Chem. 1990;265:15713–15723. [PubMed] [Google Scholar]
- 27.Mendelsohn J, Masui H, Goldenberg A. Trans Assoc Am Physicians. 1987;100:173–178. [PubMed] [Google Scholar]
- 28.Forsten K E, Lauffenburger D A. J Comp Biol. 1994;1:15–23. doi: 10.1089/cmb.1994.1.15. [DOI] [PubMed] [Google Scholar]
- 29.Peipkorn M, Lo C, Plowman G. J Cell Physiol. 1994;159:114–120. doi: 10.1002/jcp.1041590115. [DOI] [PubMed] [Google Scholar]
- 30.Higashiyama S, Iwamoto R, Goishi K, Raab G, Taniguchi N, Klagsbrun M, Mekada E. J Cell Biol. 1995;128:929–938. doi: 10.1083/jcb.128.5.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthay M A, Thiery J P, Lafont F, Stampfer M F, Boyer B. J Cell Sci. 1993;106:869–878. doi: 10.1242/jcs.106.3.869. [DOI] [PubMed] [Google Scholar]
- 32.Miettinen P J, Berger J E, Meneses J, Phung Y, Pedersen R A, Werb Z, Derynck R. Nature (London) 1995;376:337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 33.Snedeker S M, Brown C F, DiAugustine R P. Proc Natl Acad Sci USA. 1991;88:276–280. doi: 10.1073/pnas.88.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Opresko L K, Chang C P, Will B H, Burke P M, Gill G N, Wiley H S. J Biol Chem. 1995;270:4325–4333. doi: 10.1074/jbc.270.9.4325. [DOI] [PubMed] [Google Scholar]