

Abstract
Activation of B cells in the airways is now believed to be of great importance in immunity to pathogens, and it participates in the pathogenesis of airway diseases. However, little is known about the mechanisms of local activation of B cells in airway mucosa. We investigated the expression of members of the B cell-activating TNF superfamily (B cell-activating factor of TNF family (BAFF) and a proliferation-inducing ligand (APRIL)) in resting and TLR ligand-treated BEAS-2B cells and primary human bronchial epithelial cells (PBEC). In unstimulated cells, expression of BAFF and APRIL was minimal. However, BAFF mRNA was significantly up-regulated by TLR3 ligand (dsRNA), but not by other TLR ligands, in both BEAS-2B cells (376-fold) and PBEC (224-fold). APRIL mRNA was up-regulated by dsRNA in PBEC (7-fold), but not in BEAS-2B cells. Membrane-bound BAFF protein was detectable after stimulation with dsRNA. Soluble BAFF protein was also induced by dsRNA (>200 pg/ml). The biological activity of the epithelial cell-produced BAFF was verified using a B cell survival assay. BAFF was also strongly induced by IFN-β, a cytokine induced by dsRNA. Induction of BAFF by dsRNA was dependent upon protein synthesis and IFN-αβ receptor-JAK-STAT signaling, as indicated by studies with cycloheximide, the JAK inhibitor I, and small interfering RNA against STAT1 and IFN-αβ receptor 2. These results suggest that BAFF is induced by dsRNA in airway epithelial cells and that the response results via an autocrine pathway involving IFN-β. The production of BAFF and APRIL by epithelial cells may contribute to local accumulation, activation, class switch recombination, and Ig synthesis by B cells in the airways.
Airway epithelial cells form a complex physical barrier that defends against harmful substances and microbial pathogens. Airway epithelial cells also function in the regulation of immune responses through production of cytokines and chemokines and via interactions with cells of the immune system. Epithelial activation results from direct exposure of epithelial cells to injurious material, exposure to pathogen-associated molecular patterns (PAMPs)3 that trigger TLRs, or exposure to activating cytokines derived from tissue cells or infiltrating leukocytes (1–6). Activation of epithelial cells can result in immediate host defense responses, as they are induced to produce host defense molecules. In addition, prolonged or robust epithelial activation can result in the release of proinflammatory cytokines and chemokines that attract inflammatory cells. Activation of epithelium and production of cytokines and chemokines have been of particular interest in allergic conditions such as asthma and allergic rhinitis (7, 8). It has been studied widely because these mediators influence the activity of inflammatory cells such as eosinophils, T cells, B cells, and mast cells, which are the characteristic infiltrating cells in these disorders (7, 8).
The production of IgA and IgE from B cells has been known to be critical in allergic diseases. Diaz-Sanchez et al. (9) demonstrated local IgE production in the upper airways of human subjects. Recent evidence has demonstrated that extensive Ig class switch recombination (CSR) and IgE production occur in the nasal mucosa of patients with allergic rhinitis and in the bronchial mucosa of asthmatics (10, 11). In addition, activated IgA- and IgE-expressing B cells have been found in the airway mucosa of patients with these diseases. Due to the recognized importance of IgE in asthma and rhinitis, the local activation of B cells in the airway has been proposed to be an important event in the pathogenesis of allergic airway diseases.
CSR is a process by which B cells shift from expression of the IgM H chain C region (Cμ) to another C region to make a new isotype of Ab (e.g., switching from IgM to IgA or IgE). Ags elicit CSR in germinal center B cells through interactions between CD40 on B cells and the TNF ligand superfamily (TNFSF) member CD40L (also known as CD154 and TNFSF5) on activating CD4+ T cells (12, 13). Although CSR is generally thought to be highly dependent on CD40, it is also reported that viral and bacterial products can induce Ig production in the absence of T cells or CD40-dependent signaling (14–17).
A proliferation-inducing ligand (APRIL; also known as TNFSF13) and B cell-activating factor of the TNF family (BAFF; also known as BLyS, TNFSF13B, TALL-1, and THANK) are recently identified members of the TNFSF that play important roles in B cell maturation and function (18–21). BAFF promotes B cell survival and proliferation (21–23). BAFF and APRIL also promote CD40-independent, T cell-independent CSR and Ig production (15, 24). BAFF binds to three receptors that are selectively expressed on B cells, including transmembrane activator and CAML interactor (TACI), B cell maturation Ag (BCMA), and BAFF receptor (BAFF-R) (21–23). APRIL also binds to TACI and BCMA, but not BAFF-R, which only binds BAFF (21–23). BAFF-R is a potent regulator of mature B cell survival (25, 26). In contrast, TACI has been considered to suppress B cell proliferation and survival. BCMA does not appear to be important for B cell homeostasis, but is required for optimal survival of plasma cells. BAFF-R and TACI are also able to signal class switching to IgG and IgE, but the class switch to IgA is regulated by TACI only (24). It has been reported that elevated amounts of BAFF can be found in the serum of autoimmune patients, such as those with systemic lupus erythematosus, rheumatoid arthritis, and Sjogren’s syndrome, and the potential therapeutic efficacy of Abs against BAFF has been investigated (22, 27, 28). BAFF transgenic mice have high levels of total Igs, rheumatoid factors, and circulating immune complexes in their serum, whereas animals or humans lacking BAFF or BAFF-Rs have minimal B cells and Ig responses (29, 30). Thus, it has been suggested that BAFF plays an important role in both normal immune responses and autoimmune disease pathogenesis. In contrast to autoimmune disease, little is known about the role of BAFF and APRIL in inflammatory diseases of the airways.
Functional TLR family members are known to be expressed on airway epithelial cells (6, 31–34). In the present study, we investigated whether members of the B cell-activating TNFSFs, CD40L, APRIL, and BAFF were expressed or induced by TLR ligands in airway epithelial cells. We discovered that BAFF was strongly and significantly induced by the TLR3 ligand dsRNA and that expression of BAFF is critically regulated by TLR3- and IFN-β-dependent signaling in airway epithelial cells. These findings raise the possibility of an important interaction between airway epithelial cells and B cells and lead to the hypothesis that the production of BAFF by epithelial cells may contribute to local accumulation, activation, CSR, and Ig synthesis by B cells in the airway.
Materials and Methods
Reagents
Zymosan A from Saccharomyces cerevisiae, LPS from Escherichia coli, serotype 0111:B4, and fluticasone propionate (FP) were purchased from Sigma-Aldrich. Poly(I:C) (dsRNA) (Amersham Biosciences), peptidoglycan (PGN) from Staphylococcus aureus (Fluka), flagellin isolated from Salmonella typhimurium strain 14,028 (Apotech), and R-848 (InvivoGen) were purchased from the indicated sources. CpG oligodeoxynucleotide 2216 (CpG-A; 5′-ggGGGACGATCGTCgggggG-3′, small letters, phosphorothioate linkage; capital letters, phosphodiester linkage 3′ of the base) was synthesized at Sigma Genosys. Recombinant human IL-1β, IL-4, IL-6, IL-10, IL-12, IL-15, TNF-α, IFN-β, IFN-γ, G-CSF, GM-CSF, BAFF, and the IgG1 Fc fusion proteins of BAFF-R (BAFF-R:Fc) and TACI (TACI: Fc) were purchased from R&D Systems. The small interfering RNAs (siR-NAs) against STAT1 (Hs_STAT1_6 HP validated siRNA; SI02662324), IFN-αβ receptor 2 (IFNAR2) (Hs_IFNAR2_2 HP siRNA; SI00017500), and the no effect control were obtained from Qiagen.
Cell culture, treatments, and transfection
The adeno 12 SV40-transformed human bronchial epithelial cell line, BEAS-2B, was a gift from C. Harris (National Cancer Institute, Bethesda, MD) (35) and cultured in DMEM/F12 (Invitrogen Life Technologies) supplemented with 5% heat-inactivated FBS (Invitrogen Life Technologies), 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen Life Technologies). Human primary bronchial epithelial cells (PBEC) were obtained, as described previously (36). PBEC were maintained in serum-free LHC-9 medium (BioSource International). PBEC were plated in 12- or 24-well culture plates coated with collagen (Vitrogen; Collagen Biomaterials). Before stimulation, PBEC were cultured in serum-free LHC-8 medium without hydrocortisone (BioSource International) for at least 2 days. Either type of airway epithelial cells was stimulated with one of the following at the indicated concentration: 10 μg/ml PGN, 100 μg/ml zymosan, 25 μg/ml dsRNA, 1 μg/ml LPS, 10 ng/ml flagellin, 1 μg/ml R-848, and 3 μg/ml CpG-A for 18 h; or 100 ng/ml IL-1β, 100 ng/ml IL-4, 100 ng/ml IL-6, 100 ng/ml IL-10, 100 ng/ml IL-12, 100 ng/ml IL-15, 100 ng/ml TNF-α, 1000 U/ml IFN-β, 100 ng/ml IFN-γ, 100 ng/ml TGF-β, 100 ng/ml G-CSF, and 100 ng/ml GM-CSF for 6 h.
BEAS-2B cells were seeded at low density (5 × 104 cells/well) overnight in DMEM/F12 supplemented with 5% heat-inactivated FBS. At 30–50% confluence, cells were transfected with siRNA against STAT1, IF-NAR2, or control RNA at 5 or 20 nM using HiPerFect transfection reagent (Qiagen), following the manufacturer’s instruction. The transfected cells were further grown for 72 h, and then stimulated with 1000 U/ml IFN-β and 25 μg/ml dsRNA for 6 h.
Real-time PCR and RT-PCR
Total RNA was extracted using RNeasy (Qiagen) and was digested with DNase I (Qiagen), according to the manufacturer’s instructions. Single-strand cDNA was synthesized with SuperScript II reverse transcriptase (Invitrogen Life Technologies) and random primers. Real-time RT-PCR was performed with a TaqMan method using an Applied Biosystems 7500 Sequence Detection System (Applied Biosystems) in 20 μl reactions (10 μl of 2× TaqMan Master mix (Applied Biosystems), 400 nM each primer, and 200 nM TaqMan probe plus cDNA). Primer and probe sets for four genes, BAFF (sense, 5′-TCGATGTATTCAAAATATGCCTGAAA-3′; antisense, 5′-TGCAATGCCAGCTGAATAGC-3′; MGB-probe, 5′-CTA CCCAATAATTCC-3′), APRIL (sense, 5′-AGAGCAGTGCTCACCCA AAAAC-3′; antisense, 5′-GCCACATCACCTCTGTCACATC-3′; MGB-probe, 5′-CACCTGGTTCCCATTAA-3′), CD40L (sense, 5′-CTCTATTA TATCTATGCCCAAGTCACCTT-3′; antisense, 5′-GACTTTAGGCAGA GGCTGGCTA-3′; MGB-probe, 5′-CGAGTCAAGCTCC-3′), and GAP DH (sense, 5′-GAAGGTGAAGGTCGGAGTC-3′; antisense, 5′-GAAGA TGGTGATGGGATTTC-3′; FAM/TAMRA-labeled probe, 5′-CAAGCT TCCCGTTCTCAGCC-3′) were synthesized at Applied Biosystems. Real-time PCR for IFN-β was performed with a SYBR Green I method, as described previously (37). To determine the exact copy number of the target genes, quantified aliquots of purified PCR fragments of target genes were serially diluted and used as standards in each experiment. Aliquots of cDNA equivalent to 10 ng of total RNA (PCR for IFN-β was 125 ng) were used for real-time PCR. The expression levels of mRNA were normalized to the median expression of a housekeeping gene (GAPDH). Conventional RT-PCR was performed using Platinum TaqDNA Polymerase in 50 μl reactions (45 μl Platinum PCR SuperMix (Invitrogen Life Technologies) and 200 nM each primer plus cDNA). BAFF primers for RT-PCR (sense, 5′-GAAATAAGCGTGCCGTTCAGG-3′; antisense, 5′-TTAGATGTCCC ATGGCGTAGGTC-3′) were synthesized at Applied Biosystems.
GeneChip expression analysis
GeneChip analysis was performed, as described previously (38). Gene expression was measured using the GeneChip Human Genome U133 plus 2.0 probe array (Affymetrix). Data analysis was performed with the GeneChip Operating Software (Affymetrix) and the GeneSpring software version 7.2 (Agilent).
Flow cytometry
BEAS-2B cells were dispersed into a single-cell suspension by incubation with Versene (Invitrogen Life Technologies) for 15 min. Cells were suspended in PBS containing 1.5% BSA and then incubated with either PE-conjugated mouse anti-human BAFF mAb (clone: 1D6, IgG1; eBioscience) or PE-conjugated mouse control IgG1 Ab (clone: MOPC-31C; BD Pharmingen) for 30 min in the dark at 4°C. Cells were washed and resuspended in PBS containing 0.1% BSA and immediately analyzed with a BD FACSArray Bioanalyzer (BD Biosciences). Flow cytometry data were analyzed using FlowJo software version 5.6.1 (Tree Star).
ELISA
The concentrations of BAFF protein in cell-free supernatants were measured with a specific ELISA kit (R&D Systems). The detection limit for this kit is 31.25 pg/ml.
B cell isolation
Human peripheral blood buffy coats were obtained from LifeSource Blood Services. Human PBMC were isolated by centrifugation on a Ficoll-sodium diatrizoate density gradient (d = 1.077 g/ml, Ficoll-Paque plus; Amersham Biosciences) from buffy coat, according to the manufacturer’s directions. After lysing erythrocytes, B cells were purified using the MACS system (Miltenyi Biotec) with anti-CD19 MicroBeads (Miltenyi Biotec). The purity of the CD20-positive B cells prepared was always >99%.
MTT assay
For the analysis of the biological activity of secreted BAFF, BEAS-2B cells were cultured in serum-free DMEM/F12 medium and then stimulated with 1000 U/ml IFN-β for 72 h. The supernatant was concentrated 150–200 times using Centriplus YM-10 (Millipore) and Microcon YM-10 (Millipore). B cells were cultured at a density of 2 × 105 cells/100 μl/well of a 96-well flat-bottom microtiter plate in RPMI 1640 (Invitrogen Life Technologies) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were stimulated as indicated with 5 ng/ml BAFF, 10 ng/ml IL-4, 10 μg/ml mouse anti-human CD40 mAb (clone: 5C3, IgG1; eBioscience), concentrated supernatant, or combinations of the above for 5 days in the presence or absence of 5 μg/ml control IgG1, 5 μg/ml BAFF-R:Fc, and 5 μg/ml TACI:Fc. To detect intracellular enzyme activity, 10 μl of MTT reagent (American Type Culture Collection) was added for the last 4 h of incubation. After 4 h, 100 μl of detergent reagent (American Type Culture Collection) was added to all wells, and the plate was incubated in the dark overnight at room temperature. Absorbance at 570 nm was measured using a microtiter plate reader.
Statistical analysis
All data are presented as the mean ± SEM, unless otherwise noted. Differences between groups were analyzed using the paired Student’s t test and considered to be significant if p < 0.05.
Results
Effect of TLR ligands on BAFF and APRIL expression in airway epithelial cells
Growing evidence indicates that B cell proliferation and differentiation occur at mucosal surfaces. To test the possible role of epithelial cells, we first set out to determine the spontaneous expression of mRNA for members of the B cell-activating TNF superfamily ligand such as CD40L, BAFF, and APRIL in unstimulated bronchial epithelial cells using GeneChip and real-time PCR. Neither CD40L (ID: 207892_at), BAFF (ID: 223501_at and 223502_at), or APRIL (ID: 229326_at) was detected in unstimulated BEAS-2B cells using the GeneChip (data not shown). Using real-time PCR, mRNA for BAFF and APRIL were detected, but very weakly expressed in both resting BEAS-2B cells and resting PBEC (Fig. 1, A and B). The level of mRNA for CD40L was below the detection limit of real-time PCR (data not shown).
FIGURE 1.

Effect of TLR ligands on the up-regulation of BAFF and APRIL in airway epithelial cells. BEAS-2B cells (A) or PBEC (B) were incubated for 18 h with 10 μg/ml PGN, 100 μg/ml zymosan, 25 μg/ml dsRNA, 1 μg/ml LPS, 10 ng/ml flagellin, 1 μg/ml R-848, or 3 μg/ml CpG-A, as indicated, and then mRNA was extracted and analyzed for BAFF and APRIL using real-time PCR. C, BEAS-2B cells or PBEC were incubated for 18 h with 25 μg/ml dsRNA, and then expression of mRNA for full-length BAFF (246 bp), ΔBAFF (189 bp), and GAPDH (226 bp) was analyzed by conventional RT-PCR. D, BEAS-2B cells were incubated for 6 h with 0.25–25 μg/ml dsRNA, and then expression of mRNA for BAFF was analyzed by real-time PCR. E, BEAS-2B cells were incubated with 25 μg/ml dsRNA (circle) or vehicle control (square) for 0.5–18 h, and then expression of mRNA for BAFF was analyzed by real-time PCR. The copy number is expressed as the number of transcripts/ng total RNA. The results are shown as the mean ± SEM of three to five independent experiments. *, p < 0.05.
To assess whether or not TLR ligands induce CD40L, BAFF, and APRIL expression, BEAS-2B cells and PBEC were treated with PGN (TLR2 ligand), zymosan (TLR2 ligand), dsRNA (TLR3 ligand), LPS (TLR4 ligand), flagellin (TLR5 ligand), R-848 (TLR7/8 ligand), and CpG-A (TLR9 ligand) for 18 h. mRNA for BAFF was significantly induced by stimulation with dsRNA in BEAS-2B cells (376-fold; n = 4; p < 0.05) (Fig. 1A). LPS induced a nonsignificant increase in BAFF expression in BEAS-2B cells (4-fold; n = 4; p = 0.068), whereas ligands for TLR 2, 5, 7/8, and 9 had no effect. In contrast, mRNA for APRIL and CD40L were not induced by any of the TLR ligands tested in BEAS-2B cells, even after stimulation up to 72 h (Fig. 1A and data not shown). Results were similar in PBEC, with the exception that BAFF was not induced by LPS, and APRIL was significantly induced only by dsRNA (7-fold; n = 4; p < 0.05) (Fig. 1B). BAFF mRNA was significantly up-regulated by dsRNA in PBEC (224-fold; n = 4; p < 0.05). We also examined the effect of TLR ligands on the induction of BAFF and APRIL in human primary nasal epithelial cells. mRNA for BAFF and APRIL was also significantly induced by dsRNA in human primary nasal epithelial cells, and these effects were the same as those observed in PBEC (data not shown). Because the absolute levels of mRNA and relative increase induced by dsRNA were higher for BAFF than for APRIL, we have focused our studies in BAFF. Recently, ΔBAFF, which has been shown to be a natural negative regulator of BAFF, has been identified. ΔBAFF is a spliced isoform of BAFF and it lacks exon 3 containing 57 bp (39, 40). We examined whether the form of BAFF induced by dsRNA was the active form or the inactive δ form using conventional RT-PCR. Although the ΔBAFF isoform was found to be expressed in the dsRNA-treated BEAS-2B and PBEC, it was only a minor isoform (Fig. 1C). To further examine the details of the induction of mRNA for BAFF by dsRNA, we determined the concentration and the time dependence of the response in BEAS-2B cells. BAFF mRNA was induced by concentrations of dsRNA as low as 0.25 μg/ml and with a time lag of ~3 h (Fig. 1, D and E).
BAFF exists as a type II membrane protein as well as a soluble protein derived from the membrane-bound form by cleavage with a putative furin family protease (20, 21, 41). To confirm epithelial expression of BAFF protein, we assayed the cell surface expression using flow cytometry and measured the concentration in the culture supernatant using a specific ELISA. Low levels of membrane-bound BAFF (mBAFF) were detectable only after stimulation of BEAS-2B cells with dsRNA (Fig. 2A). In contrast, significant levels of soluble BAFF (sBAFF) were detected in the supernatant after stimulation with dsRNA. Appearance of sBAFF occurred in a time-dependent manner in BEAS-2B cells (maximum at 72 h of culture: control, undetectable; dsRNA treated, 228 ± 8 pg/ml; n = 3; Fig. 2B). In PBEC, sBAFF was also strongly induced by dsRNA (72-h culture: control, undetectable; dsRNA treated, 97 ± 15 pg/ml; n = 3; Fig. 2C). These results indicate that stimulation of airway epithelial cells with dsRNA induces significant expression of both BAFF mRNA and BAFF protein. To investigate the polarity of BAFF production, we used a trans-well system. After confluence, BEAS-2B cells were stimulated with 25 μg/ml dsRNA for 48 h. The supernatant was concentrated to 100 μl using Microcon YM-10. These showed that airway epithelial cells produced BAFF protein on both the apical and basolateral sides of the monolayer after stimulation with dsRNA (apical, 401 ± 63 pg/ml; basolateral, 234 ± 17 pg/ml; n = 4).
FIGURE 2.

Detection of BAFF protein in airway epithelial cells. A, Representative histograms from flow cytometry demonstrate evidence of mBAFF on BEAS-2B cells. Cells were stained with mAb specific for BAFF (dark line) or an isotope control mAb (shaded histogram) after incubation with or without (untreated) 25 μg/ml dsRNA for 24 h. Detection of sBAFF protein by ELISA in the culture supernatant of BEAS-2B (B) and PBEC (C, 72 h) stimulated with dsRNA for the indicated time. Results shown are mean ± SEM of three to four independent experiments. **, Not detectable. *, p < 0.05.
Effect of cytokines on BAFF expression in airway epithelial cells
Published studies indicate that BAFF is induced in monocytes, macrophages, dendritic cells, neutrophils, nurse-like cells, astrocytes, and salivary gland epithelial cells by several cytokines, including IL-10, IFN-α, IFN-β, IFN-γ, and G-CSF (15, 41–45). We therefore examined whether cytokines were able to induce BAFF expression in airway epithelial cells. BEAS-2B cells and PBEC were incubated with IL-1β, IL-4, IL-6, IL-10, IL-12, IL-15, TNF-α, IFN-β, IFN-γ, TGF-β, G-CSF, and GM-CSF for 6 h. mRNA for BAFF was significantly up-regulated by TNF-α (6-fold; n = 4; p < 0.05), IFN-β (485-fold; n = 4; p < 0.05), and IFN-γ (26-fold; n = 4; p < 0.05), but not by the other cytokines tested in BEAS-2B cells (Fig. 3A). Similar results were obtained in PBEC, with the exception that TNF-α did not significantly induce BAFF (Fig. 3B). BAFF was induced by IFN-β (21-fold; n = 4; p < 0.05) and IFN-γ (2-fold; n = 4; p < 0.05) in PBEC (Fig. 3B).
FIGURE 3.
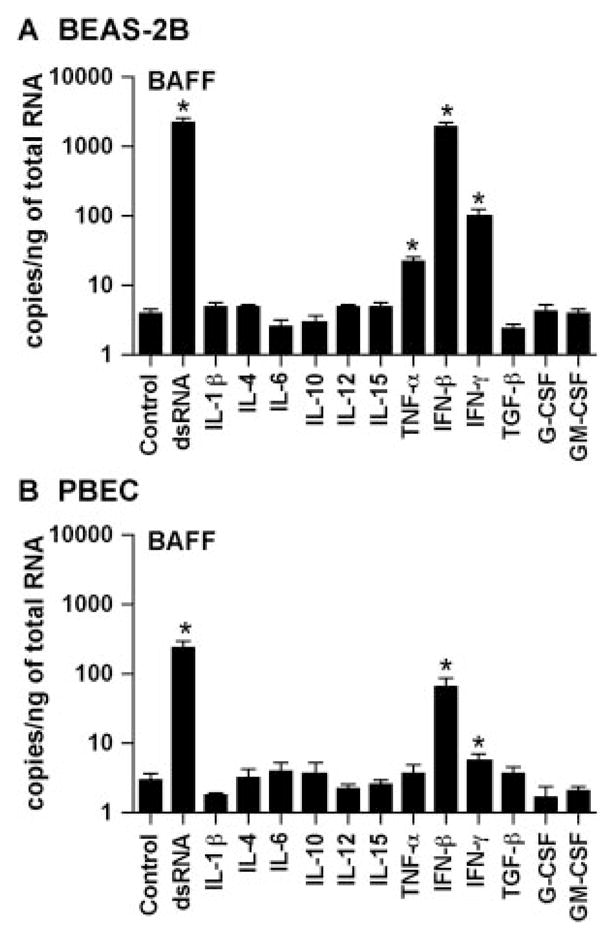
Effect of cytokines on the expression of BAFF in airway epithelial cells. BEAS-2B cells (A) and PBEC (B) were incubated for 6 h with 25 μg/ml dsRNA, 100 ng/ml IL-1β, 100 ng/ml IL-4, 100 ng/ml IL-6, 100 ng/ml IL-10, 100 ng/ml IL-12, 100 ng/ml IL-15, 100 ng/ml TNF-α, 1000 U/ml IFN-β, 100 ng/ml IFN-γ, 100 ng/ml TGF-β, 100 ng/ml G-CSF, or 100 ng/ml GM-CSF, as indicated. The level of BAFF mRNA was determined by real-time PCR. The results are shown as the mean ± SEM of three to four independent experiments. *, p < 0.05.
Role of IFN-β in dsRNA-induced BAFF expression
Among the cytokines tested, IFN-β induced the highest level of BAFF expression. We therefore investigated further the potential role that IFN-β may play in the induction of mRNA for BAFF. BEAS-2B cells were incubated with different concentrations of IFN-β for up to 6 h. BAFF mRNA was strongly induced by IFN-β in a dose-dependent manner (Fig. 4A). Surprisingly, concentrations of IFN-β as low as 1 U/ml (~1 pg/ml) induced BAFF expression (Fig. 4A). Induction of mRNA for BAFF by IFN-β occurred very rapidly, as quickly as 1 h after stimulation with IFN-β (Fig. 4B). The response to IFN-β was more rapid than the response to dsRNA, which required 3 h to be apparent (Figs. 1E and 4B). These results indicate that IFN-β is a potent and rapid stimulator of BAFF expression in airway epithelial cells.
FIGURE 4.

Effect of IFN-β on the up-regulation of BAFF by dsRNA in BEAS-2B cells. A, BEAS-2B cells were incubated for 6 h with 1–1000 U/ml IFN-β. B, BEAS-2B cells were incubated with 100 U/ml IFN-β for up to 6 h. C, BEAS-2B cells were incubated with 25 μg/ml dsRNA or control medium for up to 6 h. The levels of BAFF and IFN-β mRNA were determined by real-time PCR. The results are shown as the mean ± SEM of three independent experiments.
It has been reported that IFN-β expression is also strongly induced by virus infection or dsRNA stimulation of airway epithelial cells (46, 47). We examined the influence of dsRNA on expression of IFN-β in epithelial cells. Stimulation with dsRNA rapidly and strongly induced IFN-β mRNA (Fig. 4C). If BAFF expression in response to dsRNA occurs as a result of induction of IFN-β, then inhibition of protein synthesis should prevent the expression of mRNA for BAFF, but not IFN-β. BEAS-2B cells were treated with DMSO (vehicle control) or the protein synthesis inhibitor cycloheximide for 1 h, and then stimulated with dsRNA for 3 h. Up-regulation of mRNA for BAFF was significantly and dose dependently inhibited by cycloheximide (Fig. 5A). In contrast, induction of mRNA for IFN-β by dsRNA was not inhibited by cycloheximide (Fig. 5A). In fact, induction of IFN-β mRNA was enhanced by cycloheximide, suggesting the presence of a short-lived or inducible inhibitor of the induction of IFN-β. Although these results do not prove that BAFF expression is secondary to the expression of IFN-β, they do demonstrate that BAFF is not an immediate early gene in this response, despite the fact that it is rapidly induced. It has been reported that the JAK inhibitor, referred to as JAK inhibitor I (JAKI), inhibits activation of STAT proteins by several JAK family kinases, including JAK1, JAK2, JAK3, and tyrosine kinase 2 (Tyk2) (48, 49). Signaling induced by IFN-β via IFNAR1 and IFNAR2 is known to involve the activation of STAT1 and STAT2 by JAK1 and Tyk2. To determine whether induction of BAFF by dsRNA was mediated by an autocrine/paracrine pathway using IFN-β, we treated BEAS-2B cells with DMSO or JAKI for 1 h, and then stimulated with dsRNA for 6 h. Induction of mRNA for BAFF by dsRNA was dose dependently and significantly inhibited by JAKI (Fig. 5B). Finally, to investigate the mechanism of BAFF induction by dsRNA more carefully, we knocked down IFN-signaling molecules. BEAS-2B cells were transfected with siRNA against control RNA, STAT1, or IFNAR2, and then stimulated with IFN-β and dsRNA for 6 h. Induction of BAFF by IFN-β and dsRNA was significantly inhibited by siRNA against STAT1 and IFNAR2, but not by control RNA (Fig. 5C). These results suggest that BAFF is induced by dsRNA in airway epithelial cells, and that the response results via an autocrine/paracrine pathway involving IFN-β.
FIGURE 5.

Effect of the IFN-STAT pathway on the expression of BAFF induced by dsRNA in BEAS-2B cells. A, BEAS-2B cells were pre-incubated with 0.01% DMSO or 0.1–10 mg/ml cycloheximide (CHX) for 1 h, and then stimulated with 25 μg/ml dsRNA for 3 h. B, BEAS-2B cells were preincubated with 0.1% DMSO or 0.1–10 μM JAKI for 1 h, and then stimulated with 25 μg/ml dsRNA for 6 h. C, BEAS-2B cells were transfected with siRNA against STAT1, IFNAR2, or control RNA at 5 or 20 nM for 72 h, and then stimulated with 1000 U/ml IFN-β and 25 μg/ml dsRNA for 6 h. The levels of BAFF and IFN-β mRNA were determined by real-time PCR. The results are shown as the mean ± SEM of four independent experiments. *, p < 0.05 vs dsRNA or IFN-β stimulated.
Effect of glucocorticoids on induction of BAFF
Glucocorticoids are a mainstay in the treatment of diseases characterized by airway inflammation such as asthma, chronic obstructive pulmonary disease, and chronic rhinosinusitis (50). We investigated whether glucocorticoids were able to suppress the dsRNA-dependent production of BAFF in airway epithelial cells. BEAS-2B cells were treated with DMSO or the potent topical glucocorticoid FP for 2 h, and then stimulated with dsRNA for 18 h. Induction of mRNA for BAFF was significantly and dose dependently inhibited by FP (Fig. 6, A and C). To confirm this phenomenon at the protein level, we measured the production of sBAFF (72-h culture) and mBAFF (24-h culture). We found that induction of both sBAFF and mBAFF by dsRNA was strongly inhibited by FP in BEAS-2B cells (Fig. 6, D and E). In PBEC, similar results were obtained when we analyzed mRNA for BAFF. Induction of mRNA for BAFF was significantly inhibited by FP in PBEC, but to a lesser extent than in BEAS-2B cells (Fig. 6B).
FIGURE 6.

Effect of glucocorticoids on the up-regulation of BAFF by dsRNA in airway epithelial cells. BEAS-2B cells (A and C–E) and PBEC (B) were preincubated with 0.001% DMSO, 100 nM FP (A, B, D, and E), or 10−11–10−6 M FP (C) for 2 h, and then stimulated with 25 μg/ml dsRNA for 18 h (A–C), 24 h (E), or 72 h (D). The level of BAFF mRNA was determined by real-time PCR (A–C). Concentrations of sBAFF protein in the culture supernatant were measured by ELISA (D). Cells were stained with mAb specific for BAFF (dark line) or an isotope control mAb (shaded histogram) and analyzed by flow cytometry (E). The results are shown as the mean ± SEM of three to four independent experiments. *, p < 0.05 vs dsRNA stimulated. **, Not detectable.
Functional activity of epithelial cell-derived BAFF
It has been reported that BAFF directly promotes proliferation, cell survival, CSR, and Ig production in B cells through a CD40-independent and T cell-independent pathway (15, 19–21, 24). We therefore investigated whether epithelial cell-derived BAFF was able to induce B cell survival using a MTT assay. We used supernatants from IFN-β-treated cells in a functional assay based upon B cell proliferation. Purified human B cells were stimulated with BAFF or concentrated supernatant in the presence or absence of IL-4 for 5 days. Interestingly, control supernatants from epithelial cells significantly suppressed B cell proliferation. Compared with supernatants from unstimulated epithelial cells, IFN-β-treated supernatants increased B cell survival (Fig. 7A). To determine whether this effect depended on epithelial cell-derived BAFF, we used Fc fusion proteins of the BAFF-Rs, BAFF-R and TACI, to absorb BAFF. Enhancement of cell survival by supernatants of IFN-β-treated epithelial cells was significantly inhibited by BAFF-R:Fc and TACI:Fc, but not by control IgG1 (Fig. 7B). In contrast, anti-CD40-dependent and CpG-dependent B cell proliferation was not inhibited by either BAFF-R:Fc or TACI:Fc (Fig. 7B and data not shown). These results suggest that epithelial cell-derived BAFF is functionally active.
FIGURE 7.

Effect of epithelial cell-derived BAFF on the cell survival of B cells. A, Purified human B cells were stimulated with 5 ng/ml BAFF or concentrated BEAS-2B supernatant for 5 days. B, Purified human B cells were stimulated with 5 ng/ml BAFF, 10 ng/ml IL-4, 10 μg/ml mouse anti-human CD40 mAb, concentrated supernatant, or the indicated combinations for 5 days in the presence or absence of 5 μg/ml control IgG1, 5 μg/ml BAFF-R:Fc, or 5 μg/ml TACI:Fc. MTT reagent was added for the last 4 h of incubation. The concentrated BEAS-2B supernatant was added at a final dilution of 1/20 to the B cell culture. Absorbance at 570 nm was measured using a microtiter plate reader. The results are shown as the mean ± SEM of five independent experiments.*, p < 0.05.
Discussion
This study provides the first direct demonstration that airway epithelial cells can produce the B cell-activating TNF family members, BAFF and APRIL (Figs. 1 and 2), although BAFF production by epithelial cells from salivary glands has been published recently (45). BAFF is a centrally important regulator and activator of B cells and has been recognized to be a main product of myeloid cells such as monocytes, macrophages, dendritic cells, neutrophils, and nurse-like cells. The quantity of BAFF produced by airway epithelial cells in these experiments (~100–200 pg/106 cells) is of the same order of magnitude as those produced by myeloid cells, which have been reported to generate 100–500 pg/106 cells in a 72-h culture (15, 41–43).
TLRs play important roles linking innate immunity and adaptive immunity, and airway epithelial cells have been shown to express functional TLR, notably TLR2–6 (6, 33, 34). Among the TLR ligands that we tested in this study, only dsRNA, a TLR3 ligand and mimic of viral RNA and viral replication, induced BAFF expression in primary bronchial epithelial cells. BAFF was also induced by LPS in the BEAS-2B-immortalized cell line, but not in PBEC (Fig. 1). Although epithelial cells express TLR4, and we might have expected a more robust response to LPS, previous studies have shown that production of IFN-β by LPS is regulated by serum LPS-binding protein (37). In addition, soluble CD14 is required for activation of epithelial cells by LPS due to the lack of membrane-bound CD14 on airway epithelial cells (33, 51). In the present experiments, we used serum-free medium in the culture of primary bronchial epithelial cells, to avoid contamination with fibroblasts. This may explain the lack of production of IFN-β and BAFF by PBEC after stimulation with LPS. The failure of other TLR ligands tested to induce expression of BAFF may reflect low levels of receptor expression or adaptor proteins involved in the respective responses.
IFN-β is known to be an immediate early gene, i.e., one in which mRNA expression is induced without the requirement for protein synthesis. IFN-β is directly induced by bacterial and viral PAMPs such as LPS, dsRNA, ssRNA, and CpG motif-containing DNA via TLR-dependent or TLR-independent signaling (37, 38, 52–54). The IFNARs are composed of two subunits, IFNAR1 and IFNAR2, which are associated with members of the JAK family, specifically Tyk2 and JAK1, respectively (52, 55, 56). Activation by IFN-β of the JAKs that are associated with the IFNAR results in tyrosine phosphorylation of STAT1 and STAT2. This leads to the formation of a STAT1-STAT2-IFN regulatory factor 9 complex that is known as IFN-stimulated gene factor 3, and formation of STAT1-STAT1 homodimers. These complexes translocate to the nucleus, bind to IFN-stimulated response elements (in the case of IFN-stimulated gene factor 3) and IFN-γ-activated sites (in the case of STAT1 homodimer) in the promoter regions of relevant genes, and initiate gene transcription (52, 55, 56). Several lines of evidence indicate that IFN-β played a critical role in the induction of BAFF by dsRNA in the present study: 1) dsRNA induced the expression of IFN-β more rapidly than it induced expression of BAFF (Figs. 1E and 4C); 2) the addition of IFN-β induced BAFF more rapidly than dsRNA induced BAFF (Fig. 4B); 3) inhibition of protein synthesis by cycloheximide reduced induction of BAFF by dsRNA suggestion that it is indirect (Fig. 5A); 4) inhibition of JAK kinase by JAKI suppressed induction of BAFF by dsRNA (Fig. 5B); and 5) induction of BAFF by dsRNA was significantly suppressed by siRNA knockdown of STAT1 and IFNAR2 (Fig. 5C). Taken together, these results suggest that dsRNA induces expression of IFN-β, which in turn activates expression of BAFF. Although this mechanism has been demonstrated for other IFN target genes (52), it has not been shown for induction of BAFF by dsRNA. Litinskiy et al. (15) have shown that human dendritic cells and monocytes up-regulate BAFF and APRIL upon stimulation with IFN-α, IFN-γ, and LPS; however, they have not described the mechanism. This suggests that induction of BAFF expression by dsRNA and other PAMPs that induce IFN may be a secondary effect of expression of IFN-α or IFN-β.
It has been reported that sBAFF is derived from the membrane-bound form by cleavage with a putative furin family protease or derived from intracellular processing of the longer form by the same enzyme (20, 21, 41, 42). In airway epithelial cells, production of sBAFF by dsRNA occurred in a time-dependent manner (Fig. 2). In contrast, expression of mBAFF peaked at 12–24 h after stimulation (Fig. 2 and data not shown). Production of sBAFF by dsRNA was completely suppressed by decanoyl-Arg-Val-Lys-Arg-chloromethylketone, a specific furin convertase inhibitor (42), in BEAS-2B cells (data not shown). Furin mRNA (ID: 201945_at) was detected in BEAS-2B and primary epithelial cells in our GeneChip analysis (data not shown). These results suggest that the mechanism of production of sBAFF in airway epithelial cells may be similar to that found in myeloid cells.
BAFF is a B cell survival factor and a costimulator of B cell proliferation and Ab secretion. Recently, Gavin et al. (39, 40) have identified a splice isoform of BAFF that is called ΔBAFF in both mouse and human. The ΔBAFF transcripts of human and mouse are devoid of exons 3 and 4, respectively. ΔBAFF may oppose the effects of BAFF, as ΔBAFF transgenic mice have been shown to have a significant reduction in mature B cell numbers, whereas mature B cells are expanded in BAFF transgenic mice (40). The ΔBAFF isoform is detected in human myeloid cells, but it is a minor isoform (39). In airway epithelial cells, the ΔBAFF isoform was present and induced by dsRNA, but it was only a minor isoform (Fig. 1C). Future studies are required to determine whether epithelial cells produce ΔBAFF protein.
Functional analysis showed that concentrated supernatants from IFN-β-treated BEAS-2B cells stimulated B cell proliferation and/or survival through a mechanism that partially involves BAFF. This activity was observed despite the fact that supernatants from unstimulated BEAS-2B cells suppressed B cell survival (Fig. 7A). The cell survival-inducing activity found in IFN-β-treated supernatants was probably due to BAFF based upon inhibition of the response with fusion proteins specific for BAFF-Rs (BAFF-R:Fc and TACI:Fc), but not control IgG1 (Fig. 7B). BAFF-R:Fc and TACI:Fc did not inhibit CD40-dependent proliferation and/or cell survival, establishing specificity (Fig. 7B). These results suggest that the BAFF produced by dsRNA- and IFN-β-stimulated airway epithelial cells is functionally active. The identity of the suppressive activity found in supernatants of unstimulated BEAS-2B is worthy of future investigation.
We examined whether glucocorticoids can inhibit dsRNA-dependent BAFF production, because glucocorticoids are widely used in the therapeutic management of inflammatory airway diseases. In vivo studies in human subjects have demonstrated that glucocorticoids inhibit the seasonal increase in IgE in the airways and in the serum of allergic individuals (57, 58). The prevailing view is that glucocorticoids achieve these effects by inhibiting the production of cytokines such as IL-4 and IL-13 that induce CSR, because B cell responses in vitro are resistant to glucocorticoids. As shown in Fig. 6, the potent topical glucocorticoid FP significantly inhibited induction of both BAFF mRNA and BAFF protein by dsRNA in BEAS-2B cells. Expression of BAFF mRNA induced by dsRNA in PBEC was also inhibited by FP (Fig. 6B). Future clinical studies are required to investigate whether glucocorticoids suppress BAFF induction in vivo.
Local activation of B cell proliferation, CSR, and Ab production is likely to be of great importance in immunity of the airways to pathogens. In asthma, disease exacerbations are usually triggered by infection with airway-targeting viruses, especially rhinovirus, respiratory syncytial virus, influenza virus, and adenovirus (59–61). The ability of the local tissue to mount a rapid and effective local Ig response against virus is most likely to be important in the limitation of the extent and severity of virus-induced tissue damage. Local expression of germline transcripts and excision circles for IgE and activation-induced cytidine deaminase, essential regulators of CSR, has been demonstrated to occur in the airway mucosa following nasal challenge with allergen or diesel exhaust extracts (9–11, 62, 63). B cell clusters are found in the lamina propria of the nasal mucosa (11, 64). IgE-expressing B cells and plasma cells are found in the lamina propria, but not in the epithelium (64). IgA and IgE are also found in the bronchial lavage and in the nasal lavage of allergic patients (65, 66). The levels of Ag-specific IgE are frequently found to be much higher in the airways and in the nasal mucosa than they are in the serum (65–67). Thus, the local CSR and activation of B cells may be an event that is important both in disease pathogenesis and protection from pathogens. Our data show that BAFF protein is detected on both the apical and basolateral sides of cultured epithelial cells. Our results suggest that local activation of B cells in the airways may depend in part on epithelial production of BAFF and APRIL. In inflammatory lung diseases, activated airway epithelial cells produce chemokines that recruit many cell types capable of producing type I IFN, including monocytes, macrophages, and dendritic cells (50, 68). These recruited cells produce IFNs directly and also produce BAFF via autocrine pathways involving IFNs when stimulated with PAMPs and cytokines (15). Production of IFN by infiltrating inflammatory cells could activate epithelial cells to produce BAFF in vivo.
Recently, Sutherland et al. (69) have shown that BAFF transgenic mice have suppressed OVA-induced allergic airway responses, including reduced eosinophils in BALF and infiltrating leukocytes around airways and pulmonary blood vessels. The studies of Sutherland et al. (69) indicate that high levels of systemic BAFF alter T cell homing to the airways, not T cell priming or B cell responses. Studies targeting BAFF production in the airways will be necessary to determine whether local BAFF produced in the lungs or upper airways can contribute meaningfully to local B cell responses. In a separate clinical study, we have detected mRNA for BAFF and APRIL in nasal scraping epithelial cells and found increased expression of BAFF protein in nasal tissue extracts and nasal lavage from patients with chronic rhinosinusitis (A. Kato, A. T. Peters, R. Kern, D. Conley, L. C. Grammer, and R. P. Schleimer, manuscript in preparation).
In summary, we report in this study that airway epithelial cells produce the B cell-activating factors, BAFF and APRIL. Our findings indicate that BAFF is induced by cytokines and dsRNA in airway epithelial cells, and that the response to the latter results from an IFNAR-mediated autocrine/paracrine pathway of IFN-β. The production of BAFF and APRIL by epithelial cells may contribute to local accumulation, activation, CSR, and Ig synthesis by B cells in the airway.
Acknowledgments
We thank Drs. Marianna Kulka and Ning Zhang and Brian Tancowny of Northwestern University Feinberg School of Medicine for technical advice with flow cytometry, real-time PCR, and PBEC preparation. We thank Anne Jedlicka and the Malaria Research Institute Gene Array Core Facility at the Johns Hopkins University Bloomberg School of Public Health for their roles in the gene array experiments.
Footnotes
Abbreviations used in this paper: PAMP, pathogen-associated molecular pattern; APRIL, a proliferation-inducing ligand; BAFF, B cell-activating factor of TNF family; BAFF-R, BAFF receptor; BCMA, B cell maturation Ag; CSR, class switch recombination; FP, fluticasone propionate; IFNAR, IFN-αβ receptor; JAKI, JAK inhibitor I; mBAFF, membrane-bound BAFF; PBEC, primary human bronchial epithelial cell; PGN, peptidoglycan; sBAFF, soluble BAFF; siRNA, small interfering RNA; TACI, transmembrane activator and CAML interactor; TNFSF, TNF ligand superfamily; Tyk2, tyrosine kinase 2.
Disclosures
Dr. Schleimer has received honoraria from all major manufacturers of inhaled glucocorticoids, including GlaxoSmithKline, the manufacturer of fluticasone propionate.
This work was supported in part by National Institutes of Health Grants R01 HL068546 and R01 HL078860 and by a grant from the Ernest S. Bazley Trust.
References
- 1.Hogg JC, Eggleston PA. Is asthma an epithelial disease? Am Rev Respir Dis. 1984;129:207–208. [PubMed] [Google Scholar]
- 2.Cohn LA, Adler KB. Interactions between airway epithelium and mediators of inflammation. Exp Lung Res. 1992;18:299–322. doi: 10.3109/01902149209031687. [DOI] [PubMed] [Google Scholar]
- 3.Rennard SI, Romberger DJ, Sisson JH, Von Essen SG, Rubinstein I, Robbins RA, Spurzem JR. Airway epithelial cells: functional roles in airway disease. Am J Respir Crit Care Med. 1994;150:S27–S30. doi: 10.1164/ajrccm/150.5_Pt_2.S27. [DOI] [PubMed] [Google Scholar]
- 4.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 5.Diamond G, Legarda D, Ryan LK. The innate immune response of the respiratory epithelium. Immunol Rev. 2000;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- 6.Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by Toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–364. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- 7.Mills PR, Davies RJ, Devalia JL. Airway epithelial cells, cytokines, and pollutants. Am J Respir Crit Care Med. 1999;160:S38–S43. doi: 10.1164/ajrccm.160.supplement_1.11. [DOI] [PubMed] [Google Scholar]
- 8.Polito AJ, Proud D. Epithelia cells as regulators of airway inflammation. J Allergy Clin Immunol. 1998;102:714–718. doi: 10.1016/s0091-6749(98)70008-9. [DOI] [PubMed] [Google Scholar]
- 9.Diaz-Sanchez D, Dotson AR, Takenaka H, Saxon A. Diesel exhaust particles induce local IgE production in vivo and alter the pattern of IgE messenger RNA isoforms. J Clin Invest. 1994;94:1417–1425. doi: 10.1172/JCI117478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ying S, Humbert M, Meng Q, Pfister R, Menz G, Gould HJ, Kay AB, Durham SR. Local expression of ε germline gene transcripts and RNA for the ε heavy chain of IgE in the bronchial mucosa in atopic and nonatopic asthma. J Allergy Clin Immunol. 2001;107:686–692. doi: 10.1067/mai.2001.114339. [DOI] [PubMed] [Google Scholar]
- 11.Takhar P, Smurthwaite L, Coker HA, Fear DJ, Banfield GK, Carr VA, Durham SR, Gould HJ. Allergen drives class switching to IgE in the nasal mucosa in allergic rhinitis. J Immunol. 2005;174:5024–5032. doi: 10.4049/jimmunol.174.8.5024. [DOI] [PubMed] [Google Scholar]
- 12.Gauchat JF, Aubry JP, Mazzei G, Life P, Jomotte T, Elson G, Bonnefoy JY. Human CD40-ligand: molecular cloning, cellular distribution and regulation of expression by factors controlling IgE production. FEBS Lett. 1993;315:259–266. doi: 10.1016/0014-5793(93)81175-y. [DOI] [PubMed] [Google Scholar]
- 13.Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. 2004;22:307–328. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- 14.Fagarasan S, Honjo T. T-Independent immune response: new aspects of B cell biology. Science. 2000;290:89–92. doi: 10.1126/science.290.5489.89. [DOI] [PubMed] [Google Scholar]
- 15.Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–829. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fagarasan S, Honjo T. Intestinal IgA synthesis: regulation of front-line body defenses. Nat Rev Immunol. 2003;3:63–72. doi: 10.1038/nri982. [DOI] [PubMed] [Google Scholar]
- 17.Hangartner L, Zinkernagel RM, Hengartner H. Antiviral antibody responses: the two extremes of a wide spectrum. Nat Rev Immunol. 2006;6:231–243. doi: 10.1038/nri1783. [DOI] [PubMed] [Google Scholar]
- 18.Hahne M, Kataoka T, Schroter M, Hofmann K, Irmler M, Bodmer JL, Schneider P, Bornand T, Holler N, French LE, et al. APRIL, a new ligand of the tumor necrosis factor family, stimulates tumor cell growth. J Exp Med. 1998;188:1185–1190. doi: 10.1084/jem.188.6.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, Soppet D, Charters M, Gentz R, Parmelee D, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–263. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- 20.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, Acha-Orbea H, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189:1747–1756. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackay F, Schneider P, Rennert P, Browning J. BAFF and APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 22.Ng LG, Mackay CR, Mackay F. The BAFF/APRIL system: life beyond B lymphocytes. Mol Immunol. 2005;42:763–772. doi: 10.1016/j.molimm.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 23.Schneider P. The role of APRIL and BAFF in lymphocyte activation. Curr Opin Immunol. 2005;17:282–289. doi: 10.1016/j.coi.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, Bram RJ, Jabara H, Geha RS. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med. 2005;201:35–39. doi: 10.1084/jem.20032000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lentz VM, Cancro MP, Nashold FE, Hayes CE. Bcmd governs recruitment of new B cells into the stable peripheral B cell pool in the A/WySnJ mouse. J Immunol. 1996;157:598–606. [PubMed] [Google Scholar]
- 26.Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173:2245–2252. doi: 10.4049/jimmunol.173.4.2245. [DOI] [PubMed] [Google Scholar]
- 27.Mariette X, Roux S, Zhang J, Bengoufa D, Lavie F, Zhou T, Kimberly R. The level of BLyS (BAFF) correlates with the titre of auto-antibodies in human Sjogren’s syndrome. Ann Rheum Dis. 2003;62:168–171. doi: 10.1136/ard.62.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker KP, Edwards BM, Main SH, Choi GH, Wager RE, Halpern WG, Lappin PB, Riccobene T, Abramian D, Sekut L, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. 2003;48:3253–3265. doi: 10.1002/art.11299. [DOI] [PubMed] [Google Scholar]
- 29.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, Peter HH, Rockstroh JK, Schneider P, Schaffer AA, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820–828. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 31.Becker MN, Diamond G, Verghese MW, Randell SH. CD14-dependent lipopolysaccharide-induced β-defensin-2 expression in human tracheobronchial epithelium. J Biol Chem. 2000;275:29731–29736. doi: 10.1074/jbc.M000184200. [DOI] [PubMed] [Google Scholar]
- 32.Shuto T, Imasato A, Jono H, Sakai A, Xu H, Watanabe T, Rixter DD, Kai H, Andalibi A, Linthicum F, et al. Glucocorticoids synergistically enhance nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via a negative cross-talk with p38 MAP kinase. J Biol Chem. 2002;277:17263–17270. doi: 10.1074/jbc.M112190200. [DOI] [PubMed] [Google Scholar]
- 33.Schulz C, Farkas L, Wolf K, Kratzel K, Eissner G, Pfeifer M. Differences in LPS-induced activation of bronchial epithelial cells (BEAS-2B) and type II-like pneumocytes (A-549) Scand J Immunol. 2002;56:294–302. doi: 10.1046/j.1365-3083.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- 34.Homma T, Kato A, Hashimoto N, Batchelor J, Yoshikawa M, Imai S, Wakiguchi H, Saito H, Matsumoto K. Corticosteroid and cytokines synergistically enhance Toll-like receptor 2 expression in respiratory epithelial cells. Am J Respir Cell Mol Biol. 2004;31:463–469. doi: 10.1165/rcmb.2004-0161OC. [DOI] [PubMed] [Google Scholar]
- 35.Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris CC. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- 36.Atsuta J, Plitt J, Bochner BS, Schleimer RP. Inhibition of VCAM-1 expression in human bronchial epithelial cells by glucocorticoids. Am J Respir Cell Mol Biol. 1999;20:643–650. doi: 10.1165/ajrcmb.20.4.3265. [DOI] [PubMed] [Google Scholar]
- 37.Kato A, Ogasawara T, Homma T, Saito H, Matsumoto K. Lipopolysaccharide-binding protein critically regulates lipopolysaccharide-induced IFN-β signaling pathway in human monocytes. J Immunol. 2004;172:6185–6194. doi: 10.4049/jimmunol.172.10.6185. [DOI] [PubMed] [Google Scholar]
- 38.Kato A, Homma T, Batchelor J, Hashimoto N, Imai S, Wakiguchi H, Saito H, Matsumoto K. Interferon-αβ receptor-mediated selective induction of a gene cluster by CpG oligodeoxynucleotide 2006. BMC Immunol. 2003;4:8. doi: 10.1186/1471-2172-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gavin AL, Ait-Azzouzene D, Ware CF, Nemazee D. δBAFF, an alternate splice isoform that regulates receptor binding and biopresentation of the B cell survival cytokine, BAFF. J Biol Chem. 2003;278:38220–38228. doi: 10.1074/jbc.M306852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gavin AL, Duong B, Skog P, Ait-Azzouzene D, Greaves DR, Scott ML, Nemazee D. δBAFF, a splice isoform of BAFF, opposes full-length BAFF activity in vivo in transgenic mouse models. J Immunol. 2005;175:319–328. doi: 10.4049/jimmunol.175.1.319. [DOI] [PubMed] [Google Scholar]
- 41.Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, Sosnovtseva S, Carrell JA, Feng P, Giri JG, Hilbert DM. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.v97.1.198. [DOI] [PubMed] [Google Scholar]
- 42.Scapini P, Nardelli B, Nadali G, Calzetti F, Pizzolo G, Montecucco C, Cassatella MA. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med. 2003;197:297–302. doi: 10.1084/jem.20021343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishio M, Endo T, Tsukada N, Ohata J, Kitada S, Reed JC, Zvaifler NJ, Kipps TJ. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1α. Blood. 2005;106:1012–1020. doi: 10.1182/blood-2004-03-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, Kalled SL, Hess DM, Serafini B, Aloisi F, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med. 2005;201:195–200. doi: 10.1084/jem.20041674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ittah M, Miceli-Richard C, Eric Gottenberg J, Lavie F, Lazure T, Ba N, Sellam J, Lepajolec C, Mariette X. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjogren’s syndrome. Arthritis Res Ther. 2006;8:R51. doi: 10.1186/ar1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ronni T, Matikainen S, Sareneva T, Melen K, Pirhonen J, Keskinen P, Julkunen I. Regulation of IFN-αβ, MxA, 2′,5′-oligoadenylate synthetase, and HLA gene expression in influenza A-infected human lung epithelial cells. J Immunol. 1997;158:2363–2374. [PubMed] [Google Scholar]
- 47.Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 48.Thompson JE, Cubbon RM, Cummings RT, Wicker LS, Frankshun R, Cunningham BR, Cameron PM, Meinke PT, Liverton N, Weng Y, DeMartino JA. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg Med Chem Lett. 2002;12:1219–1223. doi: 10.1016/s0960-894x(02)00106-3. [DOI] [PubMed] [Google Scholar]
- 49.Tliba O, Panettieri RA, Jr, Tliba S, Walseth TF, Amrani Y. Tumor necrosis factor-α differentially regulates the expression of proinflammatory genes in human airway smooth muscle cells by activation of interferon-β-dependent CD38 pathway. Mol Pharmacol. 2004;66:322–329. doi: 10.1124/mol.104.001040. [DOI] [PubMed] [Google Scholar]
- 50.Schleimer RP. Glucocorticoids: Part A: Mechanisms of action in allergic diseases. In: Yunginger JW, Adkinson NF Jr, Busse WW, Bochner BS, Holgate ST, Simons FER, editors. Middleton’s Allergy: Principles and Practice. Mosby; St. Louis: 2003. pp. 870–913. [Google Scholar]
- 51.Pugin J, Schurer-Maly CC, Leturcq D, Moriarty A, Ulevitch RJ, Tobias PS. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc Natl Acad Sci USA. 1993;90:2744–2748. doi: 10.1073/pnas.90.7.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 53.Moynagh PN. TLR signalling and activation of IRFs: revisiting old friends from the NF-κB pathway. Trends Immunol. 2005;26:469–476. doi: 10.1016/j.it.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 54.Jefferies CA, Fitzgerald KA. Interferon gene regulation: not all roads lead to Tolls. Trends Mol Med. 2005;11:403–411. doi: 10.1016/j.molmed.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 55.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 56.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 57.Naclerio RM, Adkinson NF, Jr, Moylan B, Baroody FM, Proud D, Kagey-Sobotka A, Lichtenstein LM, Hamilton R. Nasal provocation with allergen induces a secondary serum IgE antibody response. J Allergy Clin Immunol. 1997;100:505–510. doi: 10.1016/s0091-6749(97)70143-x. [DOI] [PubMed] [Google Scholar]
- 58.Naclerio RM, Adkinson NF, Jr, Creticos PS, Baroody FM, Hamilton RG, Norman PS. Intranasal steroids inhibit seasonal increases in ragweed-specific immunoglobulin E antibodies. J Allergy Clin Immunol. 1993;92:717–721. doi: 10.1016/0091-6749(93)90015-8. [DOI] [PubMed] [Google Scholar]
- 59.Avila PC. Interactions between allergic inflammation and respiratory viral infections. J Allergy Clin Immunol. 2000;106:829–831. doi: 10.1067/mai.2000.111027. [DOI] [PubMed] [Google Scholar]
- 60.Johnston SL. Overview of virus-induced airway disease. Proc Am Thorac Soc. 2005;2:150–156. doi: 10.1513/pats.200502-018AW. [DOI] [PubMed] [Google Scholar]
- 61.Contoli M, Caramori G, Mallia P, Johnston S, Papi A. Mechanisms of respiratory virus-induced asthma exacerbations. Clin Exp Allergy. 2005;35:137–145. doi: 10.1111/j.1365-2222.2005.02163.x. [DOI] [PubMed] [Google Scholar]
- 62.Coker HA, Durham SR, Gould HJ. Local somatic hypermutation and class switch recombination in the nasal mucosa of allergic rhinitis patients. J Immunol. 2003;171:5602–5610. doi: 10.4049/jimmunol.171.10.5602. [DOI] [PubMed] [Google Scholar]
- 63.Cameron L, Hamid Q, Wright E, Nakamura Y, Christodoulopoulos P, Muro S, Frenkiel S, Lavigne F, Durham S, Gould H. Local synthesis of ε germline gene transcripts, IL-4, and IL-13 in allergic nasal mucosa after ex vivo allergen exposure. J Allergy Clin Immunol. 2000;106:46–52. doi: 10.1067/mai.2000.107398. [DOI] [PubMed] [Google Scholar]
- 64.KleinJan A, Vinke JG, Severijnen LW, Fokkens WJ. Local production and detection of (specific) IgE in nasal B-cells and plasma cells of allergic rhinitis patients. Eur Respir J. 2000;15:491–497. doi: 10.1034/j.1399-3003.2000.15.11.x. [DOI] [PubMed] [Google Scholar]
- 65.Nakajima S, Gillespie DN, Gleich GJ. Differences between IgA and IgE as secretory proteins. Clin Exp Immunol. 1975;21:306–317. [PMC free article] [PubMed] [Google Scholar]
- 66.Merrett TG, Houri M, Mayer AL, Merrett J. Measurement of specific IgE antibodies in nasal secretion: evidence for local production. Clin Allergy. 1976;6:69–73. doi: 10.1111/j.1365-2222.1976.tb01413.x. [DOI] [PubMed] [Google Scholar]
- 67.Small P, Barrett D, Frenkiel S, Rochon L, Cohen C, Black M. Local specific IgE production in nasal polyps associated with negative skin tests and serum RAST. Ann Allergy. 1985;55:736–739. [PubMed] [Google Scholar]
- 68.Schleimer RP. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc Am Thorac Soc. 2004;1:222–230. doi: 10.1513/pats.200402-018MS. [DOI] [PubMed] [Google Scholar]
- 69.Sutherland AP, Ng LG, Fletcher CA, Shum B, Newton RA, Grey ST, Rolph MS, Mackay F, Mackay CR. BAFF augments certain Th1-associated inflammatory responses. J Immunol. 2005;174:5537–5544. doi: 10.4049/jimmunol.174.9.5537. [DOI] [PubMed] [Google Scholar]