

Abstract
Several ocular diseases complicated by neovascularization are being treated by repeated intraocular injections of vascular endothelial growth factor (VEGF) antagonists. While substantial benefits have been documented, there is concern that unrecognized damage may be occurring, because blockade of VEGF may damage the fenestrated vessels of the choroicapillaris and deprive retinal neurons of input from a survival factor. One report has suggested that even temporary blockade of all isoforms of VEGF-A results in significant loss of retinal ganglion cells. In this study, we utilized double transgenic mice with doxycycline-inducible expression of soluble VEGF receptor 1 coupled to an Fc fragment (sVEGFR1Fc), a potent antagonist of several VEGF family members, including VEGF-A, to test the effects of VEGF blockade in the retina. Expression of sVEGFR1Fc completely blocked VEGF-induced retinal vascular permeability and significantly suppressed the development of choroidal neovascularizaton at rupture sites in Bruch’s membrane, but did not cause regression of established choroidal neovascularization. Mice with constant expression of sVEGF1Fc in the retina for 7 months had normal electroretinograms and normal retinal and choroidal ultrastructure including normal fenestrations in the choroicapillaris. They also showed no significant difference from control mice in the number of ganglion cell axons in optic nerve cross sections and the retinal level of mRNA for 3 ganglion cell-specific genes. These data indicate that constant blockade of VEGF for up to 7 months has no identifiable deleterious effects on the retina or choroid and support the use of VEGF antagonists in the treatment of retinal diseases.
Keywords: age-related macular degeneration, angiogenesis, apoptosis, diabetic retinopathy, retinal degeneration, survival factors
Introduction
Diseases complicated by neovascularization and/or excessive vascular permeability account for the vast majority of visual loss in developed countries. Choroidal neovascularization (CNV) occurs in patients with abnormalities of Bruch’s membrane and the retinal pigmented epithelium (RPE), such as age-related macular degeneration (AMD), pathologic myopia, ocular histoplasmosis, angioid streaks, and various types of choroiditis. Both retinal neovascularization and macular edema occur in ischemic retinopathies such as diabetic retinopathy and retinal vascular occlusive diseases. A common theme has emerged in this highly diverse group of diseases; vascular endothelial growth factor (VEGF) is a major contributor to the neovascularization and leakage that occurs in all of them (for review, see (Campochiaro, 2006).
The treatment of retinal vascular diseases has been revolutionized by the identification of VEGF as an important therapeutic target and the development of potent VEGF antagonists. Whereas previous treatments for CNV due to AMD did not cause improvement and merely slowed the rate of vision loss, intraocular injections of ranibizumab, an Fab fragment that binds all isoforms of VEGF-A, caused significant improvement in 30–40% of patients that was sustained for at least 2 years (Brown et al., 2006; Rosenfeld et al., 2006). Intraocular injections of ranibizumab substantially improved vision in patients with diabetic macular edema (Nguyen et al., 2006), which has led to phase III clinical trials. Likewise, preliminary results suggest that ranibizumab or bevacizumab may cause improvement in patients with macular edema due to branch or central retinal vein occlusions (Campochiaro et al., 2007; Costa et al., 2007; Iturralde et al., 2006; Pai et al., 2007; Rabena et al., 2007; Stahl et al., 2007)..
Despite the undeniable short-term benefits seen in patients with a variety of retinal and choroidal diseases treated with VEGF antagonists, some investigators have sounded a note of caution. Fenestrated vascular beds are more dependent upon VEGF raising concern that the choroicapillaris, which is fenestrated, may be damaged over time. VEGF type 2 receptors have been identified on neurons, including retinal neurons, and it has been suggested that their activation may promote cell survival (Jin et al., 2000; Kilic et al., 2006). A recent report suggested that VEGF antagonists may cause death of retinal ganglion cells (Nishijima et al., 2007).
In this study, we have used a transgenic approach to explore the effects of long term blockade of VEGF in the retina and choroid. Transgenic mice that carry a tetracycline response element (TRE) coupled to soluble VEGF receptor 1 attached to an IgG1 Fc fragment (TRE/sVEGFR1Fc) have been previously used in combination with transgenic mice that carry a heart-specific promoter coupled to a tetracycline transactivator to generate double transgenic mice that express sVEGFR1Fc in heart or liver (Grunewald et al., 2006). The expression of sVEGFR1Fc in heart causes a progressive microvascular deficit (Eli Keshet, personal communication 10/07). We crossed TRE/sVEGFR1Fc mice with opsin promoter/reverse tetracycline transactivator (rtTA) mice (Chang et al., 2000) to generate mice with doxycycline-inducible expression of sVEGFR1Fc to determine the effect of sustained blockade of VEGF in the retina and choroid.
Materials and Methods
Double transgenic mice with inducible expression of a potent VEGF binding protein
Mice were treated in accordance with the guidelines of the Association for Research in Vision and Ophthalmology and the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. Transgenic mice carrying a fusion gene consisting of the tetracycline response element (TRE), the extracellular domain of human VEGFR1, and an IgG1 Fc tail (TRE/sVEGFR1Fc) (Grunewald et al., 2006) were crossed with opsin/rtTA or IRBP/rtTA mice (Chang et al., 2000; Ohno-Matsui et al., 2002; Oshima et al., 2005). Double transgenic opsin/rtTA-TRE/sVEGFR1Fc (Tet/opsin/sVEGFR1Fc) or IRBP/rtTA-TRE/sVEGFR1Fc (Tet/IRBP/sVEGFR1Fc) mice were identified by polymerase chain reaction (PCR) of tail DNA using previously described primers and techniques (Chang et al., 2000; Grunewald et al., 2006). Four to five week old Tet/IRBP/sVEGFR1Fc or Tet/opsin/sVEGFR1Fc mice were given drinking water containing 2 mg/ml of doxycycline and 5% dextrose for the VEGF blockade group or 5% dextrose for the control group. Doxycycline-treated (VEGF blockade) and control mice were sacrificed at various time points for each experiment.
Measurement of transgene product expression
Four week old Tet/opsin/sVEGFR1Fc (n=4) or Tet/IRBP/sVEGFR1Fc (n=4) mice were treated with 2 mg/ml of doxycycline/5% dextrose in their drinking water or unsupplemented water for 1 week and then euthanized. Retinas were removed and quick frozen in 50μL of lysis buffer (10 mM Tris, pH 7.2/0.5% Triton X-100/50 mM NaCl/1 mM EDTA) with a proteinase inhibitor mixture tablet (Roche, Indianapolis, IN). After dounce homogenization and 3 freeze/thaw cycles, homogenates were microfuged at 14,000g for 5 minutes at 4°C and the protein concentration of the supernatants was measured using a Bio-Rad Protein Assay Kit (BioRad, Hercules, CA). Samples (80μg of protein) were added to wells of a 12.5% acrylamide gel (BIO-RAD, Hercules, CA) and resolved by SDS-PAGE. The separated proteins were transferred to a nitrocellulose membrane (Hybond-ECL, Amersham Biosciences, Piscataway, NJ). Non-specific binding was blocked by incubation with 5% non-fat milk at room temperature for 1 hour and then the membranes were incubated with rabbit anti-VEGFR1 (1:5000, ab32152 Abcam Inc, Cambridge, MA) overnight at 4°C. After washing, the blots were incubated with SuperSignal western Pico lumino/Enhancer solution (Pierce, Rockford, IL) and horseradish peroxidase-linked anti-rabbit IgG and exposed to X-ray film (Eastman-Kodak, Rochester, NY).
In some experiments, transgene expression was assessed by ELISA for human VEGFR1 using the Quantikine VEGFR1 assay kit (R & D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Serial dilutions of recombinant VEGFR1 were used to generate standard curves. Statistical analysis was done using unpaired Students t-test.
Assessment of breakdown of the blood-retinal barrier by immunoblotting for albumin
Tet/IRBP/sVEGFR1Fc mice (n=6 in each group) were treated with or without 2 mg/ml of doxycycline for 2 weeks and then 1 μl of PBS or PBS containing 10−6 M VEGF was injected into the vitreous cavity. Intravitreous injections were given with a Harvard pump apparatus (Harvard Apparatus, Holliston, MA) and pulled-glass micropipettes, as previously described (Derevjanik et al., 2002). Six hours after intraocular injections, the mice were anesthetized and perfused with phosphate-buffered saline. Western blotting was done as described above. The primary antibodies were rabbit anti-rat albumin (1:5000, Nordic Immunological Labs, Tilburg, Netherlands) and rabbit anti-beta actin (Cell Signaling, Danvers, MA)
Murine model of choroidal neovascularization (CNV)
The murine model of laser-induced CNV has been described and utilized in several prior studies (Kwak et al., 2000; Saishin et al., 2003a; Tobe et al., 1998). Tet/IRBP/sVEGFR1Fc double transgenic mice were given 2 mg/ml doxycycline and 5% dextrose (VEGF blockade, n=12) or 5% dextrose (controls, n=11) in their drinking water and after 1 day, Bruch’s membrane was ruptured with laser photocoagulation in 3 locations in each eye as previously described (Tobe et al., 1998). Fourteen days after rupture of Bruch’s membrane, the mice were perfused with fluorescein-labeled dextran and choroidal flat mounts were prepared from one eye. The area of CNV was measured at Bruch’s membrane rupture sites on choroidal flat mounts by image analysis (see below). The fellow eyes were used for measurement of sVEGFR1Fc level by ELISA as described above.
To determine the effect of sVEGFR1Fc on established CNV, laser-induced rupture of Bruch’s membrane was done in Tet/opsin/sVEGFR1Fc mice and after 7 days some of the mice were perfused with fluorescein-labeled dextran and the baseline area of CNV was measured by image analysis. The remainder of the mice were divided into 2 groups; one received 2 mg/ml of doxycycline and 5% dextrose in drinking water and the other received only 5% dextrose. One or 4 weeks later the mice were perfused with fluorescein-labeled dextran and the area of CNV at Bruch’s membrane rupture sites was measured in one eye, and the level of sVEGFR1Fc was measured in the fellow eye by ELISA.
Measurement of CNV
Mice were anesthetized and perfused with 50 mg/ml fluorescein-labeled dextran (Sigma, St. Louis, MO) and choroidal flat mounts were prepared. Flat mounts were examined by fluorescence microscopy on an Axioskop microscope (Zeiss, Thornwood, NY) and images were digitized using a 3-color CCD video camera (IK-TU40A, Toshiba, Tokyo, Japan) and a frame grabber. Image-Pro Plus software (Media Cybernetics, Silver Spring, MD) was used to measure the total area of choroidal neovascularization associated with each burn with the operator masked with respect to treatment group. Statistical comparisons were done using a linear mixed model (Verbeke and Molenberghs, 2000). This model is analogous to analysis of variance (ANOVA), but allows analysis of all CNV area measurements from each mouse rather than average CNV area per mouse by accounting for correlation between measurements from the same mouse. P-values for comparisons of treatments were adjusted for multiple comparisons using Dunnett’s method.
ERG recordings
Tet/IRBP/sVEGFR1Fc double transgenic mice were treated with or without 2 mg/ml of doxycycline for 7 months. Scotopic and photopic ERGs were recorded on 12 doxycycline-treated and eight control mice using an Espion ERG machine (Diagnosys LLL, Littleton, MA) as previously described (Komeima et al., 2007; Okoye et al., 2003). For scotopic recordings, mice were dark adapted overnight, and for photopic recordings, mice were adapted for 10 minutes to background white light at an intensity of 30 cd/m2. The mice were anesthetized with an intraperitoneal injection of ketamine hydrochloride (100 mg/kg body weight) and xylazine (5mg/kg body weight). Corneas were further anesthetized with proparacaine (0.5%) and the pupils dilated with 1% tropicamide (Alcon Labs, Forth Worth, TX). The mice were placed on a heating pad for the duration of ERG recording and platinum loop electrodes were placed on each cornea after application of gonioscopic prism solution (Alcon Labs, Forth Worth, TX). A reference electrode was placed subcutaneously in the anterior scalp between the eyes, and a ground electrode was inserted into the tail. Both eyes were recorded simultaneously. Scotopic ERGs were recorded at 11 intensity levels of white light ranging from −3.00 to 1.40 log cd-s/m2. Six measurements were averaged at each flash intensity. Photopic ERGs were recorded at three intensity levels of white light ranging from 0.60 to 1.40 log cd-s/m2. Twenty measurements were averaged at each flash intensity. Intensity response functions were analyzed using ANOVA.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP-Biotin Nick-End Labeling (TUNEL)
Tet/IRBP/sVEGFR1Fc double transgenic mice were treated with or without 2 mg/ml of doxycycline in their water for 7 months (n=3 for each) and then euthanized. Eyes were removed and embedded in optimal cutting temperature compound (OCT)and 10 μm serial sections were cut and fixed with 1% paraformaldehyde in PBS for 10 minutes at room temperature. Apoptotic cells were detected by TUNEL with an apoptosis detection kit (Apoptag Red In Situ Apoptosis Detection Kit; Chemicon International Inc., Temecula, CA). Sections were counterstained with Hoechst 33258 (1:1200; Sigma-Aldrich, St Louis, MO) and examined with a fluorescence microscope (Nikon, Tokyo, Japan). Negative controls had omission of the terminal deoxynucleotidyl transferase enzyme from the labeling solution. Positive control slides were pretreated for 15 minutes with 3 U/ml DNAse I (Invitrogen, Carlsbad, CA) in buffer (20 mM Tris–HCl, pH 8.4, 2mM MgCl2 500 mM KCL) to induce DNA strand breaks.
Light and Electron Microscopy
Tet/IRBP/sVEGFR1Fc double transgenic mice were treated with or without 2 mg/ml of doxycycline in their drinking water for 7 months (n=3 for each group). The mice were perfused with phosphate-buffered saline (PBS) followed by 2% paraformaldehyde/2% glutaraldehyde in 0.1M cacodylate buffer (pH 7.4). After perfusion, eyes were removed and immersed in the same fixative for 24 hours at 4°C. The anterior segments were removed and the posterior eyecups were divided into 4 parts, postfixed with 1% osmium tetroxide/cacodylate buffer (pH 7.4), dehydrated through a series of graded alcohols, and embedded in Poly/Bed 812 resin (Polysciences, Warrington, PA). One micron sections were cut, stained with toluidine blue, and examined with an Axioskop microscope and optimal areas were selected to cut ultrathin sections for electron microscopy. The utlrathin sections were counterstained with 2.0% uranyl acetate and 0.3% lead citrate, and examined with a transmission electron microscope (H-7600; Hitachi Co. Ltd., Tokyo, Japan).
Counting of ganglion cell axons
Tet/IRBP/sVEGFR1Fc double transgenic mice were treated with or without 2 mg/ml of doxycycline in their drinking water for 7 months (n=6, VEGF blockade group; n=5, control group). Mice were euthanized by exsanguination under deep ketamine/xylazine/acepromazine anesthesia and perfused through the heart with 2% paraformaldehyde/2% glutaraldehyde in 0.1M phosphate buffer (pH 7.2). Eyes and attached optic nerves were removed and a small segment of the optic nerve 1 mm posterior to the globe was post fixed in 1% osmium tetroxide in phosphate buffer. Nerves were embedded in epoxy resin and 1 μm sections were stained with 1% toluidine blue. A digital image of each nerve cross-section was captured and its border outlined using MetaMorph image acquisition and analysis software (Molecular Devices Corporation, Downingtown PA). Three nerve area measurements were taken and the mean was calculated for each nerve. To measure the density and fiber diameter distribution, images were captured with a 100X phase contrast objective and green filter from 5 randomly selected 40 μm2 areas of the nerve. The software was used to remove non-neural structures, and the size of each axon internal to its myelin sheath (minimum diameter) and the density of axons per square millimeter were calculated for each image and for the entire nerve. The mean density was multiplied by the nerve area to determine the fiber number for each nerve. The number of axons counted among the 5 images of each nerve was approximately 10% of the total fiber number. The observer was masked as to the status of each sample. Since the two eyes of each animal did not represent independent data, the fiber number was averaged to generate a mean fiber number per mouse. For each treatment group, the group mean fiber number and standard deviation were then calculated for comparison. Statistical analysis was done by unpaired Students t-test.
Measurement of ganglion cell-specific mRNAs by quantitative real-time RT-PCR
Tet/IRBP/sVEGFR1Fc double transgenic mice were treated with or without doxycycline (n=7, doxycycline group; n=6 control group) for 7 months and euthanized. One retina was removed for ELISA and the other was used for quantitative real-time RT PCR. Total retinal RNA was isolated using an RNeasy kit (QIAGEN, Valencia, CA). After quantification of RNA concentration using Gene Spec III (Hitachi, Japan), 1 μg was treated with DNase I (invitrogen, Kingston, ON, Canada) to remove any contaminating genomic DNA and cDNA was synthesized with reverse transcriptase (SuperScript III; Invitrogen, Carlsbad, CA) and 5 μM random hexamer. Samples of cDNA were divided into aliquots and stored at −80°C. Quantitative PCR was performed with the following primers and iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) using iQ5 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). The mRNA levels were measured for 3 ganglion cell-specific genes, Thy1, neuritin 1 (Nrn1), and neurofilament 3 (Nef3) (Ivanov et al., 2006) using previously published primers (Kim et al., 2006): Thy1 forward primer 5′-AAC TCT TGG CAC GAC CAT GAA CC-3′ and reverse primer 5′-CTC GGG ACA CCT GCA AGA-3′; Nrn1 forward primer 5′-CGC TCC CTC TCT TTCTCT CC -3′ and reverse primer 5′-CTT CCT GGC AAT CCG TAA GA-3′; Nef3 forward primer 5′-CGT GTC CTC CTC CTA CAA GC-3′ and reverse primer 5′-GGT GGA TGT CTT CCT CCA AG-3′. Cyclophilin A was used as an internal control; forward primer 5′-CAG ACG CCA CTG TCGCTT T-3′ and reverse primer 5′-TGT CTT TGG AAC TTT GTC TGC AA-3′. One RNA sample from a control retina was selected to serve as a standard for comparison and was used to generate standard curves for each gene of interest and cyclophilin A. Each of the VEGF blockade and control samples were run in triplicate with each of the 4 sets of primers and the 3 resulting ratios relative to the standard were used to calculate a mean. The mean value for gene of interest was divided by the mean value for cyclophilin A to give the relative expression level for gene of interest for each sample. Statistical comparisons were made using Students unpaired t-test.
Results
Double transgenic mice with inducible blockade of VEGF family members in the retina
Crossing TRE/sVEGFR1Fc transgenic mice (Grunewald et al., 2006) with the heart-specific driver line, MHCα-tTA (Yu et al., 1996) or the liver-specific driver line, PLAP-tTA (Kistner et al., 1996) results in mice with inducible blockade of VEGF family members in heart or liver, respectively (Grunewald et al., 2006). TRE/sVEGFR1Fc transgenic mice were crossed with the retina-specific driver lines, opsin/rtTA or IRBP/rtTA (Chang et al., 2000) to generate Tet/opsin/sVEGFR1Fc and Tet/IRBP/sVEGFR1Fc double transgenic lines. Treatment of these double transgenic mice with doxycycline induced high level expression of the sVEGFR1Fc transgene product in the retina (Figure 1). Injection of VEGF into the vitreous cavity of wild type mice causes leakage of systemically-injected radiolabeled markers and endogenous serum albumin into the retina (Derevjanik et al., 2002; Saishin et al., 2003b). In Tet/IRBP/sVEGFR1Fc mice that were treated with doxycycline causing expression of sVEGFR1Fc, there were low levels of albumin in the retina after intraocular injection of PBS (Figure 2, lane1) and the levels were essentially identical after injection of VEGF (Figure 2, lane 2). Tet/IRBP/sVEGFR1Fc mice that were not treated with doxycycline showed very high levels of albumin in the retina after intraocular injection of VEGF (Figure 2, lane 3), but when injected with PBS the levels of albumin were low (Figure 2, lane 4). Thus, doxycycline treatment of the double transgenic mice resulted in complete blockade of VEGF-induced permeability.
Figure 1. Double transgenic mice with doxycycline-inducible expression of sVEGFR1Fc in the retina.
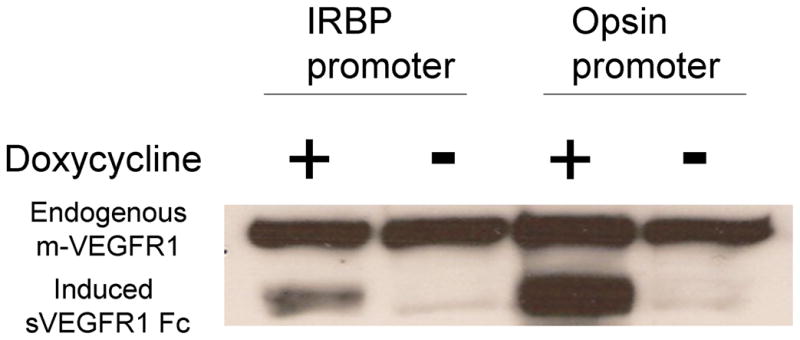
Double transgenic Tet/IRBP/sVEGFR1Fc or Tet/opsin/sVEGFR1Fc mice were given 2 mg/ml of doxycycline in their drinking water for 2 weeks and then euthanized and retinal homogenates were run in immunoblots using an anti-VEGFR1 antibody. Both Tet/IRBP/VEGFR1Fc and Tet/opsin/VEGFR1Fc mice treated with doxycycline showed bands for sVEGFR1Fc. There was minimal leakiness in the absence of doxycycline. All of the retinas showed similar bands for endogenous murine VEGFR1 indicating that the differences in sVEGFR1Fc are not due to loading differences.
Figure 2. Induced expression of sVEGFR1Fc blocks VEGF-induced breakdown of the blood-retinal barrier.
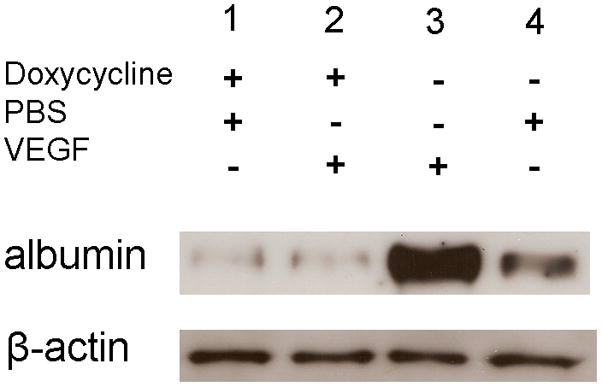
Tet/IRBP/sVEGFR1Fc mice were given 2 mg/ml of doxycycline in their drinking water or given normal water for 2 weeks and then 1 μl of PBS or PBS containing 10−6 M VEGF was injected into the vitreous cavity. Six hours after injection the mice were perfused with PBS and then the retinas were isolated and retinal homogenates were run in immunoblots using an anti-albumin antibody. The blot was stripped and incubated with anti-β-actin. Retinas from doxycycline-treated mice showed a faint band for albumin whether they received an intraocular injection of PBS (lane 1) or VEGF (lane 2). Mice that were not given doxycycline showed a very strong band for albumin when they received an intraocular Injection of VEGF (lane 3) and a weak band when given an intraocular injection of PBS (lane 4). These results are representative of 3 different experiments.
Blockade of VEGF causes suppression but not regression of CNV
Antagonists of VEGF cause significant suppression of laser-induced CNV in mice (Saishin et al., 2003a). Intraocular injection of VEGF antagonists causes rapid reduction of excessive vascular permeability and slows or prevents further growth of CNV in patients with AMD, but it does not cause the CNV to be eliminated (Brown et al., 2006; Rosenfeld et al., 2006). We sought to determine if sustained expression of a VEGF antagonist could cause CNV to regress. Compared to control mice, Tet/IRBP/sVEGFR1Fc mice treated with doxycycline for 2 weeks prior to rupture of Bruch’s membrane had significantly smaller areas of CNV (Figure 3), but when doxycycline was initiated 1 week after laser when CNV was established, neither 1 (Figure 4A) nor 4 weeks (Figure 4B) of doxycycline treatment caused significant regression of CNV.
Figure 3. Blockade of VEGF suppresses the development of choroidal neovascularization (CNV).

Tet/IRBP/VEGFR1Fc mice or littermate controls were given 2 mg/ml of doxycycline in their drinking water and after 2 weeks Bruch’s membrane was ruptured with laser photocoagulation at 3 locations in one eye. Two weeks after laser treatment the mice were perfused with fluorescein-labeled dextran and the area of CNV was measured at Bruch’s membrane rupture sites by image analysis on choroidal flat mounts (A). The other retina was used to measure sVEGFR1Fc by ELISA (B).
(A) Doxycycline-induced expression of sVEGFR1 Fc caused significant suppression of CNV. Each bar represents the mean (±SEM) area of CNV, calculated from the total number of rupture sites for which measurements were taken in each group. *p < 0.01 for difference from control by linear mixed model. (B) ELISA showed two weeks of doxycycline treatment induced a large increase in sVEGFR1 Fc. Each bar represents the mean (±SEM) of sVEGFR1Fc per mg of retinal protein. **p < 0.001 for difference from control by unpaired t-test.
Figure 4. Blockade of VEGF does not cause regression of established choroidal neovascularization (CNV).

(A) Tet/opsin/sVEGFR1Fc mice had laser-induced rupture of Bruch’s membrane at 3 locations in each eye and after 7 days some mice were perfused with fluorescein-labeled dextran and the baseline area of CNV was measured. The remaining mice were given 2 mg/ml of doxycycline and dextrose or dextrose alone in their drinking water and one (A) or four (B) weeks later the area of CNV was measured and the retina in the fellow eye was used to measure sVEGFR1Fc by ELISA. Each point represents the mean (±SEM) of CNV size (left side) and each bar represents the mean (±SEM) of sVEGFR1Fc per mg retinal protein (right side). Expression of sVEGFR1Fc for 1 or 4 weeks did not cause significant reduction in the area of CNV compared to that seen in control mice or the baseline amount of CNV present at the outset of treatment. Statistical comparisons were made by general linear model.
Prolonged blockade of VEGF does not reduce retinal function as assessed by ERGs
Scotopic ERGs are performed in dark-adapted animals with dim illumination and a wide range of stimuli. They provide a quantitative measure of global function of rod photoreceptor signaling pathways in the retina. Photopic ERGs are performed in light-adapted animals with a range of strong stimuli and provide quantitative assessment of cone photoreceptor signaling pathways. Scotopic ERGs in Tet/IRBP/sVEGFR1Fc mice that were treated with 2 mg/ml of doxycycline in their drinking water for 7 months showed waveforms that looked very similar to those seen in control mice (Figure 5A, top row). Plots of a-wave and b-wave amplitudes versus stimulus intensity looked almost identical in mice with long term VEGF blockade and control mice and there were no significant differences in amplitudes at any stimulus intensity (Figure 5A, bottom row). Waveforms of photopic ERGs also looked similar in doxycycline-treated and control mice (Figure 5B, left), and there were no significant differences in b-wave amplitudes (Figure 5B, right).
Figure 5. Long term blockade of VEGF does not alter electroretinograms (ERGs).

Tet/IRBP/sVEGFR1Fc mice or littermate controls were given 2 mg/kg of doxycycline in their drinking water and after 7 months scoptopic (A) and photopic (B) ERGs were performed. The scotopic ERG wave forms appeared very similar in mice with chronic blockade of VEGF in the retina and controls (A, top row). There was no significant difference in mean a-wave or b-wave amplitude at any of the flash intensities tested (A, bottom row). Photopic ERG wave forms also appeared similar in mice with chronic blockade of VEGF in the retina and controls (B, left) and there was no significant difference in mean photopic b-wave amplitudes (B, right).
Absence of ultrastructural changes in the retinas of mice with long term blockade of VEGF
Light microscopy showed normal appearing retinas in mice after 7 months of blockade of VEGF (Figure 6A) that looked the same as those from control mice (Figure 6B). Transmission electron microscopy of the inner surface of retinas of mice with long term blockade of VEGF showed normal vessels, astrocytes, and ganglion cells (Figure 6C). The deeper parts of the retina including the inner nuclear layer, outer plexiform layer, and outer nuclear layer had a normal architecture and contained cells with completely normal ultrastructural appearance (Figure 6D). High magnification views of retinal vessels showed tight junctions between normal endothelial cells surrounded by normal pericytes (Figure 6E). Along the outer surface of the retina, photoreceptors and RPE cells appeared healthy with normally stacked outer segment disks (Figure 6F, inset). The ultrastructure of the choriocapillaris showed a normal appearance with numerous fenestrations and endocytic vesicles (Figure 6G–I). In every respect, the fine structure of cells in the retinas of mice exposed to long term blockade of VEGF were identical to that seen in control mice (not shown).
Figure 6. Long term blockade of VEGF does not cause identifiable morphologic changes in the retina, retinal pigmented epithelium (RPE), or choriocapillaris.

Tet/IRBP/sVEGFR1Fc mice or littermate controls were given 2 mg/ml of doxycycline in their drinking water for 7 months. One micron ocular sections stained with toluidine blue appeared normal in both Tet/IRBP/sVEGFR1Fc (A) and control (B) mice. Electron micrographs of the retina, RPE, or choroid of Tet/IRBP/sVEGFR1Fc mice are shown in C–I.
(C) Along the inner surface of the retina, there were normal vessels (V) with normal pericytes (PC), ganglion cells (GC) and astrocytes (AC).
(D) The inner nuclear layer (INL), outer plexiform layer (OPL), and outer nuclear layer (ONL) appeared normal.
(E) High magnification views of retinal vessels showed tight junctions (arrowheads) between normal endothelial cells (EC) surrounded by normal pericytes (PC).
(F) The photoreceptors (PR) and RPE appeared normal and high magnification showed normally stacked disks in photoreceptor outer segments (inset).
(G) The RPE, choriocapillaris (CC) and large choroidal vessels (CDV) appeared normal.
(H) High magnification of the boxed area in (G) shows normal Bruch’s membrane (BrM) with numerous fenestrations in the endothelial cells of the choriocapillaris (asterisks).
(I) High magnification of a choroidal endothelial cell (En) shows numerous vesicles (arrowheads) in the cytoplasm indicative of active vesicular transport. The illustrations are representative of 6 sections from 3 Tet/IRBP/sVEGFR1Fc mice that were examined. The ultrastructure of control mice appeared identical and is not illustrated.
Lack of apoptotic cells in retinas of mice with chronic VEGF blockade in the retina
Tet/IRBP/sVEGFR1Fc mice or littermate controls were treated with 2 mg/ml of doxycycline in their drinking water for 7 months and TUNEL staining was done on retinal sections. While numerous TUNEL-positive nuclei were seen in positive control retinas treated with DNAase (Figure 7A), negative control retinas (Figure 7B), retinas of Tet/IRBP/SVEGFR1Fc mice treated with doxycycline for 7 months (Figure C), and retinas of wild type mice (Figure 7D) all showed no TUNEL-positive nuclei.
Figure 7. Lack of apoptotic cells in retinas of mice with chronic VEGF blockade in the retina.

Tet/IRBP/sVEGFR1Fc mice or littermate controls were treated with 2 mg/ml of doxycycline in their drinking water for 7 months and TUNEL staining was done on retinal sections. Ocular sections from control mice treated with DNAase showed numerous TUNEL-positive nuclei (A). Sections stained without terminal deoxynucleotidyl transferase enzyme as a negative control showed only background fluorescence (B). A representative image of a section from Tet/IRBP/SVEGFR1Fc mouse treated with doxycycline (VEGF blockade) for 7 months (C) and a section from a control mouse (D) showed no TUNEL-positive nuclei in the retina.
Chronic blockade of VEGF does not cause any reduction in ganglion cell axons
There is a one-to-one correspondence between ganglion cells and their axons in the optic nerve and therefore quantification of the number of axons provides an assessment of the number of ganglion cells. Cross-sections of optic nerves from Tet/IRBP/sVEGFR1Fc mice treated with doxycycline for 7 months (Figure 8A and C) looked similar to those from littermate controls (Figure 8B and D) with no visible abnormalities in either group. Counting of the number of axons per optic nerve by image analysis with the investigator masked with respect to treatment group showed no significant difference in the mean number of axons per optic nerve in mice with long term VEGF blockade versus controls (Figure 8E).
Figure 8. Chronic blockade of VEGF does not cause any reduction in ganglion cell axons.

Representative low (A,B) and high (C,D) magnification light micrographs of optic nerves from a Tet/IRBP/sVEGFR1Fc mouse treated with doxycycline for 7 months (VEGF blockade; A,C) and a littermate control mouse (B,D). No visible abnormalities were detected in either group. The number of axons per optic nerve was counted by image analysis and the bars represent the mean (± SEM) for each group (E). Statistical comparison by general linear model showed no difference.
The mRNA levels for ganglion-cell specific genes were not reduced in retinas of mice exposed to chronic blockade of VEGF
Another objective way to assess ganglion cell number and function is by quantifying the mRNA level of ganglion cell-specific genes in the retina. Compared to matched controls, Tet/IRBP/sVEGFR1Fc double transgenic mice treated with doxycycline for 7 months showed high levels of transgene product in the retina (Figure 9A) and no significant difference in the level of mRNA for three ganglion cell-specific genes, Thy1 (Figure 9B), neurofilament 3 (Nef3, Figure 9C), and neuritin 1 (Nrn1, Figure 9D).
Figure 9. The mRNA levels for ganglion-cell specific genes were not reduced in retinas exposed to chronic blockade of VEGF.

Tet/IRBP/sVEGFR1Fc double transgenic mice were given drinking water containing 2 mg/ml of doxycycline and dextrose (VEGF blockade, n=7) or dextrose alone (control, n=6) for 7 months. The mean (±SEM) level of sVEGFR1Fc per total retinal protein measured by ELISA showed high levels in the doxycycline treated group. **p < 0.001 for difference from control by unpaired t-test.
The level of mRNA for three ganglion cell-specific genes, Thy1 (B), neurofilament 3 (Nef3, C), and neuritin 1 (Nrn1, D) was measured by real time RT-PCR. Each bar represents the mean (±SD) ratio of target gene mRNA to cyclophilin A relative expression of m-RNA based on a standard sample.
Discussion
In this study, we have used a transgenic approach to achieve inducible, complete blockade of VEGF in the eye. Treatment of our transgenic mice with doxycycline resulted in high intraocular levels of sVEGFR1Fc, a potent VEGF binding protein, and complete blockade of VEGF-induced vascular permeability. It also caused substantial, but not complete prevention of CNV at Bruch’s membrane rupture sites. This result is consistent with previous results, because the same level of suppression of CNV was achieved with systemic or intraocular injection of VEGF Trap, an extremely potent VEGF binding protein (Saishin et al., 2003a). This suggests that factors other than VEGF contribute to CNV in this model and one of them is likely to be PDGF-B, because agents that block both VEGF and PDGF signaling cause near complete suppression of CNV (Seo et al., 1999; Kwak et al., 2000).
In this study, we have also shown that CNV occurring at laser-induced rupture sites in Bruch’s membrane become independent of VEGF within a week of its development and the CNV does not regress despite continuous blockade of VEGF. This is similar to what has been found in clinical trials in which intraocular injections of ranibizumab eliminate leakage from CNV in patients with AMD and prevent further growth, but do not cause the CNV to regress (Rosenfeld et al., 2006). Endothelial cells receive survival signals from surrounding cells and extracellular matrix (ECM), mediated by secreted proteins or through integrin binding to ECM and cells. A major secreted survival-promoting molecule for endothelial cells is VEGF. Retinal endothelial cells of newly formed vessels which have not developed mature connections with surrounding cells and ECM are completely dependent upon VEGF and undergo apoptosis when VEGF is withdrawn or blocked (Alon et al., 1995). As vessels become more mature, endothelial cells receive survival signals from multiple sources and tend to become less susceptible to blockade of VEGF, but this varies among vascular beds. In mice, development of the retinal vasculature starts at birth and is completed by postnatal day (P) 18. During that time, the newly developed vessels depend upon VEGF and regress if it is withdrawn, but after P18 they are no longer affected by blockade of VEGF. In contrast, retinal neovascularization which grows along the surface of the retina and into the vitreous maintains VEGF-dependence for a long period of time. Scatter photocoagulation throughout the peripheral retina reduces retinal ischemia, decreases secretion of VEGF, and results in regression of retinal or disc neovascularization (Pournaras et al., 1990). Likewise, intraocular injection of VEGF antagonists results in rapid regression of retinal neovascularization (Avery et al., 2006; Bakri et al., 2006; Friedlander and Welch, 2006; Mason et al., 2006). Endothelial cells in longstanding retinal neovascularization recruit other cells resulting in fibrovascular tissue and neovascularization within extensive networks of cells and ECM on the surface of the retina is less susceptible to VEGF withdrawal and generally does not completely regress after scatter photocoagulation.
In patients with neovascular AMD and in our model of CNV, there is exuberant ECM associated with the CNV. It appears likely that the new vessels establish connections with surrounding cells and ECM much earlier than occurs in retinal neovascularization. However, this may not occur in all types of CNV, such as that occurring in young patients with pathologic myopia or ocular histoplasmosis, because in several such patients systemic infusions of bevacizumab resulted in partial regression of CNV (Nguyen et al., 2007). This illustrates that differences in the microenvironment of blood vessels contributes to how vessels respond to VEGF withdrawal.
Fenestrated capillary beds of some endocrine glands undergo partial regression when exposed to VEGF blockade (Baffert et al., 2006; Kamba et al., 2005). Since the choriocapillaris is fenestrated, we thought that there might be some alterations in the choroicapillaris after long term VEGF blockade, especially in view of a report suggesting loss of fenestrations and other substantial ultrastructural changes such as giant melanosomes, photoreceptor damage, and collections of leukocytes after a single intraocular injection of bevacizumab in primates (Peters et al., 2007). However, we did not see any alterations and our findings suggest that endothelial cells of normal choroidal vessels do not depend upon VEGF for survival and do not undergo apoptosis when subjected to long term blockade of VEGF. The changes described in primates after a single injection of bevacizumab may be related to trauma, because it is hard to explain photoreceptor damage, leukocytes, and giant melanosomes after transient blockade of VEGF and the controls used for comparison in that study were uninjected eyes. Additional studies are needed to resolve these discrepancies.
Thus, different types of endothelial cells in various vascular beds respond differently to blockade of VEGF. The same may be true for neurons. Some neurons, such as motor neurons in the spinal cord, are strongly dependent upon VEGF for survival (Oosthuyse et al., 2001; Storkebaum et al., 2005; Wang et al., 2007). Our data suggest that unlike motor neurons of the spinal cord, retinal neurons are not dependent upon VEGF, because prolonged blockade of VEGF caused no reduction in retinal function assessed by ERGs nor loss of retinal neurons, including ganglion cells. Our data differ from those of Nishijima et al. (Nishijima et al., 2007) who reported a 50–60% loss of retrogradely labeled ganglion cells in mice treated systemically three times a week for 8 weeks with sVEGFR1 or rats given weekly intraocular injections for 6 weeks of an antibody directed against VEGF. We cannot explain this difference, because our mice showed high levels of sVEGFR1Fc in the retina, certainly much higher than could have been achieved by systemic injections. The expression of sVEGFR1Fc completely blocked the ability of VEGF to cause leakage in the retina and suppressed the development of CNV by 50%; this is similar to the range of 40–60% achieved with intraocular or repeated systemic injections of VEGF Trap (Saishin et al., 2003a), a recombinant protein consisting of the VEGF binding domains of VEGFR1 and VEGFR2 and an Fc domain that is an extremely potent antagonist of multiple VEGF family members (Holash et al., 2002). Our data had sufficient power to detect a 50% reduction in ganglion cell axons if such a reduction was present. Thus, high levels of sVEGFR1Fc were present in the retina and the levels were sufficient to block VEGF-induced effects on retinal and choroidal vessels, but we could not detect any loss of ganglion cell axons. Another study showed no reduction in ganglion cells three days after injection of an antibody directed against rat VEGF (Iriyama et al., 2007); this follow up period is too short to provide evidence one way or the other. Additional independent studies in which VEGF blockade is maintained for at least 6 weeks are needed to reconcile these differences.
If the findings of our study are correct and can be generalized to humans, they suggest that repeated intraocular injections of randibizumab, bevacizumab, or VEGF Trap are not likely to cause local toxicity. Likewise, gene therapy resulting in sustained expression of a VEGF antagonist should not be eliminated from consideration. In view of the benefits achieved in several clinical trials and case series, it is likely that blockade of VEGF will continue to be an important component of the treatment of several retinal and choroidal diseases for many years to come. Our study suggests that long term follow-up of patients treated with aggressive inhibition of VEGF is not likely to uncover retinal or choroidal toxicity secondary to the blockade, but careful long term follow-up is needed. In addition, we strongly recommend additional animal studies by other investigators to fully evaluate the effects of long term blockade of VEGF in the retina and choroid.
Acknowledgments
Supported by grant EY05951 from the National Eye Institute and a senior scientist award from Research to Prevent Blindness
PAC is the George S. and Dolores Dore Eccles Professor of Ophthalmology and Neuroscience.
Dr. Campochiaro has received support for clinical trials from Genentech, is on the DSMC for a trial investigating VEGF Trap, and consults for LPath. These activities are monitored by the conflict of interest committee of the Johns Hopkins University School of Medicine.
References
- Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nature Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- Avery RL, Pearlman J, Pieramici DJ, Rabena MD, Castellarin AA, Nasir MA, Giust MJ, Wendel R, Patel A. Intravitreal bevacizumab (Avastin) in the treatment of proliferative diabetic retinopathy. Ophthalmolgy. 2006;113:1695. e1–15. doi: 10.1016/j.ophtha.2006.05.064. [DOI] [PubMed] [Google Scholar]
- Baffert F, Le T, Sennino B, Thruston F, Kuo C, Hu-Lowe D, McDonald DM. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol. 2006;290:H547–H559. doi: 10.1152/ajpheart.00616.2005. [DOI] [PubMed] [Google Scholar]
- Bakri SJ, Donaldson MJ, Link TP. Rapid regression of disc neovascularization in a patient with proliferative diabetic retinopathy following adjunctive intravitreal bevacizumab. Eye. 2006;20:1474–1475. doi: 10.1038/sj.eye.6702364. [DOI] [PubMed] [Google Scholar]
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, Sy JP, Schneider S, Group AS. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Eng J Med. 2006;355:1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA. Molecular targets for retinal vascular diseases. J Cell Physiol. 2006;210:575–581. doi: 10.1002/jcp.20893. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Shah SM, Hafiz G, Quinlan E, Zimmer-Galler I, Nguyen QD, Do DV, Ying H, Sung JU. Ranibizumab for macular edema due to retinal vein occlusions. Invest Ophthalmol Vis Sci ARVO. 2007 doi: 10.1038/mt.2008.10. online abstract 1545. [DOI] [PubMed] [Google Scholar]
- Chang MA, Horner JW, Conklin BR, DePinho RA, Bok D, Zack DJ. Tetracycline-inducible system for photoreceptor-specific gene expression. Invest Ophthalmol Vis Sci. 2000;41:4281–4287. [PubMed] [Google Scholar]
- Costa RA, Jorge LA, Calucci D, Melo LAJ, Cardillo JA, Scott IU. Intravitreal bevacizumab (Avastin) for central and hemicentral retinal vein occlusions IBeVO Study. Retina. 2007;27:141–149. doi: 10.1097/IAE.0b013e31802eff83. [DOI] [PubMed] [Google Scholar]
- Derevjanik NL, Vinores SA, Xiao W-H, Mori K, Turon T, Hudish T, Dong S, Campochiaro PA. Quantitative assessment of the integrity of the blood-retinal barrier in mice. Invest Ophthalmol Vis Sci. 2002;43:2462–2467. [PubMed] [Google Scholar]
- Friedlander SM, Welch RM. Vanishing disc neovascularization following intravitreal bevacizumab (avastin) injection. Arch Ophthalmol. 2006;124:1365. doi: 10.1001/archopht.124.9.1365. [DOI] [PubMed] [Google Scholar]
- Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Yung S, Chimenti S, Landsman L, Abramaovitch R, Keshet E. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–189. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Holash J, Davis S, Papadoupoulos N, Croll SD, Ho L, Russell M, Boland P, Ledich R, Hylton D, Burova E, Ioffe E, Huang T, Radziejewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriyama A, Chen Y-N, Tamaki Y, Yanagi Y. Effect of anti-VEGF antibody on retinal ganglion cells in rats. Br J Ophthalmol. 2007;91:1230–1233. doi: 10.1136/bjo.2007.117309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturralde D, Spaide RF, Meyerle CB, Klancnik JM, Yannuzzi LA, Fisher YL, Sorenson J, Slakter JS, Freund KB, Cooney M, Fine HF. Intravitreal bevacizumab (Avastin) treatment of macular edema in central retinal vein occlusion: a short-term study. Retina. 2006;26:279–284. doi: 10.1097/00006982-200603000-00005. [DOI] [PubMed] [Google Scholar]
- Ivanov D, Dvoriantchikova G, Nathanson L, McKinnon SJ, Shestopalov VI. Microarray analysis of gene expression in adult retinal ganglion cells. FEBS Lett. 2006;580:331–5. doi: 10.1016/j.febslet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: Direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci U S A. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamba T, Tam BYY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O’Brien SM, Davis RB, Gowen LC, Anderson KD, Thurston G, Joho S, Springer ML, Kuo CJ, McDonald DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adulat microvasculature. Am J Physiol Heart Circ Physiol. 2005;290:H560–H576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- Kilic U, Kilic E, Jarve A, Guo Z, Spudich A, Bieber K, Barzena U, Bassetti CL, Marti HH, Hermann DM. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating ERK-1/2 and Akt pathways. J Neuosci. 2006;26:12439–12446. doi: 10.1523/JNEUROSCI.0434-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CY, Kuehn MH, Clark AF, Kwon YH. Gene expression profile of the adult human retinal ganglion cell layer. Mol Vis. 2006;12:1640–1648. [PubMed] [Google Scholar]
- Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H. Doxycycline-mediated quantitative and tissue specific control of gene expression in transgenic mice. Proc Natl Acad Sci USA. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeima K, Rogers BS, Campochiaro PA. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007 May 22; doi: 10.1002/jcp.21152. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Kwak N, Okamoto N, Wood JM, Campochiaro PA. VEGF is an important stimulator in a model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2000;41:3158–3164. [PubMed] [Google Scholar]
- Mason JOr, Nixon PA, White MF. Intravitreal injection of bevacizumab (Avastin) as adjunctive treatment of proliferative diabetic retinopathy. Am J Ophthalmol. 2006;142:685–688. doi: 10.1016/j.ajo.2006.04.058. [DOI] [PubMed] [Google Scholar]
- Nguyen QD, Shah SM, Hafiz G, Do DV, Haller JA, Pili R, Zimmer-Galler I, Janjua K, Symons RCA, Campochiaro PA. Intravenous bevacizumab causes regression of choroidal neovascularization secondary to diseases other than age-related macular degeneration. Am J Ophthalmol. 2007 2007 Dec 3; doi: 10.1016/j.ajo.2007.09.025. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QD, Tatlipinar S, Shah SM, Haller JA, Quinlan E, Sung J, Zimmer-Galler I, Do DV, Campochiaro PA. Vascular endothelial growth factor is a critical stimulus for diabetic macular edema. Am J Ophthalmol. 2006;142:961–969. doi: 10.1016/j.ajo.2006.06.068. [DOI] [PubMed] [Google Scholar]
- Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, Shima DT. Vascular endothelial growth factor-A is a survivial factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007 Apr 26;170 doi: 10.2353/ajpath.2007.061237. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Matsui K, Hirose A, Yamamoto S, Saikia J, Okamoto N, Gehlbach P, Duh EJ, Hackett SF, Chang M, Bok D, Zack DJ, Campochiaro PA. Inducible expression of vascular endothelial growth factor in photoreceptors of adult mice causes severe proliferative retinopathy and retinal detachment. Am J Pathol. 2002;160:711–719. doi: 10.1016/S0002-9440(10)64891-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye G, Zimmer J, Sung JPG, Deering T, Nambu N, Hackett SF, Melia M, Esumi N, Zack DJ, Campochiaro PA. Increased expression of BDNF preserves retinal function and slows cell death from rhodopsin mutation or oxidative damage. J Neuosci. 2003;23:4164–4172. doi: 10.1523/JNEUROSCI.23-10-04164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Dorpe JV, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert J-M, Collen D, Carmeliet P. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- Oshima Y, Oshima S, Nambu H, Kachi S, Takahashi K, Umeda N, Shen J, Dong A, Apte RS, Duh E, Hackett SF, Okoye G, Ishibashi K, Handa J, Melia M, Wiegand S, Yancopoulos G, Zack DJ, Campochiaro PA. Different effects of angiopoietin 2 in different vascular beds in the eye; new vessels are most sensitive. FASEB J. 2005;19:963–965. doi: 10.1096/fj.04-2209fje. [DOI] [PubMed] [Google Scholar]
- Pai SA, Shetty R, Vijayan PB, Venkatasubramaniam G, Yadav NK, Shetty BK, Babu RB, Narayana KM. Clinical, anatomic, and electrophysiologic evaluation following intravitreal bevacizumab for macular edema in retinal vein occlusion. Am J Ophthalmol. 2007;143:601–606. doi: 10.1016/j.ajo.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Peters S, Heiduschka P, Julien S, Ziemssen F, Fietz H, Bartz-Schmidt KU, Group TBS, Schraermeyer U. Ultrastructural findings in the primate eye after intravitreal injection of bevacizumab. Am J Ophthalmol. 2007;143:995–1002. doi: 10.1016/j.ajo.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Pournaras CJ, Tsacopoulos M, Strommer K, Gilodi N, Leuenberger PM. Scatter photocoagulation restores tissue hypoxia in experimental vasoproliferative microangiopathy in miniature pigs. Ophthalmology. 1990;97:1329–1333. doi: 10.1016/s0161-6420(90)32414-4. [DOI] [PubMed] [Google Scholar]
- Rabena MD, Pieramici DJ, Castellarin AA, Nair MA, Avery RL. Intravitreal bevacizumab (Avastin) in the treatment of macular edema secondary to branch retinal vein occlusion. Retina. 2007;27:419–425. doi: 10.1097/IAE.0b013e318030e77e. [DOI] [PubMed] [Google Scholar]
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY, Group MS. Ranibizumab for neovascular age-related macular degeneration. N Eng J Med. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- Saishin Y, Saishin Y, Takahashi K, Lima Silva R, Hylton D, Rudge J, JWS, Campochiaro PA. VEGF-TRAPR1R2 suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003a;195:241–248. doi: 10.1002/jcp.10246. [DOI] [PubMed] [Google Scholar]
- Saishin Y, Saishin Y, Takahashi K, Melia M, Vinores SA, Campochiaro PA. Inhibition of protein kinase C decreases prostaglandin-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003b;195:210–219. doi: 10.1002/jcp.10238. [DOI] [PubMed] [Google Scholar]
- Seo M-S, Kwak N, Ozaki H, Yamada H, Okamoto N, Fabbro D, Hofmann F, Wood JM, Campochiaro PA. Dramatic inhibition of retinal and choroidal neovascularization by oral administration of a kinase inhibitor. Am J Pathol. 1999;154:1743–1753. doi: 10.1016/S0002-9440(10)65430-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl A, Agostini H, Lutz HL, Feltgen N. Bevacizumab in retinal vein occlusion-resutls of a prospective case series. Graefe’s Arch Clin Exp Ophthalmol. 2007 doi: 10.1007/s00417-007-0569-6. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Scmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Tobe T, Ortega S, Luna JD, Ozaki H, Okamoto N, Derevjanik NL, Vinores SA, Basilico C, Campochiaro PA. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am J Pathol. 1998;153:1641–1646. doi: 10.1016/S0002-9440(10)65753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke G, Molenberghs G. Linear Mixed Models for Longitudinal Data. Springer-Verlag, Inc; New York: 2000. [Google Scholar]
- Wang Y, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA, Jin K. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival i amyotrophic lateral sclerosis mice. J Neuosci. 2007;27:304–307. doi: 10.1523/JNEUROSCI.4433-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Redfern CS, Fishman GI. Conditional transgene expression in the heart. Circ Res. 1996;79:691–697. doi: 10.1161/01.res.79.4.691. [DOI] [PubMed] [Google Scholar]