

Abstract

Allylic phenyl ethers serve as electrophiles towards Pd(0) en route to a variety of allylic silanes. The reactions can be run at room temperature in water as the only medium using micellar catalysis.
Allylic silanes are among the most important and widely used reagents in organic synthesis.1 Well known transformations, such as the Hosomi-Sakurai reaction,2 or Hiyama couplings,3 underscore their value as building blocks, in particular for complex natural product syntheses.1 Although a vast array of synthetic methods for the preparation of allylic silanes now exists,4,5 further development of selective entries to allylic silanes are still of considerable interest. Tsuji described a general silylation reaction of allylic esters with organodisilanes in the presence of a palladium catalyst at 100 °C.6 Terao and Kambe reported the synthesis of allylic silanes from chlorosilanes and allylic ethers using palladium or nickel catalysts and an excess of Grignard reagents.7 Takaki utilized allylic ethers en route to allylic silanes via allylic samarium reagents.8 Recently Woerpel reported metal-catalyzed insertions of highly reactive silylenes into allylic ethers to provide allylic silanes.9
Despite the advantages of less reactive allylic ethers, e.g. stability towards nucleophilic attack and resistance to strongly acidic and basic conditions, their use in allylation chemistry remains quite rare.10,11,12 Moreover, such silylation reactions reported to date all require strictly anhydrous conditions.
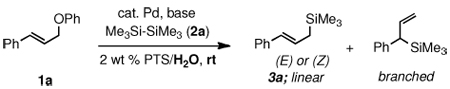
Herein we disclose an exceptionally mild entry to allylic silanes made possible by micellar catalysis13 in water at room temperature from the combination of allylic ethers and disilanes (Scheme 1).
Scheme 1.

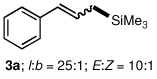
An initial investigation focused on the reaction of cinnamyl phenyl ether (1a) with hexamethyldisilane (2a) in 2 wt % PTS/H2O at room temperature (PTS = Polyoxyethanyl α-Tocopheryl Sebacate).14 Among several palladium catalysts screened, those bearing monodentate phosphine ligands or ligandless sources gave poor results; only PdCl2(PPh3)2 led to a moderate level of conversion (Table 1, entry 1). However, reaction in the presence of bidentate ligands afforded generally better results (entries 2, 3), and in the case of pre-formed complex PdCl2(DPEphos), full conversion with associated high isolated yield and excellent regioselectivity could be realized (entry 4). In previously reported silylations of more reactive allylic acetates, heating in organic solvents (e.g., DMF)6 was essential for activation of the disilane; under these new conditions, catalysis between cinnamyl phenyl ether (1a) and hexamethyldisilane (2a) proceeds at ambient temperature in the complete absence of organic solvent. Consistent results were best achieved in the presence of excess Et3N (4 equiv; entries 5, 6), although the amounts of base could be reduced without significant loss in reactivity. Curiously, other amine bases led to little-to-no conversion (entry 7), whereas K2CO3 gave reasonable results (entry 8). Control experiments in the absence of PTS, or in Et3N as solvent, confirmed the essential role of the surfactant.
Table 1.
Optimization of Pd-catalyzed silylation reactions.a
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | base (equiv) | convn (%)b | l:bb | E:Zb |
| 1 | PdCl2(PPh3)2 | Et3N (6) | 44 | 25:1 | 19:1 |
| 2 | PdCl2(DtBPF) | Et3N (6) | 40 | 7:1 | 25:1 |
| 3 | PdCl2(DiPPF) | Et3N (6) | 60 | 24:1 | 25:1 |
| 4 | PdCl2(DPEphos) | Et3N (6) | 100 (91)c | 25:1 | 10:1 |
| 5 | PdCl2(DPEphos) | Et3N (4) | 95 | 25:1 | 10:1 |
| 6 | PdCl2(DPEphos) | Et3N (2) | 95 | 25:1 | 10:1 |
| 7 | PdCl2(DPEphos) | aminesd | 0 | ||
| 8 | PdCl2(DPEphos) | K2CO3 (3) | 73 | 25:1 | 13:1 |
Reaction conditions: 1a (1.0 equiv, 0.25 mmol), 2a (1.5 equiv), catalyst (3 mol %), base, 2 wt % PTS/H2O (1.5 mL), rt, 20 h.
Determined by GCMS and 1H NMR.
Isolated yield.
nBu3N and EtiPr2N were used.
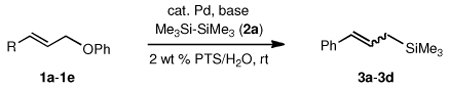
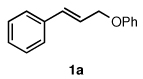
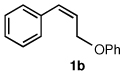
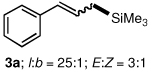
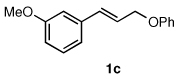
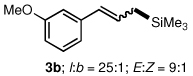
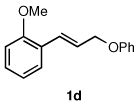
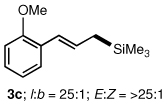
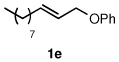
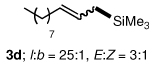
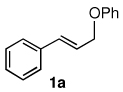
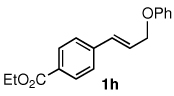
To explore the scope of this Pd-catalyzed silylation, various allylic phenyl ethers were examined (Table 2). While (E)-cinnamyl phenyl ether (1a) gave mainly the E-olefinic product 3a (entry 1; also Table 1, entry 4), the (Z)-educt 1b afforded the corresponding cinnamyl silane 3a in high yield with decreased stereoselectivity, favoring the (E)-isomer (entry 2). Cinnamyl derivatives 1c and 1d, e.g., those bearing an electron-donating methoxy substituent in either the meta or ortho position readily underwent the silylation reaction, the latter substitution pattern showing a strong preference for the E-product 3c (entries 3, 4). Surprisingly, alkyl substituted allylic phenyl ether 1e did not undergo full conversion under the optimized conditions. Ultimately, use of a mixed catalyst system consisting of PdCl2(PPh3)2 and PdCl2(DPEphos)15 gave the long chain allylic silane 3d in high yield, again favoring the E-isomer (entry 5). Each of these Pd-catalyzed allylic silylation reactions occurred with excellent regioselectivity favoring the corresponding linear products.
Table 2.
Pd-catalyzed silylation reactions with hexamethyldisilane. a
All reactions were carried out on 0.25 mmol scale.
l:b and E:Z ratio determined by GCMS and 1H NMR methods.
Isolated yield after column chromatography.
6 mol % catalyst and 4 equiv NEt3 were used.
1.5 mol % PdCl2(PPh3)2 and 1.5 mol % PdCl2(DPEphos) were used.
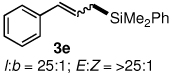
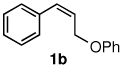
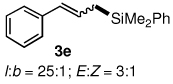
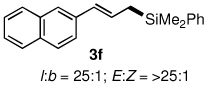
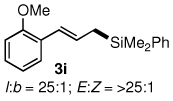
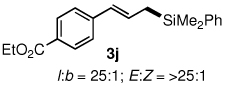
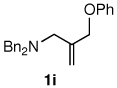
The alternative allylic dimethylphenylsilanes 3e–k (Table 3) could also be prepared in an analogous fashion, using commercially available 1,2-diphenyltetramethyl-disilane (2b). In general, these cross-couplings are both efficient and more selective than those forming the corresponding TMS derivatives. For example, (E)-cinnamyl phenyl ether (1a) gave product 3e in high yield with an E:Z ratio of > 25:1 (entry 1), although the (Z)-isomer 1b gave the same 3:1 preference favoring the (E)-derivative (entry 2; compare with Table 2, entry 2). Substitution on the aryl ring in cinnamyl phenyl ethers, whether electron-donating (entries 4–6) or -withdrawing (entry 7), and seemingly independent of location on the ring, gave the corresponding allylic silanes in high yields and with excellent regio- and stereoselectivity. Dibenzylamine-substituted methallyl substrate 1i also smoothly undergoes silylation (entry 8).
Table 3.
Pd-catalyzed silylations with 1,2-diphenyltetramethyldisilane 2b.a
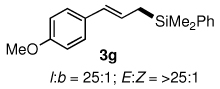
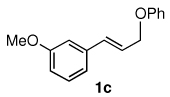
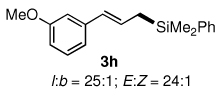
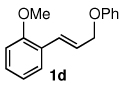
All reactions were carried out on 0.25 mmol scale.
l:b and E:Z ratio determined by GCMS and 1H NMR methods.
Isolated yield after column chromatography.
10 mol % PdCl2(DPEphos) used.
When the silylation reaction of alkyl substituted allylic phenyl ether 1e was carried out in pure MeOH, essentially no conversion was observed (Scheme 2). In PTS/H2O, however, the desired product allylic dimethylphenylsilane 3l was isolated in high yield (cat Pd = 3 mol % PdCl2(DPEphos); 4:1 E:Z). Thus, while nanomicelles are clearly involved when water is the medium,14 it is not known how PTS behaves in alcoholic solvent.
Scheme 2.

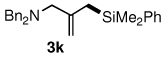
The relative reactivity of an allylic acetate compared to an allylic phenyl ether is as expected: reaction first occurs at the former site. Under optimized conditions, the combination of catalytic [allylPdCl]2 ligated by Xantphos gave chemoselective amination of allylic acetate 4 (Scheme 3). Without isolation, introduction of PdCl2(DPEphos) and disilane 2b resulted in silylation at the allylic ether fragment. This 1-pot amination/silylation sequence, done in its entirety in water at room temperature, could be applied to generation of dibenzylaminated methallylsilane 3k, and amino acid-substituted allyic silane 3m in good overall yields.
Scheme 3.

Scheme 4 illustrates one pathway accounting for the observed silylations, although details on the mechanism await further investigation. Initial oxidative addition of Pd(0) to allylic ether 1, aided by presumably high concentrations within micellar nanoreactors,16 produces allyl palladium intermediates 5, 5’ and 5”, each containing the bidentate ligand. Slow transmetallation of a π-allyl palladium complex 5”17 suggests that σ-allyl palladium species 5 and/or 5’ more likely undergoes transmetallation with disilane to give allyl silyl palladium 6 and/or 6’, leading to the corresponding branched and/or linear products, respectively, strongly favoring the latter.
Scheme 4.

In conclusion, new technology has been developed for Pd-catalyzed formation of allylic silanes from allylic ethers. These cross-couplings are very efficient, and occur under “green” conditions: without heat, without organic solvent, and within a micellar environment where water is the only external medium. Readily available disilanes serve as the stoichiometric source of silicon. Both the regio- as well as stereoselectivity associated with the catalysis are well-controlled. Further applications of micellar catalysis to transition metal chemistry are currently underway and will be reported in due course.
Supplementary Material
Acknowledgement
We thank the NIH (GM 86485) for support, and Johnson Matthey for supplying Pd catalysts.
Footnotes
Supporting Information Available: Experimental procedures and characterization of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews see: Curtis-Long MJ, Aye Y. Chem. Eur. J. 2009;15:5402. doi: 10.1002/chem.200900337. Gawronski J, Wascinska N, Gajewy J. Chem. Rev. 2008;108:5227. doi: 10.1021/cr800421c. Chabaud L, James P, Landais Y. Eur. J. Org. Chem. 2004:3173. Denmark SE, Fu J. Chem. Rev. 2003;103:2763. doi: 10.1021/cr020050h. Kennedy JWJ, Hall DG. Angew. Chem. Int. Ed. 2003;42:4732. doi: 10.1002/anie.200301632. For recent applications to syntheses of natural products, see: Lowe JT, Panek JS. Org. Lett. 2008;10:3813. doi: 10.1021/ol801499s. Youngsaye W, Lowe JT, Pohlki F, Ralifo P, Panek JS. Angew. Chem. Int. Ed. 2007;46:9211. doi: 10.1002/anie.200704122.
- 2.Hosomi A, Miura K. Bull. Chem. Soc. Jpn. 2004;77:835. [Google Scholar]
- 3.(a) Denmark SE, Werner NS. J. Am. Chem. Soc. 2008;130:16382. doi: 10.1021/ja805951j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hatanaka Y, Yasuo E, Hiyama T. J. Am. Chem. Soc. 1991;113:7075. [Google Scholar]
- 4.(a) Barbero A, Pulido FJ. Acc. Chem. Res. 2004;37:817. doi: 10.1021/ar0400490. [DOI] [PubMed] [Google Scholar]; (b) Sarkar TK. Science of Synthesis. Vol. 4. Stuttgart: Georg Thieme Verlag; 2002. pp. 837–925. [Google Scholar]; (c) Sarkar TK. Synthesis. 1990:969. [Google Scholar]; (d) Sarkar TK. Synthesis. 1990:1101. [Google Scholar]
- 5.Selected metal catalyzed silylations: Simon M-O, Martinez R, Genêt J-P, Darses S. Adv. Synth. Catal. 2009;351:153. Tobisu M, Kita Y, Ano Y, Chatani N. J. Am. Chem. Soc. 2008;130:15982. doi: 10.1021/ja804992n. Li Z, Yang C, Zheng H, Qiu H, Lai G. J. Organomet. Chem. 2008;693:3771. Hayashi S, Hirano K, Yorimitsu H, Oshima K. J. Am. Chem. Soc. 2007;129:12650. doi: 10.1021/ja0755111. Shintani R, Ichikawa Y, Hayashi T, Chen J, Nakao Y, Hiyama T. Org. Lett. 2007;9:4643. doi: 10.1021/ol702125q. Kabalka GW, Venkataiah B, Dong G. Organometallics. 2005;24:762. Suginome M, Ito Y. J. Organomet. Chem. 2003;680:43. Obora Y, Tsuji Y, Kawamura T. J. Am. Chem. Soc. 1995;117:9814. doi: 10.1021/ja010674p. Obora Y, Tsuji Y, Kawamura T. J. Am. Chem. Soc. 1993;115:10414. doi: 10.1021/ja010674p. Matsumoto H, Yako T, Nagashima S, Motegi T, Nagai Y. J. Organomet. Chem. 1978;148:97.
- 6.(a) Tsuji Y, Funato M, Ozawa M, Ogiyama H, Kajita S, Kawamura T. J. Org. Chem. 1996;61:5779. [Google Scholar]; (b) Tsuji Y, Kajita S, Isobe S, Funato M. J. Org. Chem. 1993;58:3607. [Google Scholar]
- 7.(a) Naitoh Y, Bando F, Terao J, Otsuki K, Kuniyasu H, Kambe N. Chem. Lett. 2007;36:236. [Google Scholar]; (b) Terao J, Watabe H, Watanabe H, Kambe N. Adv. Synth. Catal. 2004;346:1674. [Google Scholar]
- 8.Takaki K, Kusudo T, Uebori S, Nishiyama T, Kamata T, Yokoyama M, Takehira K, Makioka Y, Fujiwara Y. J. Org. Chem. 1998;63:4299. [Google Scholar]
- 9.(a) Bourque LE, Haile PA, Loy JMN, Woerpel KA. Tetrahedron. 2009;65:5608. doi: 10.1016/j.tet.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bourque LE, Cleary PA, Woerpel KA. J. Am. Chem. Soc. 2007;127:12602. doi: 10.1021/ja073758s. [DOI] [PubMed] [Google Scholar]
- 10.Allylic amination: Mora G, Piechaczyk O, Le Goff XF, Le Floch P. Organometallics. 2008;27:2565. Murakami H, Minami T, Ozawa F. J. Org. Chem. 2004;69:4482. doi: 10.1021/jo049732q. Takahashi K, Miyake A, Hata G. Bull. Chem. Soc. Jpn. 1972;45:230. Cross-couplings: Yasui H, Mizutani K, Yorimitsu H, Oshima K. Tetrahedron. 2006;62:1410. Alkylation: Onoue H, Moritani I, Murahashi S-I. Tetrahedron Lett. 1973;14:121. Addition: Masuyama Y, Marukawa M. Tetrahedron Lett. 2007;48:5963.
- 11.(a) Tamaru Y. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. New York: Wiley; 2002. p. 1917. [Google Scholar]; (b) Tamaru Y. In: Perspectives in Organopalladium Chemistry for the XXI Century. Tsuji J, editor. Switzerland: Elsevier Science; 1999. p. 215. [Google Scholar]; (c) Tsuji J. Palladium Reagents and Catalysts, Innovations in Organic Synthesis. New York: Wiley; 1995. p. 290. [Google Scholar]; (d) Masuyama Y. In: Advances in Metal-Organic Chemistry. Liebeskind LS, editor. Vol. 3. Greenwich, CT: JAI Press; 1994. p. 255. [Google Scholar]; (e) Trost BM, Verhoeven TR. Comprehensive Organometallic Chemistry. Vol. 8. Oxford: Pergamon Press; 1982. p. 799. [Google Scholar]
- 12.Li C-J. Chem. Rev. 2005;105:3095. doi: 10.1021/cr030009u. [DOI] [PubMed] [Google Scholar]
- 13.Dwars T, Paetzold E, Oehme G. Angew. Chem. Int. Ed. 2005;44:7174. doi: 10.1002/anie.200501365. [DOI] [PubMed] [Google Scholar]
- 14. Nishikata T, Lipshutz BH. Org. Lett. 2009;11:2377. doi: 10.1021/ol900235s. For a review, see: Lipshutz BH, Ghorai S. Aldrichim. Acta. 2008;41:58. and references cited therein.
- 15.We cannot explain this synergistic effect at this stage.
- 16.Zana R, editor. Dynamics of Surfactant Self-Assemblies; Micelles, Microemulsions, Vesicles, and Lyotropic Phases. Boca Raton, FL: Taylor & Francis; 2005. Surfactant Science Series 125. [Google Scholar]
- 17.Hegedus LS. In: Organometallics in Synthesis. Second Edition. Schlosser M, editor. Wiley; 2004. p. 1185. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.