

Abstract
The nucleus tractus solitarius (NTS) is essential for orchestrating baroreflex control of blood pressure. When a change in blood pressure occurs, the information is transmitted by baroreceptor afferent fibers to the central network by glutamate binding to ionotropic glutamate receptors on second-order baroreceptor neurons. Glutamate also activates presynaptic group II and III metabotropic glutamate receptors (mGluRs), depressing both glutamate and GABA release to modulate baroreceptor signal transmission. Here we present a novel role for postsynaptic group II mGluRs to further fine-tune baroreceptor signal transmission at the first central synapses. In a brainstem slice with ionotropic glutamate and GABA receptors blocked, whole-cell patch-clamp recordings of second-order baroreceptor neurons revealed that two group II mGluR agonists evoked concentration-dependent membrane hyperpolarizations. The hyperpolarization remained when a presynaptic contribution was prevented with Cd2+, was blocked by a postsynaptic intervention of intracellular dialysis of the G-protein signaling inhibitor, was mimicked by endogenous release of glutamate by tractus solitarius stimulation, and was prevented by a group II mGluR antagonist. Postsynaptic localization of group II mGluRs was confirmed by fluorescent confocal immunohistochemistry and light microscopy. Group II mGluR induced-currents consisted of voltage-dependent outward and inward components, prevented by tetraethylammonium chloride and tetrodotoxin, respectively. In contrast to group II mGluR-induced hyperpolarization, there was no effect on intrinsic excitability as determined by action potential shape or firing in response to depolarizing current injections. The data suggest a novel mechanism for postsynaptic group II mGluRs to fine-tune baroreceptor signal transmission in the NTS.
Introduction
The nucleus tractus solitarius (NTS) is the principal brainstem site for coordinating CNS control of blood pressure. When a change in blood pressure occurs, the blood pressure-related sensory information is rapidly and efficiently transmitted by primary baroreceptor afferent fibers to the central baroreflex network by glutamate binding to ionotropic glutamate receptors on the second-order baroreceptor neurons in the NTS (Talman, 1989; Ohta and Talman, 1994; Aylwin et al., 1997). At these synapses, the information is fine-tuned by presynaptic and postsynaptic influences, originating from local networks and other brain regions, so that the baroreceptor signal output to sympathetic and parasympathetic premotoneuronal pools is optimized to regulate blood pressure. Dysregulation of baroreceptor signal transmission in the NTS disrupts cardiovascular homeostasis and contributes to the consequences of hypertension and heart failure (Timmers et al., 2004).
The G-protein-linked metabotropic glutamate receptors (mGluRs) provide acute and long-term modulation of glutamatergic transmission in various neural networks (Anwyl, 1999). Eight subtypes of mGluRs divide into three groups based on signal transduction pathways, pharmacology, and genetic sequence. Group I mGluRs are located predominantly on cell soma in which in most instances they couple to Gq/11-proteins to increase neuronal excitability, essentially increasing the excitatory response to glutamate activation of the ionotropic glutamate receptors. Group II and III mGluRs are located predominantly on presynaptic terminals in which they inhibit adenylyl cyclase to decrease transmitter release.
In the NTS, glutamate binding to group II and III mGluRs inhibits baroreceptor signal transmission by limiting glutamate release from the primary baroreceptor afferent fibers (Liu et al., 1998; Chen et al., 2002). Group II mGluRs can also increase baroreceptor signal transmission by decreasing GABA release at the first central baroreceptor synapses (Chen and Bonham, 2005). Glutamate binding to postsynaptic group I mGluRs depolarizes the second-order baroreceptor neurons, thus enhancing baroreceptor signal transmission. There are very few reports of functional postsynaptic mGluRs in the NTS, but a recent study in the neocortex has implicated a potential inhibitory postsynaptic role for group II mGluRs in regulating glutamatergic transmission. Lee and Sherman (2009) reported that mGluR agonist application to layer 4 cortical neurons elicited a hyperpolarization that was abolished by a group II mGluR antagonist and mimicked by electrical and photostimulation of the intracortical inputs to these neurons (Lee and Sherman, 2009).
To the extent that postsynaptic group II mGluRs exhibit this role in the baroreceptor network, they would provide a novel postsynaptic mechanism to further regulate baroreceptor synaptic transmission during fluctuations in blood pressure by blunting the excitability of the second-order neurons. Thus, we asked the following questions: (1) are postsynaptic group II mGluRs functional on second-order baroreceptor neurons, and (2) are they stimulated by endogenous glutamate release? Because neurons in various autonomic pathways are intermingled in the NTS, we used whole-cell patch clamping on second-order NTS baroreceptor neurons anatomically identified by the presence of attached boutons of the primary baroreceptor fibers to specifically determine the contribution of mGluRs to baroreceptor signal transmission. Postsynaptic localization of group II mGluRs was confirmed by immunohistochemical fluorescent confocal and light microscopy.
Materials and Methods
All experimental protocols in this work were reviewed and approved by the Institutional Animal Care and Use Committee in compliance with the Animal Welfare Act and in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Labeling second-order NTS baroreceptor neurons.
Male Sprague Dawley rats (11 weeks old, 332 ± 22 g, means ± SD) were anesthetized by intramuscular injections of ketamine (40 mg/kg) and xylazine (8 mg/kg). As described previously (Chen et al., 2002; Chen and Bonham, 2005; Sekizawa and Bonham, 2006), both aortic depressor nerves (ADNs) were carefully isolated, and a 4–5 mm segment was placed on a section of Parafilm for application of the crystal form of the fluorescent tracer 1,1′-dilinoleyl-3,3,3′,3′ tetra-methylindocarbo-cyanine, 4-chlorobenzenesulfonate [FAST DiI solid; DiI Δ 9,12-C18(3)] with subsequent coating of the nerve with Polyvinylsiloxane gel. DiI is transported anterogradely to label the terminal boutons, without being transported transynaptically. To allow for transport of the dye to the terminal boutons, the rats were allowed to recover for 2 weeks before the experimental protocols were performed (Chen et al., 2002; Chen and Bonham, 2005; Sekizawa and Bonham, 2006).
Brainstem slice preparation and electrophysiology.
Rats were anesthetized with a combination of ketamine (20 mg/kg) and xylazine (2 mg/kg) and decapitated. As in previous studies (Chen et al., 2002; Chen and Bonham, 2005; Sekizawa and Bonham, 2006), the brain was rapidly exposed and submerged in ice-cold (<4°C) high-sucrose artificial CSF (aCSF) that contained the following (in mm): 3 KCl, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 220 sucrose, and 2 CaCl2 (300 mOsm); the pH was 7.4 when continuously bubbled with 95% O2/5% CO2. Brainstem coronal slices (250 μm thick) containing the intermediate to caudal NTS and the tractus solitarius (TS) were cut with the Vibratome 1000 (Technical Products International). After incubation for 45 min at 37°C in high-sucrose aCSF, the slices were placed in normal aCSF that contained the following (in mm): 125 NaCl, 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, and 2 CaCl2 (300 mOsm); the pH was 7.4 when continuously bubbled with 95% O2/5% CO2. Four or five slices were obtained from each animal. During the experiments, a single slice was transferred to the recording chamber, held in place with a silk mesh, and continuously perfused with oxygenated aCSF at a rate of ∼4 ml/min. All experiments were performed at 33–34°C.
All recordings were taken from second-order NTS baroreceptor neurons with attached ADN boutons. The neurons were visualized with Nomarski infrared differential interference contrast (IR-DIC), and the fluorescent boutons were visualized with an optical filter set for DiI (XF108; Omega Optical) and an image integrating system (InstaGater; Dage-MTI). All images were captured with a charge-coupled device camera (CCD-100; Dage-MTI), displayed on a television monitor, and stored in a personal computer using Computer Eyes software (Winnov). Recordings were made with the Axopatch 1D patch-clamp amplifier (Molecular Devices). Currents were filtered at 2 kHz, digitized at 10 kHz with the DigiData1322A interface (Molecular Devices), and stored in an IBM-compatible computer. Data were analyzed offline using the pClamp9 software (Molecular Devices). After establishing the cell-attached configuration with a seal resistance of >1 GΩ, whole-cell currents were recorded using borosilicate glass pipettes (2.2–5 MΩ; 4.0 ± 0.7 MΩ, mean ± SD) filled with a potassium gluconate (K-gluconate)-based solution containing the following (in mm): 130 KC6H11O7, 1 NaCl, 1 MgCl2, 2 K-ATP, 0.3 Na-GTP, 10 EGTA, and 10 HEPES (300 mOsm); pH was adjusted to 7.4 with KOH. In some experiments, Na-GTP was replaced with 1 mm guanosine-5′-O-(2-thiodiphosphate) (GDPβS) to examine G-protein participation. In these experiments, we established the whole-cell configuration on the same neuron with two different but identical pipettes, in which any pipette-related parameters and whole-cell response were similar when the same control pipette solution was used for both two pipettes (Sekizawa and Bonham, 2006). In some experiments, biocytin (0.5%) was added to normal pipette solution to perform immunohistochemical analysis after the electrophysiological recording. The series resistance was <22 MΩ (11.1 ± 3.4 MΩ, mean ± SD). After establishing the whole-cell configuration, neurons were tested for TS input with the membrane voltage clamped at −60 mV. All experiments were performed with 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo(t)quinoxaline-7-sulfonamide disodium salt (NBQX) (10 μm), dl-2-amino-5-phosphonopentanoic acid (AP-5) (50 μm), and the GABAA receptor antagonist (−)-bicuculline methiodide (bicuculline) (10 μm) to exclude ionotropic glutamate and GABAA receptor inputs under current- or voltage-clamp conditions unless otherwise stated. Membrane potential was adjusted to −50 mV, which was close to resting membrane potential, in current-clamp mode before each recording. Liquid junction potential was −9.8 mV and not corrected in this study. When a brainstem slice was once used for mGluR agonists and/or antagonist, the slice was not used further.
Agonist studies.
The selective group II mGluR agonists (2S,3S,4S)-CCG/(2S,1′S,2′S)-2-(carboxycyclopropyl)glycine (l-CCG-1) (up to 100 μm) and LY 379268 (1R,4R,5S,6R-2-oxa-4-aminobicyclo[3.1.0]hexane-4,6-dicarboxylate) (up to 300 nm) were used under current- or voltage-clamp conditions and added to the perfusion for 1 min unless otherwise stated. Under the current-clamp condition, a brief negative current (200 ms, −5 to −20 pA) was injected at frequency of 0.1 Hz to monitor input resistance continuously. When concentration-dependent responses were examined, up to three different concentrations of drugs were applied per cell in random order with a washout period of at least 8 min. In some experiments, agonist perfusion was repeated in the presence of group II mGluR antagonists, Cd2+, or after the replacement of intracellular GTP to GDPβS in the pipette solution.
Endogenous glutamate release.
To test whether synaptically released glutamate activated postsynaptic mGluRs, the TS was stimulated by bipolar tungsten electrodes (1 μm tips separated by 80 μm; FHC). Paired stimuli separated by 28 ms intervals were delivered every 167 ms (six paired stimuli per second) (see Fig. 4A, inset) for 5 min, to mimic phasic baroreceptor afferent signaling in rats with a heart rate of ∼360 beats per minute. Square-wave 0.1 ms pulses were applied with a microelectrode stimulator (SD9; Grass Instruments). The neurons were initially held at −50 mV, and recordings were made in current-clamp mode. After 5 min of continuous TS stimulation, the neurons were maintained in current-clamp mode for the recovery for at least 5 min for 80% recovery from changes in membrane potential, and then the group II mGluR antagonist LY 341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth- 9-yl) propanoic acid] (200 nm) was added to the perfusate. At least 5 min later during the antagonist perfusion, the TS stimulation protocol was repeated in the presence of antagonist.
Figure 4.
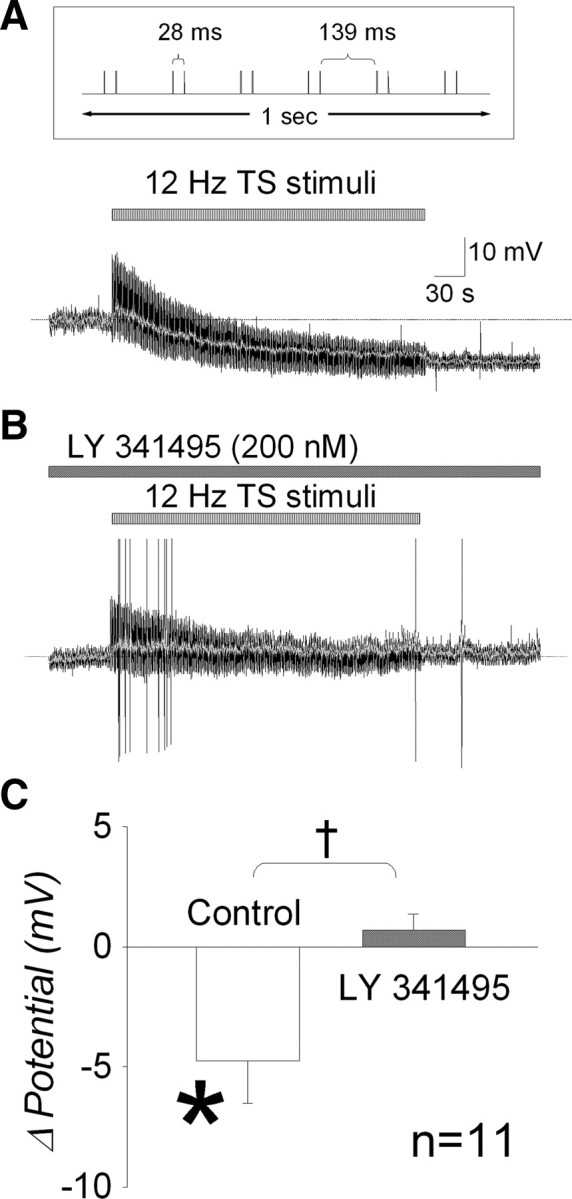
Endogenous glutamate can activate postsynaptic group II mGluRs on NTS baroreceptor neurons. A, TS was stimulated at an overall frequency of 12 Hz for 5 min to increase endogenous glutamate in the synaptic cleft. Inset (top) shows stimulus protocol in 1 s section. Neurons were initially held at −50 mV of resting membrane potential under the current-clamp mode. The membrane potential gradually hyperpolarized for the first 2–3 min and reached to a plateau level (10.7 mV hyperpolarization) by the end of 5 min stimulation. Because recordings are very noisy during TS stimulation, the membrane potentials without stimulus artifacts are indicated by gray line. Baseline membrane potential is indicated by the dashed line. B, TS stimulation was repeated in the presence of the group II mGluR antagonist LY 341495 (200 nm) in the same neuron. Membrane potential is barely changed even at the end of TS stimulation (2.0 mV depolarization). C, Group data show that continuous TS stimulation significantly hyperpolarizes membrane potential (*p = 0.025) (control). However, hyperpolarization response was blockade by LY 341495 (†p = 0.017).
Voltage dependency of group II mGluR currents.
The current- and membrane-voltage (I–V) relationship was obtained in voltage-clamp mode by using a slow voltage ramp-down protocol from +20 to −120 mV, 2 s duration from the holding potential of −60 mV before and at the peak of the LY 379268-induced current. The relationship of the LY 379268-induced current to membrane voltage was obtained by subtracting the I–V relationship before LY 379268 application from the I–V relationship during LY 379268 application. To further investigate the ionic components of the LY 379268-induced current, the voltage ramp-down protocol was repeated in the presence of the voltage-sensitive Na+ channel blocker tetrodotoxin (TTX) (1 μm) or the broad-range K+ channel blocker tetraethylammonium chloride (TEA) (10 mm).
Immunohistochemistry for group II mGluR.
After completing the electrophysiological recordings, some neurons were filled with biocytin. After electrophysiological recording, brainstem slices were fixed in 4% paraformaldehyde for 60 min at 4°C and then stored in PBS until processing for immunohistochemistry for group II mGluR (mGluR2/3). Before processing, brainstem sections were rinsed in PBS. All incubation and rinsing steps (three times for 10 min) between each subsequent step were at room temperature on a laboratory shaker unless otherwise noted. Slices were blocked in 10% normal goat serum (NGS) (Vector Laboratories) and 1% tyramide signal amplification (TSA) blocking agent (TSA kit; Invitrogen) for 60 min and then incubated in a rabbit anti-mGluR2/3 primary antibody (1:1000; Affinity Bioreagents) in PBS containing 1% NGS and 1% TSA blocking reagent for 60–72 h at 4°C. Slices were rinsed and incubated for 2 h in horse radish peroxidase-conjugated goat anti-rabbit secondary antibody (1:100; Invitrogen) for amplification of mGluR2/3 immunoreactivity and Alexa Fluro-568-conjugated streptavidin (1:100; Invitrogen) for visualizing intracellular biocytin in the recorded neuron. To visualize mGluR2/3, slices were then incubated in Alexa Fluro-488 conjugated tyramide (1:100; Invitrogen) and 0.0015% hydrogen peroxide for 20–25 min. Slices were rinsed, mounted on lysine-coated slides, dried, and coverslipped in anti-fade mounting media (Vector Laboratories). Tissue was viewed and images were captured with a Carl Zeiss LSM-5 inverted confocal microscope. The recorded neurons were examined with a 488 nm (mGluR2/3) and 568 nm (biocytin) laser lines under a 20× objective. Images were scanned and captured at a resolution of 1024 × 1024 pixels. Confocal images were acquired at 1-μm-thick optical slices.
Immunohistochemistry for light microscopy.
Immunohistochemistry for mGluR2/3 was also performed in naive Sprague Dawley rats (n = 3) to confirm postsynaptic localization of group II mGluR. Animals were deeply anesthetized with pentobarbital (75 mg/kg) and then perfused transcardially with 200 ml of PBS, followed by 500 ml of 4% paraformaldehyde in PBS. Brains were rapidly removed and postfixed for 2–4 h in fixative and stored overnight in PBS. Brainstems were sectioned on a vibratome and collected into PBS. Sections were immediately processed for mGluR 2/3 immunohistochemistry without freezing to prevent loss of immunoreactivity on the soma. Slices were first blocked in 10% NGS and 1% TSA blocking reagent for 60 min and then incubated in a rabbit anti-mGluR2/3 primary antibody in PBS containing 3% NGS and 1% BSA for 36 h at 4°C. Slices were thoroughly rinsed, incubated in biotinylated goat anti-rabbit secondary antibody (1:500; Vector Laboratories) for 2 h followed by a 1 h incubation in avidin-biotyinylated peroxidase complex for 1 h (ABC method), and processed to reveal peroxidase using 3,3′-diaminobenzidine (DAB) (Vector Laboratories) as the chromogen. Control sections, processed as above except for the omission of the primary antibody, did not contain DAB label on microscopic examination (negative control). Slices were mounted on lysine-coated slides, dried, and coverslipped in anti-fade mounting media. Tissue was viewed and images were captured with a Carl Zeiss Axioplan2 light microscope. Images were captured using a digital camera (AxioCam HRc).
Data analysis.
Data are expressed as means ± SEM unless otherwise stated. Peak voltage changes induced by group II mGluR agonist were obtained from greatest changes in hyperpolarizing direction from a stable control level (at least 30 s before any drug application) for current-clamp recordings, and input resistance was also obtained at each control or peak response period. For 5 min continuous TS stimulation, average voltage changes were obtained from last 30 s of stimulation. Changes in membrane voltage and input resistance were compared before and during extracellular application of group II mGluR antagonists, Cd2+, or intracellular application of GDPβS using the paired t test. Group II mGluR agonist concentration–response curves were fitted to a sigmoid function to obtain the EC50. Intrinsic cell excitability was characterized as spiking responses to 20 pA incremental steps of depolarizing current injections up to 120 pA. Action potential shapes were also characterized by their threshold, width, peak amplitude, and afterhyperpolarization (AHP). Threshold was defined and measured as the membrane potential at which the action potential rose steeply (Nakajima and Onodera, 1969), and width was measured at the level of threshold potential. Action potential characteristics were compared before and during group II mGluR agonist application using the paired t test. The paired t test was used for statistical analysis for all data except for the spiking responses before and during group II mGluR agonist application that were compared with two-way repeated measures (RM)-ANOVA. p values <0.05 were considered significant.
Drugs.
Drugs were dissolved in aCSF just before application. Ketamine and xylazine were obtained from Vedco. DiI was obtained from Invitrogen. Polyvinylsiloxane gel was obtained from Charles Laboratories. LY 379268, LY 341495, and l-CCG-1 were obtained from Tocris Cookson. K-Gluconate, bicuculline, Mg-ATP, Na-ATP, EGTA, HEPES, CaCl2, CdCl2, TEA, biocytin, NBQX, AP-5, TTX, and GDPβS were obtained from Sigma. All other chemicals were obtained from Thermo Fisher Scientific.
Results
Whole-cell patch-clamp recordings were performed on 177 second-order NTS baroreceptor neurons, which were identified by the presence of fluorescent boutons and were located at 0–1250 μm caudal to obex in the NTS in accordance with the atlas by Paxinos and Watson (1986). The averaged resting membrane potential, input resistance, and membrane capacitance were −50 ± 5.4 mV, 410 ± 249 MΩ, and 84 ± 36 pF (mean ± SD), respectively. Figure 1 shows an example neuron viewed under IR-DIC, then under fluorescence to visualize presynaptic baroreceptor afferent fiber terminal boutons, as an overlay of fluorescence and IR-DIC images, and with the patch electrode attached to cell for whole-cell recording. The averaged length of long axis of second-order NTS baroreceptor neurons was 20.0 ± 3.8 μm (mean ± SD). All results were obtained in the presence of the ionotropic glutamate receptor antagonists NBQX (10 μm) and AP-5 (50 μm) and the GABAA receptor antagonist bicuculline methiodide (10 μm) to exclude ionotropic glutamate and GABAA receptor inputs unless otherwise stated.
Figure 1.

Second-order NTS baroreceptor neuron. A, NTS neuron viewed under IR-DIC. Scale bar, 20 μm. B, Same neuron viewed under fluorescence to visualize presynaptic baroreceptor afferent fiber terminal boutons. C, Overlay of fluorescence and IR-DIC. D, Patch electrode attached to cell for whole-cell recording.
Group II mGluR agonist hyperpolarized second-order baroreceptor neurons
As shown in the example (Fig. 2A, top trace), the group II mGluR agonist LY 379268 (30 nm) produced a slowly developing hyperpolarization of the membrane potential of a second-order baroreceptor neuron. The hyperpolarization was accompanied by a decrease in the input resistance. As shown in the bottom trace, the agonist-induced responses were nearly abolished by the group II mGluR antagonist LY 341495 (200 nm), a concentration shown previously to block group II mGluR effects in the NTS (Chen and Bonham, 2005). The group data (n = 12 from 4 animals) confirmed that the agonist-induced hyperpolarization, whose peak was observed at 99 ± 5.1 s from the beginning of agonist perfusion, was nearly abolished by the antagonist [LY 341495; −5.3 ± 0.9 to −0.8 ± 0.9 mV (19 ± 16% of control); p = 0.010], as was the decrease in input resistance (−17 ± 6.1 to −7.4 ± 6.4%; p = 0.037) (Fig. 2B).
Figure 2.

Group II mGluR effects on second-order baroreceptor neurons. A, Example traces of 1 min application of the group II mGluR agonist LY 379268 (30 nm). LY 379268 clearly hyperpolarized the membrane potential and decreased action potential firing frequency (top trace). This agonist effect was not observed in the presence of the group II mGluR antagonist LY 341495 (200 nm) in the same neuron (bottom trace). Slight notch-like membrane hyperpolarizations indicate brief 0.1 Hz negative current injections used to monitor input resistance during recording. B, Group data confirmed that LY 379268-induced membrane hyperpolarization and reduced input resistance (control) were significantly reduced by LY 341495 (*p = 0.010 and *p = 0.037, respectively).
Two agonists, LY 379268 and l-CCG-1, each evoked concentration- dependent hyperpolarizations (Fig. 3A) and decreases in input resistance (Fig. 3B). All the data were obtained from 27 animals. From the fitted sigmoid curve, the calculated EC50 of LY 379268 and l-CCG-1 was 25 nm and 3.0 μm, respectively, and the predicted maximum hyperpolarization response was −7.9 mV (r2 = 0.9314) and −4.0 mV (r2 = 0.6865), respectively. The EC50 for the reduction in input resistance was 13 nm for LY 379268 and 1.9 μm for l-CCG-1, and the predicted maximum responses were 19% (r2 = 0.9823) and 22% (r2 = 0.6198) decreases, respectively. In response to higher concentration (30 and 100 μm) of l-CCG-1, some cells showed a depolarization response that was not blocked by the group II mGluR antagonist LY 341495 (200 nm) but blocked by group I mGluR antagonist (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Therefore, these depolarization data were excluded from group II mGluR-induced response (for more details, see supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 3.

Concentration dependency of group II mGluR agonist-induced response. A, Hyperpolarization response. Numbers in parentheses indicate number of neurons. Both group II mGluR agonists LY 379268 and l-CCG-1 hyperpolarized the membrane potential in a concentration-dependent manner. Curves are fitted with a sigmoid function. EC50 of LY 379268 and l-CCG-1 were 25 nm and 3.0 μm, respectively. B, Reduced input resistance. Both LY 379268 and l-CCG-1 reduced input resistance in a concentration-dependent manner as well. Numbers of neurons are the same as in A. EC50 of LY 379268 and l-CCG-1 were 13 nm and 1.9 μm, respectively.
Endogenous glutamate hyperpolarized the second-order neurons by binding to postsynaptic receptors
The physiological relevance of the postsynaptic group II mGluRs in baroreceptor signaling depends on whether they are activated by endogenous release of glutamate. As shown in Figure 4A (inset), glutamate release was stimulated by paired electrical stimuli (separated by 28 ms) applied to the TS every 167 ms at an overall frequency of 12 Hz, to mimic phasic baroreceptor afferent signaling in rats with a heart rate of ∼360 beats per minute. As shown in the example (Fig. 4A), the neuron was gradually hyperpolarized, reaching a plateau level by the end of the 5 min TS stimulation. In the same neuron, application of the group II mGluR antagonist LY 341495 (200 nm) abolished the hyperpolarization (Fig. 4B). The group data (n = 11 from 10 animals) confirm that the membrane hyperpolarization (−4.7 ± 1.8 mV) by endogenous glutamate release from continuous TS stimulation was prevented by the group II mGluR antagonist LY 341495 (0.71 ± 0.65 mV; p = 0.017) (Fig. 4C).
Postsynaptic locus of Group II mGluR activation
To confirm that the group II mGluR-induced hyperpolarization was mediated postsynaptically, we used Cd2+ (50 μm) to block presynaptic Ca2+ channels (Sekizawa et al., 2000) to prevent glutamate release from the presynaptic terminals. As shown in Figure 5A, l-CCG-1 hyperpolarized the membrane in the presence of bicuculline and AP-5. NBQX was omitted from the bath to demonstrate the presence of a synaptically evoked EPSC evoked by stimulation of the TS. (Fig. 5A, inset). When Cd2+ was added to the extracellular bath, the TS-evoked EPSC, dependent on presynaptic glutamate release, was blocked (Fig. 5B, inset), whereas the l-CCG-1-induced hyperpolarization remained intact (Fig. 5B). The group data (n = 8 from 4 animals) (Fig. 5C) confirmed that the group II mGluR agonist-induced hyperpolarization (−7.3 ± 1.3 mV) was not altered by extracellular Cd2+, suggesting a postsynaptic locus of the hyperpolarization (−8.8 ± 1.8 mV; p = 0.350).
Figure 5.
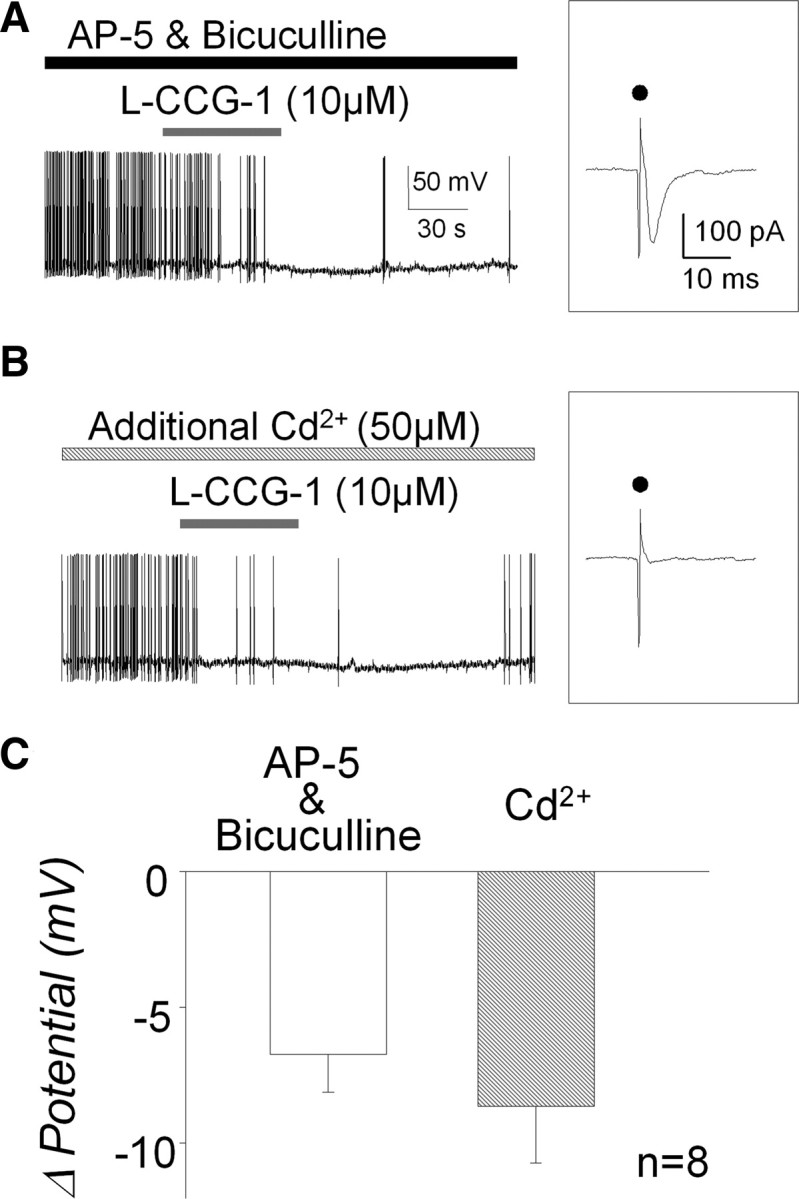
Effect of Cd2+ on group II mGluR agonist-induced hyperpolarization. A, Ten micromolar of l-CCG-1 hyperpolarized the membrane potential even in the absence of NBQX, an AMPA receptor blocker. An inset shows EPSC evoked by TS stimulation under the voltage-clamp condition in the same neuron. Dots indicate stimulus artifact. B, The presence of Cd2+ ion in the extracellular bath solution did not alter l-CCG-1-induced hyperpolarization. Note the inset in which TS stimulation failed to evoke EPSC even under the intact condition of AMPA receptors, indicating an inhibition of glutamate release attributable to a Cd2+ ion as a presynaptic Ca2+ channel blocker. C, Group data show that l-CCG-1 application can induce membrane hyperpolarization regardless of glutamate release from presynaptic terminal (p = 0.350).
To further confirm a postsynaptic site of action, we next determined whether the group II mGluR-induced hyperpolarization depended on activation of G-proteins. Blockade of G-protein function by incorporating the nonhydrolyzable GDP analog GDPβS (1 mm) into the pipette solution in which Na-GTP was omitted significantly inhibited the agonist-induced hyperpolarization by 72 ± 6.9% (n = 7 from 4 animals; −11.6 ± 2.4 to −2.9 ± 0.7 mV; p = 0.012) (Fig. 6C, left), indicating a G-protein signaling requirement. As a control, we demonstrated that group II mGluR agonist-induced hyperpolarization was the same when the first pipette and second pipette contained the same normal pipette solution (n = 4 from 4 animals; −8.2 ± 2.3 to −7.7 ± 2.4 mV; p = 0.414) (Fig. 6C, right).
Figure 6.
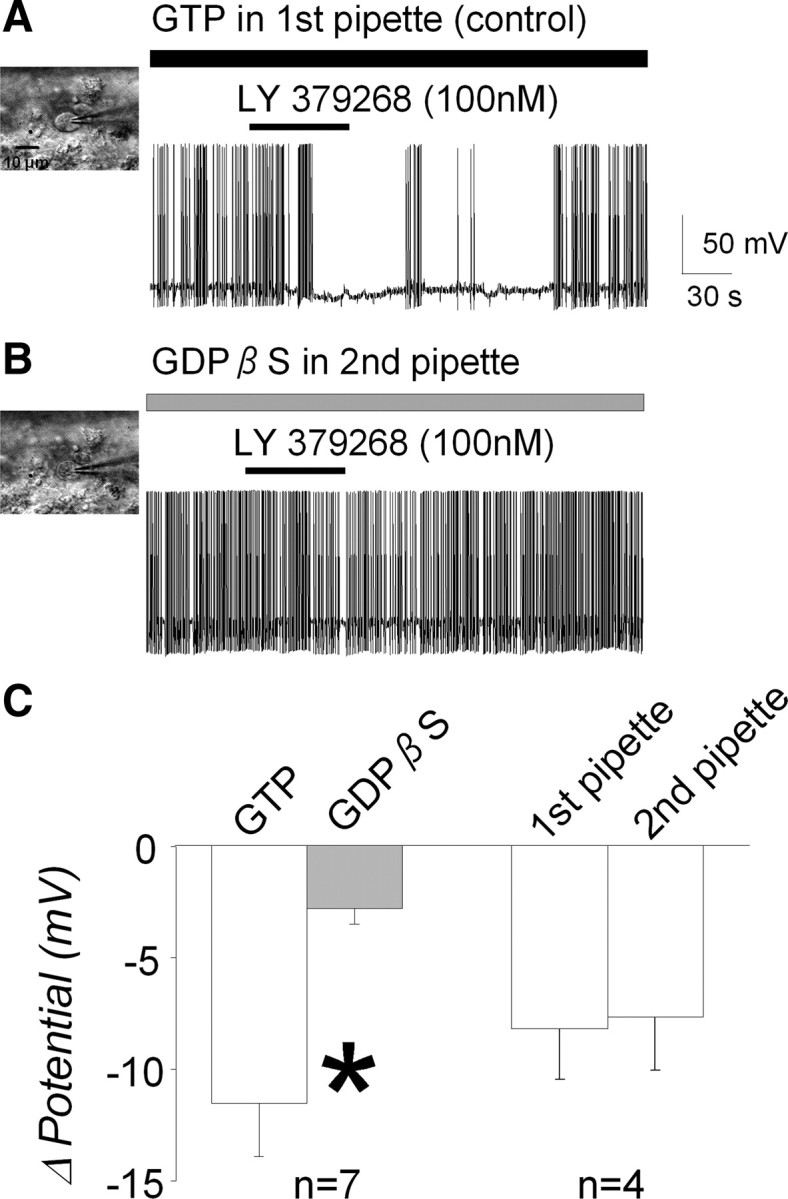
Involvements of G-protein on group II mGluR agonist-induced hyperpolarization. A, As in Figure 2A, LY 379268 induced membrane hyperpolarization with first pipette containing GTP (control). B, Inhibition of LY 379268-induced hyperpolarization by a nonhydrolyzable GDP analog, GDPβS. The recording pipette was changed between LY 379268 applications; in this case, the second pipette contained GDPβS (1 mm) without GTP. The LY 379268-induced hyperpolarization was not observed with intracellular GDPβS. C, Group data show that LY 379268-induced hyperpolarization was significantly inhibited by GDPβS (*p = 0.012), suggesting that the hyperpolarization response is G-protein dependent. Note that group II mGluR agonist-induced hyperpolarization was similar when the first pipette and second pipette contained the same normal pipette solution (p = 0.414).
Immunohistochemical studies
To localize group II mGluRs, immunohistochemical staining was performed on coronal sections from paraformaldehyde-perfused animals and slices from electrophysiological recordings of biocytin-filled neurons. Figure 7 shows an example of mGluR2/3 immunolabeling in the NTS, and a higher-power image verified soma staining in addition to extensive punctate staining (Fig. 7B,C). Postsynaptic localization was also confirmed using electron microscopy (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). To further confirm the presence of postsynaptic group II mGluRs on the recorded neurons, after completing the electrophysiological recordings of the agonist induced-hyperpolarization, we filled the neuron with biocytin, for histological confirmation of mGluR 2/3 staining. Figure 8 shows mGluR2/3 immunostaining, biocytin-filled recorded neurons, and their overlaid images from three second-order baroreceptor neurons in the NTS.
Figure 7.

Group II mGluR labeling in the rat brainstem slice. A, DAB labeling for mGluR2/3 in the medulla oblongata. A negative control is shown in the bottom photograph obtained from the same histochemistry protocol without mGluR2/3 primary antibody. B, DAB labeling for mGluR2/3 in the NTS and its adjacent area. C, High-power image of mGluR2/3 labeling. Arrows indicate soma staining. AP, Area postrema; CC, central canal; DMV, dorsal motor nucleus of vagus.
Figure 8.

Immunohistochemistry for group II mGluRs for recorded neurons. Two example neurons are shown. Green color indicates mGluR2/3 immunostaining (left), and red color shows biocytin injected from patch recording pipette (middle). Orange–yellow color in the overlaid images (right) suggest that mGluR2/3 localize on the second-order baroreceptor neurons, especially on the soma. The arrows point to mGluR2/3 staining just adjacent to biocytin-positive neurites, possibly dendrites because of the size of the processes. Scale bar is applied for all the photographs.
Voltage dependency and ionic components of the group II mGluR-induced currents
To determine the ionic components contributing to the postsynaptic response, we obtained the agonist-induced I–V relationship by using a slow voltage ramp-down protocol from +20 to −120 mV, 2 s duration from the holding potential of −60 mV before and at the peak of the LY379268-induced current (Fig. 9A). The LY 379268-induced I–V relationship was acquired by subtracting the control I–V relationship obtained before LY 379268 application from the I–V relationship obtained at the peak of LY 379268-induced current (Fig. 9B, top traces). The I–V relationship showed two voltage-dependent components: an inward component at depolarizing membrane potentials positive to approximately −40 mV and a second outward component at hyperpolarizing membrane potentials negative to approximately −40 mV (Fig. 9B, bottom trace).
Figure 9.
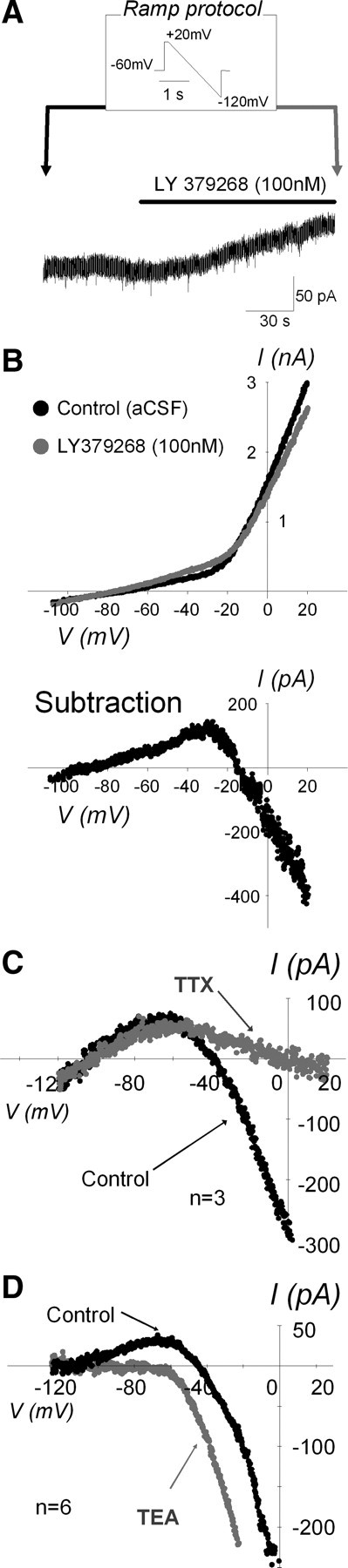
Voltage dependency and ionic components of the group II mGluR-induced current. A, A voltage trace of ramp protocol and an example trace of the LY 379268-induced outward current at membrane voltage clamped at −60 mV. Ramp protocols consisted of slow voltage shift from +20 to −120 mV during 2 s performed before and at the peak of LY 379268-induced response (arrows). B, Two I–V relationships (top graph) were obtained from ramp protocol at arrows in A. Black and gray dots are from before and during LY 379268 application, respectively. Bottom graph is the subtraction of the two I–V relationships, which is equal to LY 379268-induced current–membrane voltage relationship. Note that there are at least two voltage-dependent components, both inward and outward components. C, Averaged LY 379268-induced current–membrane voltage relationship (n = 3) were shown in the absence (black) and presence (gray) of TTX. Error bars on subtraction currents are omitted for clarity. The voltage dependence of the inward currents at potentials positive to −40 mV was greatly diminished by TTX, suggesting that the inward component is most likely attributable to facilitated Na+ channels. D, Averaged LY 379268-induced current–membrane voltage relationship (n = 6) shown in the absence (black) and presence (gray) of TEA. Error bars on subtraction currents are omitted for clarity. The voltage dependence of the outward currents at potentials negative to −40 mV was almost blocked by TEA, suggesting K+ channel contribution to outward component.
To identify the components of the inward and outward currents, first we used TTX (1 μm) to block voltage-sensitive Na+ channels and compared the LY 379268-induced I–V relationship in the absence and presence of TTX. Figure 9C shows the averaged LY 379268-induced I–V relationship (n = 3 from 3 animals) before and during TTX. TTX eliminated the inward component but had no effect on the outward component, suggesting that group II mGluR activation facilitates Na+ channels in a voltage-dependent manner. Next, we used the broad-spectrum K+ channel blocker TEA (10 mm), which has been shown to attenuate IK >80% in NTS neurons (Moak and Kunze, 1993). Figure 8D shows the averaged LY 379268-induced I–V relationship (n = 6 from 5 animals) in the absence and presence of TEA. The outward component was eliminated by TEA, indicating K+ channel contribution to group II mGluR-evoked outward currents. The reversal potential obtained from the LY 379268-induced current–membrane voltage relationship was −101 ± 24 mV (mean ± SD; n = 12), which is almost identical to equilibrium potential for K+ of −102 mV calculated from the Nernst equation.
Group II mGluR activation on postsynaptic intrinsic excitability
To determine whether group II mGluR had additional postsynaptic effects beyond enhancing postsynaptic excitability to glutamate or agonist activation, we measured the spiking responses to depolarizing current injections, delivered as a nonspecific excitatory stimulus, in the absence and presence of the group II mGluR agonist LY 379268 (100 nm) (n = 14 from 6 animals). As shown in Figure 10A, agonist activation of group II mGluR had no effect on the spiking responses to depolarizing current injections, nor on the action potential peak amplitude, width, or threshold (p > 0.05) (Fig. 10B). LY 379268 significantly reduced the input resistance by 19 ± 3.1% (342 ± 50 to 286 ± 50 MΩ; p < 0.001), as shown previously in Figure 2, and had a small 1.3 ± 0.59 mV, albeit statistically significant (p = 0.049), decrease in the AHP (Fig. 10B).
Figure 10.

Effects of group II mGluR activation on intrinsic cell excitability and action potential characteristics. Data are from 14 neurons in the absence and presence of LY 379268 application, which could induce hyperpolarization by 7.3 ± 0.98 mV. A, Intrinsic cell excitability was evaluated by measuring the spiking response to depolarizing current injection up to 120 pA from the holding potential of −50 mV in the absence (blank circles) and presence (filled circles) of the group II mGluR agonist LY 379268 (100 nm). Spiking response was slightly but not significantly attenuated by LY 379268 perfusion (two-way RM-ANOVA: interaction, p = 0.475). B, Action potential (AP) characteristics, peak amplitude, AP threshold, AHP, and AP width, in the absence and presence of LY 379268 application. Although AHP was slightly but significantly attenuated by LY 379268 (*p = 0.049), the rest of the AP characteristics were not altered by LY 379268 application (p > 0.05).
Discussion
This study provides evidence for a novel postsynaptic role for group II mGluRs in modulating baroreceptor signaling at the first central synapse in the NTS, a direct membrane hyperpolarization and reduction in input resistance via resting K+ channel mechanisms involving G-protein signaling. Activation of postsynaptic group II mGluRs with two different agonists, LY 379268 and l-CCG-1, in the presence of ionotropic glutamate and GABAA receptor antagonists evoked a concentration-dependent hyperpolarization that was blocked by the group II mGluR antagonist LY 341495.
Evidence for a postsynaptic mGluR mechanism
The hyperpolarization induced by the group II mGluR agonist LY 379268 remained intact, when Cd2+ was added to the extracellular solution at a concentration that blocked presynaptic glutamate release, as evidenced by abolition of the TS-evoked EPSCs, suggesting that the response was independent of transmitter release. The agonist-induced hyperpolarization was also abolished by a postsynaptic intervention of intracellular dialysis of the recording pipette with the GTP analog GDPβS. Voltage-clamp recordings indicated that the hyperpolarization was mediated by TEA-sensitive outward K+ that remained intact when synaptic transmission was blocked by TTX. The electrophysiological findings were further buttressed by the presence of group II mGluR immunoreactivity on the soma and adjacent to neurites of second-order baroreceptor neurons confirmed by both fluorescent confocal and light microscopy.
Activation of postsynaptic mGluRs by endogenous glutamate
The physiological relevance of activation of postsynaptic group II mGluRs in baroreceptor signal transmission will depend on the extent to which the receptors can be activated by endogenous glutamate. We generated glutamate release by electrically stimulating the TS at a frequency (12 Hz) to approximate a physiologically relevant frequency of baroreceptor afferent fiber firing. The frequency was based on data in rats showing that nonmyelinated aortic baroreceptor afferent discharge with a resting frequency of ∼2–5 Hz and a maximal frequency of 20–23 Hz and that the thinly myelinated Aδ fibers discharge with a mean frequency of ∼28–34 Hz and maximal frequency of 65–90 Hz (Thoren and Jones, 1977; Thoren et al., 1999). In the whole animal, ADN stimulation at twice the present frequency used (25 Hz) results in an ∼20 mmHg decrease in blood pressure, which is ∼66% of the maximal ADN stimulation-induced decrease in blood pressure (De Paula et al., 1999; Kobayashi et al., 1999). In the present study, TS stimulation evoked a hyperpolarization, mimicking the effect of the group II mGluR agonists. The same concentration of the group II mGluR antagonist LY 341495 prevented both TS- and agonist-induced hyperpolarizations, suggesting that endogenous glutamate can activate postsynaptic group II mGluRs to decrease the excitability of second-order baroreceptor neurons. Although activation of metabotropic receptors in general, including mGluRs, modestly modulates synaptic excitability, even small changes in excitability in the NTS are important in shaping baroreceptor signal transmission. In a previous study, in which we simultaneously recorded second-order NTS baroreceptor neuronal and sympathetic nerve activity in vivo, the data predicted that a 10% decrease in the NTS output (spike frequency) of the baroreceptor signal could lead to a 20% reduction in sympathoinhibition (Liu et al., 2000).
An inevitable limitation of TS stimulation is that it does not exclusively activate baroreceptor afferent fibers. So, the release of glutamate from other sensory afferent fibers in addition to the baroreceptor fibers undoubtedly contributed to the endogenous glutamate activation of the presynaptic mGluRs on the GABA terminals. However, multiple visceral afferent fibers of different modalities converge onto NTS neurons (Felder and Mifflin, 1988; Mifflin et al., 1988; Mifflin, 1993; Hines et al., 1994; Toney and Mifflin, 1994; Takagi et al., 1995; Mifflin, 1996; Silva-Carvalho et al., 1998), so it seems likely that, in the whole animal, endogenous glutamate could also be released from baroreceptor afferent fibers either alone or in combination with glutamate released from other peripheral afferent fiber to bind to the mGluRs.
Voltage-dependent ionic currents
The group II mGluR agonist-induced hyperpolarization was associated with voltage-dependent outward currents. At membrane potentials close to the resting potential of the neurons, the I–V relationship showed TEA-sensitive outward currents that reversed close to the equilibrium potential of K+ (−102 mV). These findings suggest that, at resting membrane potential, K+ channels contribute to the hyperpolarization. In the present study, hyperpolarization response was associated with an increase in membrane conductance, suggesting an increase in the leak current as found in thalamic reticular neurons (Cox and Sherman, 1999). At more positive membrane potentials (positive to −40 mV), the I–V relationship showed TTX-sensitive inward currents that were blocked by the voltage-sensitive Na+ channel blocker TTX, suggesting that group II mGluRs could facilitate voltage-dependent Na+ currents, when the membrane potential is depolarized. The relatively few studies on postsynaptic mGluR mechanisms generally report a hyperpolarization via G-protein-coupled K currents, including inwardly rectifying K+ channels (GIRKs), in the neocortex (Lee and Sherman, 2009), bed nucleus of the stria terminalis and basolateral amygdala (Holmes et al., 1996a; Muly et al., 2007), thalamus (Cox and Sherman, 1999), and cerebellum (Knoflach and Kemp, 1998; Watanabe and Nakanishi, 2003). Interestingly, the study by Muly et al. (2007) in the anxiety circuitry of the bed nucleus of the stria terminalis and basolateral amygdala reported the combined presynaptic and postsynaptic inhibitory effects of postsynaptic group II mGluRs that have been observed in the NTS at the baroreceptor synapses.
In contrast to the hyperpolarization effects of membrane potential, activation of group II mGluRs had no physiologically relevant effect on the ability of action potential repetitive firings of the baroreceptor neurons, as measured by spiking responses to depolarizing current injections and action potential characteristics when the cells were held at a resting potential of −50 mV. These results further corroborate the findings that, around the resting membrane potential, outward K currents are dominant compared with the inward TTX-sensitive Na currents present at more depolarized potentials.
This postsynaptic inhibitory effect of group II mGluRs adds one more mechanism for regulating baroreceptor signal transmission at the first central synapses in the central network. The synaptic organization of presynaptic and postsynaptic group II mGluRs at this pivotal site in the baroreflex network provides an intricate assembly of mechanisms for glutamate to shape baroreceptor signaling by modulating both glutamate and GABA transmission early in the circuitry through reduction of excitatory baroreceptor inputs, direct postsynaptic inhibition of the second-order baroreceptor neurons, and a counterbalancing effect of decreasing GABA release. A comparison of the EC50 values for the postsynaptic inhibitory effect of l-CCG-1 (1–3 μm) on glutamatergic transmission, the presynaptic inhibitory effect (21 μm) on glutamate release and the presynaptic inhibition of GABA release (16 μm) at these second-order neurons suggests that postsynaptic mGluRs may be more effective to inhibit signal transmission if endogenous glutamate can equally access receptors at both presynaptic and postsynaptic loci.
The question is how are these multiple effects integrated to shape the NTS output of the baroreceptor signal? At the very least, depending on other synaptic inputs or intrinsic excitability of the neurons, the excitability could be enhanced (by greater presynaptic inhibition of GABA release or increased excitability of the postsynaptic neuron by group I mGluRs) or blunted (by greater presynaptic inhibition of glutamate release by group II or III mGluRs or direct hyperpolarization of the second-order neurons by group II mGluRs). The opposing mechanisms might also provide a means of enhancing the storage capacity of the network and preventing neurons from becoming overactive and energy depleted (Vogt and Nicoll, 1999; Paton et al., 2001). Finally, understanding the full scope of mechanisms whereby mGluRs can regulate baroreceptor signaling may help to explain some of the disparate results observed in microinjection studies on blood pressure and baroreceptor function in whole animals (Foley et al., 1998; Matsumura et al., 1999; Viard and Sapru, 2002; Antunes and Machado, 2003; Simms et al., 2006).
Specificity of group II mGluR agonist
In the current study, we used two agonists to demonstrate the postsynaptic effects. Here, we showed that relatively low concentrations of l-CCG-1 can hyperpolarize neurons; however, higher concentrations of the agonist can elicit small depolarizations (Hayashi et al., 1992; Holmes et al., 1996b). We previously found that l-CCG-1 (20 μm) can slightly depolarize the membrane potential (Sekizawa and Bonham, 2006), an effect that we have since shown was mediated by group I mGluRs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Similar group I mGluR-like responses induced by l-CCG-1 (≥50 μm) have been observed in hippocampal CA1 pyramidal neurons (Davies et al., 1995). In additional studies, we also clarified that the l-CCG-1-induced-depolarization effect was limited to a subset of NTS neurons, with a specific firing type (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and that may specifically project to neurons in the caudal ventrolateral medulla (Austgen et al., 2009). Lower concentrations of l-CCG-1 (10 μm or less) consistently hyperpolarized the membrane potential of second-order baroreceptor neurons regardless of firing phenotypes.
In conclusion, the findings provide evidence for a new postsynaptic role for group II mGluRs in modulating baroreceptor signaling, which adds increasing flexibility for modulating transmission of blood pressure-related information at the first central synapse in the baroreflex network.
Footnotes
This study was supported by National Heart, Lung, and Blood Institute Grant HL-60560. We gratefully acknowledge Matthew P. Hoffman for technical help and Grete Adamson for technical help and useful suggestion.
References
- Antunes VR, Machado BH. Antagonism of glutamatergic metabotropic receptors in the NTS of awake rats does not affect the gain of the baroreflex. Auton Neurosci. 2003;103:65–71. doi: 10.1016/s1566-0702(02)00234-5. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Austgen JR, Fong AY, Foley CM, Mueller PJ, Kline DD, Heesch CM, Hasser EM. Expression of group I metabotropic glutamate receptors on phenotypically different cells within the nucleus of the solitary tract in the rat. Neuroscience. 2009;159:701–716. doi: 10.1016/j.neuroscience.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylwin ML, Horowitz JM, Bonham AC. NMDA receptors contribute to primary visceral afferent transmission in the nucleus of the solitary tract. J Neurophysiol. 1997;77:2539–2548. doi: 10.1152/jn.1997.77.5.2539. [DOI] [PubMed] [Google Scholar]
- Chen CY, Bonham AC. Glutamate suppresses GABA release via presynaptic metabotropic glutamate receptors at baroreceptor neurones in rats. J Physiol. 2005;562:535–551. doi: 10.1113/jphysiol.2004.076885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Ling Eh EH, Horowitz JM, Bonham AC. Synaptic transmission in nucleus tractus solitarius is depressed by group II and III but not group I presynaptic metabotropic glutamate receptors in rats. J Physiol. 2002;538:773–786. doi: 10.1113/jphysiol.2001.012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Sherman SM. Glutamate inhibits thalamic reticular neurons. J Neurosci. 1999;19:6694–6699. doi: 10.1523/JNEUROSCI.19-15-06694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Clarke VR, Jane DE, Collingridge GL. Pharmacology of postsynaptic metabotropic glutamate receptors in rat hippocampal CA1 pyramidal neurones. Br J Pharmacol. 1995;116:1859–1869. doi: 10.1111/j.1476-5381.1995.tb16674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paula PM, Castania JA, Bonagamba LG, Salgado HC, Machado BH. Hemodynamic responses to electrical stimulation of the aortic depressor nerve in awake rats. Am J Physiol. 1999;277:R31–R38. doi: 10.1152/ajpregu.1999.277.1.R31. [DOI] [PubMed] [Google Scholar]
- Felder RB, Mifflin SW. Modulation of carotid sinus afferent input to nucleus tractus solitarius by parabrachial nucleus stimulation. Circ Res. 1988;63:35–49. doi: 10.1161/01.res.63.1.35. [DOI] [PubMed] [Google Scholar]
- Foley CM, Moffitt JA, Hay M, Hasser EM. Glutamate in the nucleus of the solitary tract activates both ionotropic and metabotropic glutamate receptors. Am J Physiol. 1998;275:R1858–R1866. doi: 10.1152/ajpregu.1998.275.6.R1858. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Tanabe Y, Aramori I, Masu M, Shimamoto K, Ohfune Y, Nakanishi S. Agonist analysis of 2-(carboxycyclopropyl)glycine isomers for cloned metabotropic glutamate receptor subtypes expressed in Chinese hamster ovary cells. Br J Pharmacol. 1992;107:539–543. doi: 10.1111/j.1476-5381.1992.tb12780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines T, Toney GM, Mifflin SW. Responses of neurons in the nucleus tractus solitarius to stimulation of heart and lung receptors in the rat. Circ Res. 1994;74:1188–1196. doi: 10.1161/01.res.74.6.1188. [DOI] [PubMed] [Google Scholar]
- Holmes KH, Keele NB, Shinnick-Gallagher P. Loss of mGluR-mediated hyperpolarizations and increase in mGluR depolarizations in basolateral amygdala neurons in kindling-induced epilepsy. J Neurophysiol. 1996a;76:2808–2812. doi: 10.1152/jn.1996.76.4.2808. [DOI] [PubMed] [Google Scholar]
- Holmes KH, Keele NB, Arvanov VL, Shinnick-Gallagher P. Metabotropic glutamate receptor agonist-induced hyperpolarizations in rat basolateral amygdala neurons: receptor characterization and ion channels. J Neurophysiol. 1996b;76:3059–3069. doi: 10.1152/jn.1996.76.5.3059. [DOI] [PubMed] [Google Scholar]
- Knoflach F, Kemp JA. Metabotropic glutamate group II receptors activate a G protein-coupled inwardly rectifying K+ current in neurones of the rat cerebellum. J Physiol. 1998;509:347–354. doi: 10.1111/j.1469-7793.1998.347bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Cheng ZB, Tanaka K, Nosaka S. Is the aortic depressor nerve involved in arterial chemoreflexes in rats? J Auton Nerv Syst. 1999;78:38–48. doi: 10.1016/s0165-1838(99)00054-5. [DOI] [PubMed] [Google Scholar]
- Lee CC, Sherman SM. Glutamatergic inhibition in sensory neocortex. Cereb Cortex. 2009. Advance online publication. Retrieved Jan. 28, 2009. [DOI] [PMC free article] [PubMed]
- Liu Z, Chen CY, Bonham AC. Metabotropic glutamate receptors depress vagal and aortic baroreceptor signal transmission in the NTS. Am J Physiol. 1998;275:H1682–H1694. doi: 10.1152/ajpheart.1998.275.5.H1682. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chen CY, Bonham AC. Frequency limits on aortic baroreceptor input to nucleus tractus solitarii. Am J Physiol Heart Circ Physiol. 2000;278:H577–H585. doi: 10.1152/ajpheart.2000.278.2.H577. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Tsuchihashi T, Kagiyama S, Abe I, Fujishima M. Subtypes of metabotropic glutamate receptors in the nucleus of the solitary tract of rats. Brain Res. 1999;842:461–468. doi: 10.1016/s0006-8993(99)01889-2. [DOI] [PubMed] [Google Scholar]
- Mifflin SW. Inhibition of chemoreceptor inputs to nucleus of tractus solitarius neurons during baroreceptor stimulation. Am J Physiol. 1993;265:R14–R20. doi: 10.1152/ajpregu.1993.265.1.R14. [DOI] [PubMed] [Google Scholar]
- Mifflin SW. Convergent carotid sinus nerve and superior laryngeal nerve afferent inputs to neurons in the NTS. Am J Physiol. 1996;271:R870–R880. doi: 10.1152/ajpregu.1996.271.4.R870. [DOI] [PubMed] [Google Scholar]
- Mifflin SW, Spyer KM, Withington-Wray DJ. Baroreceptor inputs to the nucleus tractus solitarius in the cat: postsynaptic actions and the influence of respiration. J Physiol. 1988;399:349–367. doi: 10.1113/jphysiol.1988.sp017085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moak JP, Kunze DL. Potassium currents of neurons isolated from medical nucleus tractus solitarius. Am J Physiol. 1993;265:H1596–H1602. doi: 10.1152/ajpheart.1993.265.5.H1596. [DOI] [PubMed] [Google Scholar]
- Muly EC, Mania I, Guo JD, Rainnie DG. Group II metabotropic glutamate receptors in anxiety circuitry: correspondence of physiological response and subcellular distribution. J Comp Neurol. 2007;505:682–700. doi: 10.1002/cne.21525. [DOI] [PubMed] [Google Scholar]
- Nakajima S, Onodera K. Membrane properties of the stretch receptor neurones of crayfish with particular reference to mechanisms of sensory adaptation. J Physiol. 1969;200:161–185. doi: 10.1113/jphysiol.1969.sp008687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H, Talman WT. Both NMDA and non-NMDA receptors in the NTS participate in the baroreceptor reflex in rats. Am J Physiol. 1994;267:R1065–R1070. doi: 10.1152/ajpregu.1994.267.4.R1065. [DOI] [PubMed] [Google Scholar]
- Paton JF, Li YW, Schwaber JS. Response properties of baroreceptive NTS neurons. Ann N Y Acad Sci. 2001;940:157–168. doi: 10.1111/j.1749-6632.2001.tb03674.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Academic: San Diego; 1986. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Sekizawa S, Bonham AC. Group I metabotropic glutamate receptors on second-order baroreceptor neurons are tonically activated and induce a Na+-Ca2+ exchange current. J Neurophysiol. 2006;95:882–892. doi: 10.1152/jn.00772.2005. [DOI] [PubMed] [Google Scholar]
- Sekizawa SI, French AS, Torkkeli PH. Low-voltage-activated calcium current does not regulate the firing behavior in paired mechanosensory neurons with different adaptation properties. J Neurophysiol. 2000;83:746–753. doi: 10.1152/jn.2000.83.2.746. [DOI] [PubMed] [Google Scholar]
- Silva-Carvalho L, Paton JF, Rocha I, Goldsmith GE, Spyer KM. Convergence properties of solitary tract neurons responsive to cardiac receptor stimulation in the anesthetized cat. J Neurophysiol. 1998;79:2374–2382. doi: 10.1152/jn.1998.79.5.2374. [DOI] [PubMed] [Google Scholar]
- Simms AE, Paton JF, Pickering AE. Disinhibition of the cardiac limb of the arterial baroreflex in rat: a role for metabotropic glutamate receptors in the nucleus tractus solitarii. J Physiol. 2006;575:727–738. doi: 10.1113/jphysiol.2006.112672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi S, Umezaki T, Shin T. Convergence of laryngeal afferents with different natures upon cat NTS neurons. Brain Res Bull. 1995;38:261–268. doi: 10.1016/0361-9230(95)00098-y. [DOI] [PubMed] [Google Scholar]
- Talman WT. Kynurenic acid microinjected into the nucleus tractus solitarius of rat blocks the arterial baroreflex but not responses to glutamate. Neurosci Lett. 1989;102:247–252. doi: 10.1016/0304-3940(89)90086-4. [DOI] [PubMed] [Google Scholar]
- Thoren P, Jones JV. Characteristics of aortic baroreceptor C-fibres in the rabbit. Acta Physiol Scand. 1977;99:448–456. doi: 10.1111/j.1748-1716.1977.tb10397.x. [DOI] [PubMed] [Google Scholar]
- Thoren P, Munch PA, Brown AM. Mechanisms for activation of aortic baroreceptor C-fibres in rabbits and rats. Acta Physiol Scand. 1999;166:167–174. doi: 10.1046/j.1365-201x.1999.00556.x. [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Wieling W, Karemaker JM, Lenders JW. Baroreflex failure: a neglected type of secondary hypertension. Neth J Med. 2004;62:151–155. [PubMed] [Google Scholar]
- Toney GM, Mifflin SW. Time-dependent inhibition of hindlimb somatic afferent inputs to nucleus tractus solitarius. J Neurophysiol. 1994;72:63–71. doi: 10.1152/jn.1994.72.1.63. [DOI] [PubMed] [Google Scholar]
- Viard E, Sapru HN. Cardiovascular responses to activation of metabotropic glutamate receptors in the nTS of the rat. Brain Res. 2002;952:308–321. doi: 10.1016/s0006-8993(02)03260-2. [DOI] [PubMed] [Google Scholar]
- Vogt KE, Nicoll RA. Glutamate and gamma-aminobutyric acid mediate a heterosynaptic depression at mossy fiber synapses in the hippocampus. Proc Natl Acad Sci U S A. 1999;96:1118–1122. doi: 10.1073/pnas.96.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe D, Nakanishi S. mGluR2 postsynaptically senses granule cell inputs at Golgi cell synapses. Neuron. 2003;39:821–829. doi: 10.1016/s0896-6273(03)00530-0. [DOI] [PubMed] [Google Scholar]