

Abstract
Highly enantio- and diastereoselective methods for the synthesis of a variety of cyclopropyl alcohols are reported. These methods represent the first one-pot approaches to syn-vinyl cyclopropyl alcohols, syn-cis-disubstituted cyclopropyl alcohols, and anti-cyclopropyl alcohols from achiral precursors. The methods begin with enantioselective C–C bond formations promoted by a MIB-based zinc catalyst to generate allylic alkoxide intermediates. The intermediates are then subjected to in situ alkoxide-directed cyclopropanation to provide cyclopropyl alcohols. In the synthesis of vinyl cyclopropyl alcohols, hydroboration of enynes is followed by transmetalation of the resulting dienylborane to zinc to provide dienylzinc reagents. Enantioselective addition to aldehydes generates the requisite dienyl zinc alkoxides, which are then subjected to in situ cyclopropanation to furnish vinyl cyclopropyl alcohols. Cyclopropanation occurs at the double bond allylic to the alkoxide. Using this method, syn-vinylcyclopropyl alcohols are obtained in 65–85% yield, 76–93% ee, and >19:1 dr. To prepare anti-cyclopropanols, enantioselective addition of alkylzinc reagents to conjugated enals provides allylic zinc alkoxides. Because direct cyclopropanation provides syn-cyclopropyl alcohols, the intermediate allylic alkoxides were treated with TMSCl/Et3N to generate intermediate silyl ethers. In situ cyclopropanation of the allylic silyl ether resulted in cyclopropanation to form the anti-cyclopropyl silyl ether. Workup with TBAF affords the anti-cyclopropyl alcohols in one-pot in 60–82% yield, 89–99% ee, and ≥10:1 dr. For the synthesis of cis-disubstituted cyclopropyl alcohols, in situ generated (Z)-vinyl zinc reagents were employed in asymmetric addition to aldehydes to generate (Z)-allylic zinc alkoxides. In situ cyclopropanation provides syn-cis-disubstituted cyclopropyl alcohols in 42–70% yield, 88–97% ee, and >19:1 dr. These one-pot procedures enable the synthesis of a diverse array of cyclopropyl alcohol building blocks with high enantio- and diastereoselectivities.
1. Introduction
Cyclopropane containing compounds exhibit a broad spectrum of biological properties1-5 and are present in over 100 therapeutic agents.5-7 They are commonly encountered in natural products, including pheromones, steroids, terpenes, fatty acid metabolites and amino acids.4,5 These strained structural motifs are also valuable building blocks in organic chemistry3,8-12 that can be elaborated to provide functionalized cyclopropanes or ring-opened products.10,13-15 The synthetic utility and medicinal properties of enantioenriched cyclopropanes have inspired many investigations into their preparation, which generally follow one of three approaches.5,11,16-18 Tremendous advances have recently been documented in enantioselective organocatalytic approaches to cyclopropane synthesis,5,19-23 many of which proceed via Michael addition-initiated ring-closure sequences.5,19,24-26 Likewise, impressive progress has been made in the area of metal-catalyzed cyclopropanation of olefins with diazoesters and related precursors to afford enantio- and diastereoenriched cyclopropanes.5,27-29 The third pillar in cyclopropane synthesis is halomethylmetal-mediated cyclopropanation reactions (the Simmons–Smith reaction).12,17,30-32 In contrast to organocatalytic and diazo-based approached to cyclopropanes, progress in the catalytic asymmetric Simmons–Smith cyclopropanation of olefins33,34 and allylic alcohols11,35-41 has been limited. Catalysts for the Simmons-Smith cyclopropanation have been beset by moderate enantioselectivities, high catalyst loadings (typically 10–25 mol%)11,42 or mediocre substrate generality.43 While some exceptions are emerging,43 these problems underscore the complexity of asymmetric Simmons-Smith cyclopropanations.42
Considering the well documented difficulties in catalytic asymmetric Simmons–Smith reactions, as well as our own experiences,41 we pondered alternative approaches for the stereoselective synthesis of cyclopropanes. Rather than direct enantioselective cyclopropanation, we chose to perform a tandem reaction involving an asymmetric addition to an aldehyde as the first step. The key intermediate in our approach was envisioned to be an enantioenriched allylic zinc alkoxide that could be subjected to a diastereoselective directed cyclopropanation (Scheme 1). To generate enantioenriched allylic zinc alkoxides, two complementary carbonyl additions were studied. The first involved an asymmetric addition of alkylzinc reagents to enals followed by diastereoselective cyclopropanation to afford cyclopropyl alcohols (Scheme 1A).44 A complementary C–C bond-formation was chosen for the second approach, which entailed an enantioselective vinyl addition to a saturated aldehyde followed by cyclopropanation (Scheme 1B).44
Scheme 1.

Tandem Approaches to Enantio- and Diastereoenriched Cyclopropyl Alcohols
Nugent's amino alcohol (−)-MIB45,46 (4 mol %) was employed to generate the catalyst for both additions (Scheme 1). With one exception enantioselectivities in the carbonyl addition step exceeded 90% and the cyclopropanation proceeded with excellent diastereoselectivity (dr's >20:1). Although diastereoselective cyclopropanations of chiral allylic alcohols had been studied,47-49 these were the first examples of the assembly of enantio- and diastereoenriched cyclopropyl alcohols from achiral precursors in a one-pot procedure.44 A modified protocol based on these methods was subsequently developed for the highly enantio- and diastereoselective iodo-, bromo-, and chlorocyclopropanations of allylic zinc alkoxides to generate halocyclopropyl alcohols with up to four stereocenters.44,50
The methods illustrated in Scheme 1 enable the efficient synthesis of a variety of simple cyclopropyl alcohols with syn relationships between the hydroxyl and cyclopropane groups. In their current form, however, these methods cannot be used to access the diastereomeric anti-cyclopropyl alcohols. Both syn- and anti-cyclopropyl alcohols are encountered in natural products, as exemplified by members of the oxylipin family,51,52 which includes constanolactone A and B, solandelactones E and F, halicholactone and eicosanoid (Figure 1). Furthermore, the asymmetric vinylation method outlined in Scheme 1B provides access to only the (E)-allylic alkoxide intermediates and is not applicable to the synthesis of the (Z)-analogs. Thus, cis-disubstituted cyclopropanes are not accessible using the procedures in Scheme 1B. Finally, the utility of the method in Scheme 1B would be greatly broadened if it could be adapted to the synthesis of vinyl cyclopropanes (VCP's). VCP's are useful synthetic intermediates that undergo a variety of transformations.18,53-55 Their synthesis in highly enantio- and diastereoenriched form, however, is not trivial.
Figure 1.

Structures of members of the oxylipin natural products, which contains both syn and anti cyclopropyl alcohol motifs
Herein we introduce highly enantio- and diastereoselective methods to prepare vinyl cyclopropyl alcohols, anti-cyclopropyl alcohols, and cyclopropyl alcohols with cis-disubstituted cyclopropane motifs.
2. Experimental Section
Representative procedures and characterization of the products are described herein. Full experimental details and characterization of all compounds are provided in the Supporting Information.
General Methods
All reactions were carried out under a nitrogen atmosphere with oven-dried glassware. The progress of reactions was monitored by thin-layer chromatography on Whatman precoated silica gel 60 F-254 plates and visualized by ultraviolet light or by staining with ceric ammonium molybdate stain. All manipulations involving dialkylzinc reagents were carried out under an inert atmosphere in a Vacuum Atmosphere drybox with an attached MO-40 Dritrain or by using standard Schlenk or vacuum line techniques. Unless otherwise specified, all chemicals were obtained from Aldrich, Acros, or GFS chemicals, and solvents were purchased from Fischer Scientific. Dialkylzinc compounds, except dimethyl- and diethylzinc, which are commercially available, were prepared by literature methods.56,57 Dichloromethane and hexanes were dried through alumina columns. All aldehydes were distilled prior to use and stored under N2. The 1H and 13C{1H} NMR spectra were obtained on Bruker 500 or 300 MHz Fourier transform spectrometers at the University of Pennsylvania NMR facility. Chemical shifts are reported in units of parts per million downfield from tetramethylsilane, and all coupling constants are reported in hertz. The infrared spectra were obtained using a Perkin-Elmer 1600 series spectrometer. Mass spectra were recorded on a Waters LCTOF- Xe Premier Mass spectrometer. Silica gel (230–400 mesh, Silicycle) was used for air-flashed chromatography. Deactivated silica gel was made by combining silica gel with 2.5 wt % NEt3.
Cautionary Note
Dialkylzinc reagents, tert-butyllithium, and diethylborane are pyrophoric. Extreme caution should be used in their handling and proper laboratory attire is highly recommended.
General Procedure A
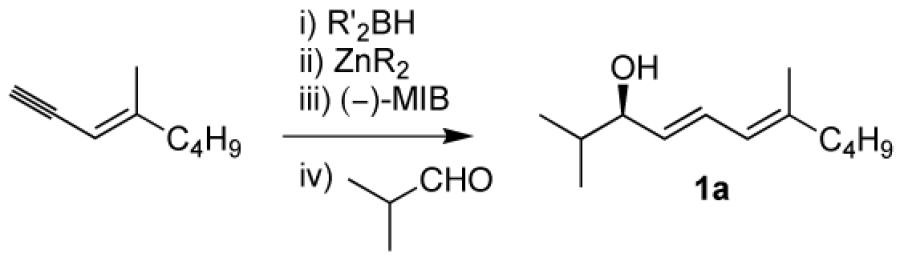
(4E,6E)-2,7-Dimethylundeca-4,6-dien-3-ol (1a)
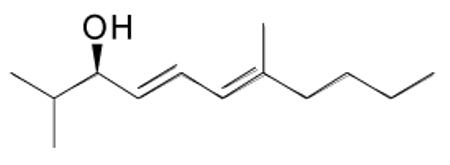 (E)-4-Methyloct-3-en-1-yne (80 mg, 0.65 mmol) and diethylborane (0.65 mL, 0.65 mmol, 1.0 M in toluene) were added to a dry flask under nitrogen and stirred at room temperature for 30 min. The reaction flask was then cooled to −78 °C and (−)-MIB (11.75 mg, 0.05 mmol, 10 mol %) was added followed by Et2Zn (0.75 mL, 1.0 M in hexanes, 0.75 mmol). The reaction mixture was then warmed to −10 °C and a solution of isobutyraldehyde (45 μL, 0.5 mmol in 3 mL hexanes) was added dropwise for 20 min. The reaction was stirred at −10 °C for 10 h until vinyl addition was complete by TLC and quenched with a saturated solution of NH4Cl (10 mL). The organic and aqueous layers were separated, and the aqueous layer was extracted with 3×15 mL dichloromethane. The combined organic layers were then washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on silica (5% ethyl acetate in hexanes) to afford the title compound as a colorless oil in 88% yield. [α]D20 = −5.3 (c = 0.5, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 6.44 (dd, 1H, J = 15.3, 11.0 Hz), 5.85 (d, 1H, J = 11.0 Hz), 5.59 (dd, 1H, J = 15.3, 7.5 Hz), 3.90 (dd, 1H, J = 7.5, 6.6 Hz), 2.07 (m, 2H), 1.90 (br s, 1H), 1.77 (m, 3H), 1.72 (m, 1H), 1.37 (m, 4H), 0.96 (d, 3H, J = 6.6 Hz), 0.92 (t, 3H, J = 7.1 Hz), 0.91 (d, 3H, J = 6.6 Hz); 13C NMR (CDCl3, 75 MHz,): δ 140.3, 132.1, 128.7, 124.2, 78.7, 40.0, 34.4, 30.4, 22.8, 18.7, 18.5, 17.0, 14.4; IR (neat): 3383 (OH), 2954, 2850, 1452, 1399, 1288, 1118, 1068, 1020, 987 cm−1; HRMS-CI m/z 178.1720 [(M-H2O)+; calcd for C13H22: 178.1722].
(E)-4-Methyloct-3-en-1-yne (80 mg, 0.65 mmol) and diethylborane (0.65 mL, 0.65 mmol, 1.0 M in toluene) were added to a dry flask under nitrogen and stirred at room temperature for 30 min. The reaction flask was then cooled to −78 °C and (−)-MIB (11.75 mg, 0.05 mmol, 10 mol %) was added followed by Et2Zn (0.75 mL, 1.0 M in hexanes, 0.75 mmol). The reaction mixture was then warmed to −10 °C and a solution of isobutyraldehyde (45 μL, 0.5 mmol in 3 mL hexanes) was added dropwise for 20 min. The reaction was stirred at −10 °C for 10 h until vinyl addition was complete by TLC and quenched with a saturated solution of NH4Cl (10 mL). The organic and aqueous layers were separated, and the aqueous layer was extracted with 3×15 mL dichloromethane. The combined organic layers were then washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on silica (5% ethyl acetate in hexanes) to afford the title compound as a colorless oil in 88% yield. [α]D20 = −5.3 (c = 0.5, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 6.44 (dd, 1H, J = 15.3, 11.0 Hz), 5.85 (d, 1H, J = 11.0 Hz), 5.59 (dd, 1H, J = 15.3, 7.5 Hz), 3.90 (dd, 1H, J = 7.5, 6.6 Hz), 2.07 (m, 2H), 1.90 (br s, 1H), 1.77 (m, 3H), 1.72 (m, 1H), 1.37 (m, 4H), 0.96 (d, 3H, J = 6.6 Hz), 0.92 (t, 3H, J = 7.1 Hz), 0.91 (d, 3H, J = 6.6 Hz); 13C NMR (CDCl3, 75 MHz,): δ 140.3, 132.1, 128.7, 124.2, 78.7, 40.0, 34.4, 30.4, 22.8, 18.7, 18.5, 17.0, 14.4; IR (neat): 3383 (OH), 2954, 2850, 1452, 1399, 1288, 1118, 1068, 1020, 987 cm−1; HRMS-CI m/z 178.1720 [(M-H2O)+; calcd for C13H22: 178.1722].
General Procedure B
2-Methyl-1-(2-((E)-2-methylhex-1-enyl)cyclopropyl)propan-1-ol (1b)
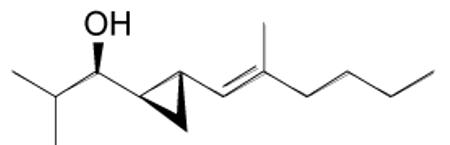 An oven-dried 10 mL Schlenk flask that had been thoroughly purged with N2 was charged with (E)-4-methyloct-3-en-1-yne (80 mg, 0.65 mmol) and diethylborane (0.65 mL, 0.65 mmol, 1.0 M in toluene) and stirred at room temperature for 30 min. After the reaction flask was cooled to −78 °C, (−)-MIB (11.75 mg, 0.05 mmol, 10 mol %) was added, followed by Et2Zn (0.75 mL, 1.0 M in hexanes, 0.75 mmol) and resulting solution stirred at this temperature for 10 min. The reaction mixture was then warmed to −10 °C and a solution of isobutyraldehyde (45 μL, 0.5 mmol in 3 mL hexanes) was added dropwise for 20 min. The reaction mixture was stirred at −10 °C for 10 h until vinyl addition was complete by TLC. The solvent and byproduct Et3B were removed in vacuo at 0 °C and 2 mL of hexanes was added. This step was done three times to remove byproduct Et3B completely. A solution of Et2Zn (1.0 mL, 1.0 M in hexanes, 1.0 mmol) and diiodomethane (81 μL, 1.0 mmol) were added at 0 °C. The reaction was stirred with light exclusion at room temperature for 10 h. A solution of Et2Zn (1.0 mL, 1.0 M in hexanes, 1.0 mmol) and diiodomethane (81 μL, 1.0 mmol) were added at 0 °C and then the reaction mixture was warmed to room temperature. The flask was covered with aluminum foil to exclude light and stirred at room temperature for 20 h. The reaction mixture was then quenched with saturated solution of NH4Cl (15 mL). The organic and aqueous layers were separated and the aqueous layer was extracted with 3×20 mL dichloromethane. The combined organic layers were then washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on silica (5% ethyl acetate in hexanes) to afford the title compound as a colorless oil in 75% yield. [α]D20 = −18.3 (c = 0.4, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 4.56 (d, 1H, J = 9.2 Hz), 2.70 (dd, 1H, J = 9.1, 6.2 Hz), 1.95 (m, 2H), 1.78 (m, 1H), 1.68 (s, 2H), 1.48 (br s, 1H), 1.32 (m, 5H), 1.02 (m, 1H), 0.98 (d, 3H, J = 7.0 Hz), 0.95 (d, 3H, J = 7.0 Hz), 0.88 (t, 3H, J = 7.2 Hz), 0.81 (m, 1H), 0.68 (m, 1H), 0.49 (m, 1H); 13C NMR (CDCl3, 75 MHz):δ 135.3, 126.7, 81.6, 39.6, 35.0, 30.5, 25.5, 22.7, 19.0, 18.9, 17.5, 16.7, 14.4, 11.4; IR (neat): 3400 (OH), 2875, 2778, 1054, 976, 897 cm−1; HRMS-CI m/z 193.1963 [(M-H2O)+; calcd for C14H24: 193.1962].
An oven-dried 10 mL Schlenk flask that had been thoroughly purged with N2 was charged with (E)-4-methyloct-3-en-1-yne (80 mg, 0.65 mmol) and diethylborane (0.65 mL, 0.65 mmol, 1.0 M in toluene) and stirred at room temperature for 30 min. After the reaction flask was cooled to −78 °C, (−)-MIB (11.75 mg, 0.05 mmol, 10 mol %) was added, followed by Et2Zn (0.75 mL, 1.0 M in hexanes, 0.75 mmol) and resulting solution stirred at this temperature for 10 min. The reaction mixture was then warmed to −10 °C and a solution of isobutyraldehyde (45 μL, 0.5 mmol in 3 mL hexanes) was added dropwise for 20 min. The reaction mixture was stirred at −10 °C for 10 h until vinyl addition was complete by TLC. The solvent and byproduct Et3B were removed in vacuo at 0 °C and 2 mL of hexanes was added. This step was done three times to remove byproduct Et3B completely. A solution of Et2Zn (1.0 mL, 1.0 M in hexanes, 1.0 mmol) and diiodomethane (81 μL, 1.0 mmol) were added at 0 °C. The reaction was stirred with light exclusion at room temperature for 10 h. A solution of Et2Zn (1.0 mL, 1.0 M in hexanes, 1.0 mmol) and diiodomethane (81 μL, 1.0 mmol) were added at 0 °C and then the reaction mixture was warmed to room temperature. The flask was covered with aluminum foil to exclude light and stirred at room temperature for 20 h. The reaction mixture was then quenched with saturated solution of NH4Cl (15 mL). The organic and aqueous layers were separated and the aqueous layer was extracted with 3×20 mL dichloromethane. The combined organic layers were then washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on silica (5% ethyl acetate in hexanes) to afford the title compound as a colorless oil in 75% yield. [α]D20 = −18.3 (c = 0.4, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 4.56 (d, 1H, J = 9.2 Hz), 2.70 (dd, 1H, J = 9.1, 6.2 Hz), 1.95 (m, 2H), 1.78 (m, 1H), 1.68 (s, 2H), 1.48 (br s, 1H), 1.32 (m, 5H), 1.02 (m, 1H), 0.98 (d, 3H, J = 7.0 Hz), 0.95 (d, 3H, J = 7.0 Hz), 0.88 (t, 3H, J = 7.2 Hz), 0.81 (m, 1H), 0.68 (m, 1H), 0.49 (m, 1H); 13C NMR (CDCl3, 75 MHz):δ 135.3, 126.7, 81.6, 39.6, 35.0, 30.5, 25.5, 22.7, 19.0, 18.9, 17.5, 16.7, 14.4, 11.4; IR (neat): 3400 (OH), 2875, 2778, 1054, 976, 897 cm−1; HRMS-CI m/z 193.1963 [(M-H2O)+; calcd for C14H24: 193.1962].
General Procedure C
1-(2-Phenylcyclopropyl)propan-1-ol (11)
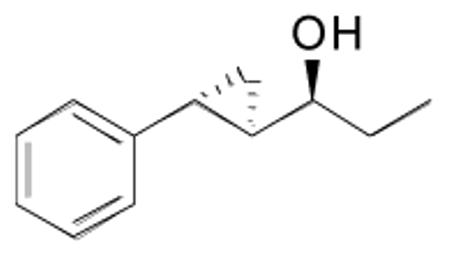 A 10 mL Schlenk flask was charged with (−)-MIB (2.9 mg, 0.012 mmol) and cooled to 0 °C. A solution of Et2Zn (0.45 mL, 1.0 M in hexanes) was added, followed by dropwise addition of trans-cinnamaldehyde (38 μL, 0.3 mmol). The reaction mixture was stirred at 0 °C for 8 h until alkyl addition was complete by TLC. 1.5 Equiv trimethylsilane chloride (0.45 mmol) and 1.5 equiv triethyl amine (0.45 mmol) were added with 2 mL dichloromethane at 0 °C. The reaction flask was slowly warmed to room temperature and stirred for 14 h. Next, 5 equiv of Et2Zn (0.75 mL, 2.0 M in dichloromethane) and 5 equiv CF3CH2OH (108 μL, 1.5 mmol) were added slowly at 0 °C. After stirring at 0 °C for 10 min, 5 equiv CH2I2 (120 μL, 1.5 mmol) was added. The reaction mixture was stirred with light exclusion at room temperature for 24 h. It was then quenched with 3-4 drops of water and 2 equiv TBAF (1M solution in THF) at 0 °C. After stirring for 1 h, 5 mL of saturated NH4Cl solution was added. The organic and aqueous layers were separated and the aqueous layer was extracted three times with 10 mL dichloromethane. The combined organic layers were then washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on deactivated silica (10% ethyl acetate in hexanes) to afford the title compound as a colorless oil in 75% yield. [α]D20 = +12.6 (c = 0.50, CHCl3); 1H NMR (CDCl3, 500 MHz): δ 7.36 (m, 2H), 7.26 (m, 1H), 7.16 (m, 2H), 3.24 (m, 1H), 1.93 (m, 1H), 1.86 (br s, 1H), 1.78 (m, 2H), 1.34 (m, 1H), 1.10 (t, 3H, J = 7.5 Hz), 1.06 (m, 2H); 13C{1H} NMR (CDCl3, 125 MHz,): δ 142.7, 128.6, 126.1, 125.9, 77.2, 30.5, 29.5, 21.4, 13.3, 10.3; IR (neat); 3385 (OH), 3057, 2950, 1459, 1299, 1071, 924, 720 cm−1.
A 10 mL Schlenk flask was charged with (−)-MIB (2.9 mg, 0.012 mmol) and cooled to 0 °C. A solution of Et2Zn (0.45 mL, 1.0 M in hexanes) was added, followed by dropwise addition of trans-cinnamaldehyde (38 μL, 0.3 mmol). The reaction mixture was stirred at 0 °C for 8 h until alkyl addition was complete by TLC. 1.5 Equiv trimethylsilane chloride (0.45 mmol) and 1.5 equiv triethyl amine (0.45 mmol) were added with 2 mL dichloromethane at 0 °C. The reaction flask was slowly warmed to room temperature and stirred for 14 h. Next, 5 equiv of Et2Zn (0.75 mL, 2.0 M in dichloromethane) and 5 equiv CF3CH2OH (108 μL, 1.5 mmol) were added slowly at 0 °C. After stirring at 0 °C for 10 min, 5 equiv CH2I2 (120 μL, 1.5 mmol) was added. The reaction mixture was stirred with light exclusion at room temperature for 24 h. It was then quenched with 3-4 drops of water and 2 equiv TBAF (1M solution in THF) at 0 °C. After stirring for 1 h, 5 mL of saturated NH4Cl solution was added. The organic and aqueous layers were separated and the aqueous layer was extracted three times with 10 mL dichloromethane. The combined organic layers were then washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on deactivated silica (10% ethyl acetate in hexanes) to afford the title compound as a colorless oil in 75% yield. [α]D20 = +12.6 (c = 0.50, CHCl3); 1H NMR (CDCl3, 500 MHz): δ 7.36 (m, 2H), 7.26 (m, 1H), 7.16 (m, 2H), 3.24 (m, 1H), 1.93 (m, 1H), 1.86 (br s, 1H), 1.78 (m, 2H), 1.34 (m, 1H), 1.10 (t, 3H, J = 7.5 Hz), 1.06 (m, 2H); 13C{1H} NMR (CDCl3, 125 MHz,): δ 142.7, 128.6, 126.1, 125.9, 77.2, 30.5, 29.5, 21.4, 13.3, 10.3; IR (neat); 3385 (OH), 3057, 2950, 1459, 1299, 1071, 924, 720 cm−1.
General Procedure D
(Z)-1-(2-(2-(tert-Butyldiphenylsilyloxy)ethyl)cyclopropyl)-2-methylpropan-1-ol (17)
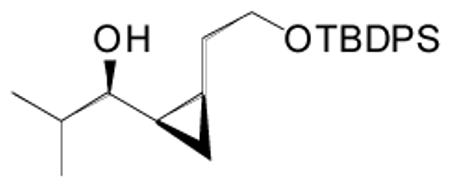 Dicyclohexylborane (88 mg, 0.5 mmol) was weighed into a Schlenk flask under nitrogen and dry t-BuOMe (1 mL) was added. tert-Butyl-(4-chloro-but-3-ynyloxy)-diphenyl-silane (160 μL, 0.5 mmol) was then added slowly to the reaction mixture at 0 °C. After 15 min the reaction mixture was warmed to room temperature and stirred for 45 min resulting in a clear solution. t-BuLi (0.365 mL, 0.55 mmol, 1.5 M solution in pentane) was added dropwise at −78 °C and the reaction mixture stirred for 60 min. The solution was warmed to room temperature and stirred for an additional 60 min during which time a precipitate formed. Diethylzinc (0.275 mL, 0.55 mmol, 2 M solution in hexanes) was slowly added to the reaction mixture at −78 °C and stirred for 20 min. Addition of TEEDA (14 μL, 0.066 mmol) and hexanes (4 mL) was performed at −78 °C. The solution was warmed to 0 °C and (−)-MIB (166 μL, 0.017 mmol) and isobutyraldehyde (30 μL, 0.332 mmol) were added. The reaction mixture was then slowly warmed to room temperature and stirred 12–16 h. After the reaction was complete by TLC analysis, the temperature was lowered to 0 °C and ZnEt2 (0.83 mL, 1.66 mmol, 2 M solution in hexanes) was added. Next, CF3CH2OH (120 μL, 1.65 mmol) was added dropwise. After stirring at 0 °C for 10 min, CH2I2 (135 μL, 1.67 mmol) was added. The reaction mixture was stirred with light exclusion at room temperature for 24 h. It was then quenched with saturated solution of NH4Cl. The organic and aqueous layers were separated and the aqueous layer was extracted with dichloromethane (3×5 mL). The combined organic layers were then washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and the crude product was purified by column chromatography on deactivated silica gel (5% ethyl acetate in hexanes) to afford the title compound (92.1 mg, 70% yield) as an oil. (c = 0.026, CHCl3); 1H NMR (CDCl3, 500 MHz): δ 0.05 (m, 1H), 0.67 (m, 1H), 0.93 (m, 2H), 0.98 (t, J = 7.9 Hz, 6H), 1.1 (m, 9H), 1.23 (m, 1H), 1.3 (d, J = 3.6 Hz, 1H), 1.74 (m, 1H), 1.91 (m, 1H), 2.96 (m, 1H), 3.76 (m, 2H), 7.42 (m, 6H), 7.7 (m, 4H); 13C{1H} NMR (CDCl3, 125 MHz): δ 8.9, 14.8, 17.6, 19.2, 19.4, 21.1, 27.1, 32.8, 34.7, 64.5, 76.7, 127.8, 129.8, 134.2, 135.8; IR (neat): 3599, 3411, 3134, 3070, 3050, 3013, 2952, 2912, 2895, 2858, 2739, 2319, 1958, 1888, 1823, 1589, 1486, 1471, 1428, 1362, 1331, 1306, 1260, 1235, 1187, 1157, 1110 1029, 1007 cm−1; HRMS calcd for C25H36O2NaSi (M+Na)+: 419.2382, found 419.2377.
Dicyclohexylborane (88 mg, 0.5 mmol) was weighed into a Schlenk flask under nitrogen and dry t-BuOMe (1 mL) was added. tert-Butyl-(4-chloro-but-3-ynyloxy)-diphenyl-silane (160 μL, 0.5 mmol) was then added slowly to the reaction mixture at 0 °C. After 15 min the reaction mixture was warmed to room temperature and stirred for 45 min resulting in a clear solution. t-BuLi (0.365 mL, 0.55 mmol, 1.5 M solution in pentane) was added dropwise at −78 °C and the reaction mixture stirred for 60 min. The solution was warmed to room temperature and stirred for an additional 60 min during which time a precipitate formed. Diethylzinc (0.275 mL, 0.55 mmol, 2 M solution in hexanes) was slowly added to the reaction mixture at −78 °C and stirred for 20 min. Addition of TEEDA (14 μL, 0.066 mmol) and hexanes (4 mL) was performed at −78 °C. The solution was warmed to 0 °C and (−)-MIB (166 μL, 0.017 mmol) and isobutyraldehyde (30 μL, 0.332 mmol) were added. The reaction mixture was then slowly warmed to room temperature and stirred 12–16 h. After the reaction was complete by TLC analysis, the temperature was lowered to 0 °C and ZnEt2 (0.83 mL, 1.66 mmol, 2 M solution in hexanes) was added. Next, CF3CH2OH (120 μL, 1.65 mmol) was added dropwise. After stirring at 0 °C for 10 min, CH2I2 (135 μL, 1.67 mmol) was added. The reaction mixture was stirred with light exclusion at room temperature for 24 h. It was then quenched with saturated solution of NH4Cl. The organic and aqueous layers were separated and the aqueous layer was extracted with dichloromethane (3×5 mL). The combined organic layers were then washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and the crude product was purified by column chromatography on deactivated silica gel (5% ethyl acetate in hexanes) to afford the title compound (92.1 mg, 70% yield) as an oil. (c = 0.026, CHCl3); 1H NMR (CDCl3, 500 MHz): δ 0.05 (m, 1H), 0.67 (m, 1H), 0.93 (m, 2H), 0.98 (t, J = 7.9 Hz, 6H), 1.1 (m, 9H), 1.23 (m, 1H), 1.3 (d, J = 3.6 Hz, 1H), 1.74 (m, 1H), 1.91 (m, 1H), 2.96 (m, 1H), 3.76 (m, 2H), 7.42 (m, 6H), 7.7 (m, 4H); 13C{1H} NMR (CDCl3, 125 MHz): δ 8.9, 14.8, 17.6, 19.2, 19.4, 21.1, 27.1, 32.8, 34.7, 64.5, 76.7, 127.8, 129.8, 134.2, 135.8; IR (neat): 3599, 3411, 3134, 3070, 3050, 3013, 2952, 2912, 2895, 2858, 2739, 2319, 1958, 1888, 1823, 1589, 1486, 1471, 1428, 1362, 1331, 1306, 1260, 1235, 1187, 1157, 1110 1029, 1007 cm−1; HRMS calcd for C25H36O2NaSi (M+Na)+: 419.2382, found 419.2377.
3. Results and Discussion
3.1. Synthesis of syn-Vinylcyclopropyl Alcohols
The chemistry of vinylcyclopropanes is very rich.10,58,59 VCP's are found in biologically active compounds1,7,60-62 and natural products,2,63-65 such as carenes, sesquicarenes, sirenines, dictyopterenes, pyrethrine and ambruticin.9,66-68 Being useful intermediates in organic synthesis,10,11,53,69-71 the chemistry of VCP's has been studied in detail in several laboratories, including those of Wender,72,73 de Meijere,74 and Trost.75,76
Accessing enantioenriched vinylcyclopropanes is often challenging.20,77,78 Davies' pioneering route involved the catalytic asymmetric cyclopropanation of alkenes with vinyldiazoacetates. Good to excellent enantioselectivities were obtained, although trans-disubstituted alkenes were inert to cyclopropanation under these conditions.79,80 Conversely, use of simple diazoesters in the cyclopropanation of dienes can lead to VCP's with high enantio- and diastereoselectivity.81 Organocatalytic routes have also been used with increasing success.19,24 The enantioselective Simmons-Smith reactions has been employed with two dienol substrates (Scheme 2). Barrett82 used Fujisawa's83 diethyl tartrate-based system to prepare, with moderate enantioselectivity, a VCP (R=CH2OTBDPS) for use in the synthesis of FR-900848. They also prepared the bis(cyclopropanes) with moderate to excellent diastereoselectivity in the cyclopropanation of the both C=C double bonds. Charette84 reported the highly enantioselective cyclopropanation of a dienol (R=Ph) using a boron-based tartrate additive (Scheme 2). Both reactions required stoichiometric tartrate-derived Lewis acids.
Scheme 2.

Asymmetric Cyclopropanation of Dienols by Charette and Barrett.
Given the importance of VCP's and the limited variety that can be easily prepared under catalytic conditions with high enantio- and diastereoselectivity, we turned our attention to their synthesis. We envisioned adapting the tandem aldehyde vinylation/cyclopropanation sequence outlined in Scheme 1B to the synthesis of VCP's,44 as shown in Scheme 3. In place of the terminal alkyne of Scheme 1B, however, an enyne would be required.
Scheme 3.

Synthesis of VCP's.
3.1.1. Synthesis of Enantioenriched Dienols
Although a variety of terminal alkynes have been employed in the asymmetric vinylation of aldehydes based on Oppolzer's procedure,85-90 related examples of dienylation of aldehydes have not been reported to our knowledge. Reductive coupling reactions of enynes with aldehydes91,92 and ketones93,94 and related reactions95-97 allow access to dienols with high enantioselectivities in some cases. In prior work from our laboratories, the vinylation and dienylation of ketones was investigated.98-100 In this case, Oppolzer's method failed to provide the desired alcohols with high levels of enantioselectivity and yield. We found it necessary to use Wipf's101 hydrozirconation/transmetalation procedure to accomplish the dienylation reaction to afford enantioenriched dienols containing tertiary alcohols.99
The first step in advancing the catalytic asymmetric synthesis of VCP's was to optimize the yield and enantioselectivity in the dienylation of aldehydes (Table 1). In our initial investigations, we applied the optimized conditions for the asymmetric vinylation outlined in Scheme 1B. Thus, using the enyne 4-methyl-3-decen-1-yne, hydroboration with diethylborane at 25 °C occurred with high chemo- and regioselectivity to form the intermediate dienylborane. Transmetalation with diethylzinc generated the dienylzinc intermediate. In the presence of catalyst derived from (−)-MIB, the dienylation of isobutyraldehyde was conducted at −10 °C (Table 1, entry 1) and afforded the dienol product with good enantioselectivity (84%). The enantioselectivity in the dienylation, however, was significantly less than observed in the vinylations in Scheme 1B (91–99% ee).44
Table 1.
Optimization of the Dienyl Addition to Aldehydes
 | |||||
|---|---|---|---|---|---|
| entry | borane | (−)-MIB (mol %) | ZnR2 | T (°C) | ee (%) |
| 1 | Et2BH | 4 | ZnEt2 | −10 | 84 |
| 2 | Et2BH | 4 | ZnEt2 | −30 | 88a |
| 3 | Et2BH | 10 | ZnEt2 | −10 | 89 |
| 4 | Cy2BH | 4 | ZnEt2 | −10 | 84 |
| 5 | Et2BH | 4 | ZnMe2 | −10 | 83 |
| 6 | Et2BH | 10 | ZnEt2 | −10 | 93b |
Over 24 h reaction time.
Slow addition of aldehyde in hexanes.
We suspected that the more electron rich π-system of the dienyl group resulted in greater nucleophilicity over vinyl groups and that the increased rate of the background reaction was eroding the enantioselectivity. To decrease the impact of the background reaction, the temperature was lowered to −30 °C. Although the enantioselectivity increased to 88%, the reaction time was greatly extended (entry 2). Raising the catalyst loading to 10 mol % resulted in slightly higher enantioselectivity (89%, entry 3) and a more rapid reaction. Use of dicyclohexyl borane in place of diethyl borane (entry 4) or dimethylzinc instead of diethylzinc (entry 5) did not result in any improvement in enantioselectivity. Finally, use of 10 mol % MIB and dropwise addition of isobutyraldehyde (in a solution of hexanes) resulted in an increase in enantioselectivity to 93% (entry 6). We proposed that the slow addition ensures that a greater percentage of the catalyst is available to promote the aldehyde dienylation, reducing the contribution of the uncatalyzed background reaction.
3.1.2. Substrate Scope of the Asymmetric Dienylation of Aldehydes
With the optimized conditions for the dienylation outlined in Table 1 (entry 6), the substrate scope was explored (Table 2). Use of 4-methyl-3-decen-1-yne with isobutyraldehyde and cyclohexane carboxaldehyde provided the dienols in 93 and 92% ee with 88 and 90% yields, respectively (entries 1 and 2). Using ethynylcyclohexene with isobutyraldehyde and cyclohexane carboxaldehyde provided products with 89% ee and 85% yield and 90% ee and 80% yield, respectively (entries 3 and 4). Pivaldehyde underwent dienylation with slightly lower ee (80%) but high yield (85%, entry 5). Aromatic aldehydes underwent addition with 84–91% yield and enantioselectivities between 93–94% (entries 6 and 7). The silyl protected enynol participated in the addition to branched aldehydes with 90% enantioselectivity and ≥90% yield (entries 8 and 9) and dihydrocinnamaldehyde with 76% enantioselectivity and 79% yield (entry 10). These dienylations were next incorporated into the tandem synthesis of VCP's.
Table 2.
Substrate Scope for the Asymmetric Addition of Dienyl Groups to Aldehydes
| entry | product | yielda (%) | eeb (%) | |
|---|---|---|---|---|
| 1 | 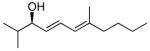 |
1a | 88 | 93 |
| 2 | 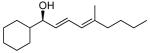 |
2a | 90 | 92 |
| 3 | 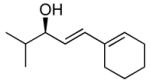 |
3a | 85 | 89 |
| 4 | 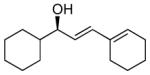 |
4a | 80 | 90 |
| 5 | 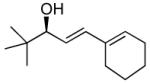 |
5a | 85 | 80 |
| 6 | 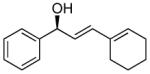 |
6a | 91 | 93 |
| 7 | 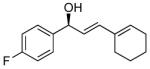 |
7a | 84 | 94 |
| 8 | 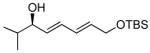 |
8a | 90 | 90 |
| 9 | 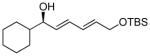 |
9a | 93 | 90 |
| 10 | 10a | 79 | 76 |
Isolated yield.
Determined by HPLC (see Supporting Information).
3.1.3. Tandem Enantioselective Dienylation/Diastereoselective Cyclopropanation: Synthesis of VCP's
With the conditions established for the asymmetric dienylation of aldehydes, we proceeded to explore the possibility of a tandem enantioselective dienylation/chemo- and diastereoselective cyclopropanation. As shown in Scheme 2, alkoxide directed cyclopropanation at allylic double bonds is faster than cyclopropanation at more remote positions.82,84 The carbenoid CF3CH2OZnCH2I was successfully employed in our cyclopropanation reactions outlined Scheme 1A.44 Unfortunately, with our dienyl zinc alkoxides (Scheme 3), the bis(cyclopropyl) alcohol was also generated with poor diastereoselectivity (1:1 dr) despite efforts to optimize the VCP synthesis. Screening other zinc carbenoids, such as IZnCH2I102 and Zn(CH2I)2 was also unsuccessful.103 To overcome these problems we examined the milder alkylzinc carbenoid, EtZnCH2I, which was employed in Scheme 1B.104,105 Reactions with 2 equiv of this carbenoid were thwarted by low conversions, possibly due to decomposition of the carbenoid, while those with 5 equiv led to generation of bis(cyclopropyl) alcohol (1:1 dr). On the basis of these results, we explored portion-wise addition of 4 equiv EtZnCH2I (see General Procedure B and the Supporting Information). This mode of addition resulted in formation of the desired vinyl cyclopropyl alcohols with excellent diastereoselectivity (dr>19:1). Under these reaction conditions, the enynes and aldehyde partners employed in Table 2 were subjected to the tandem asymmetric dienylation/diastereoselective cyclopropanation reactions (Scheme 3, Table 3). In all cases, the optimized conditions for the cyclopropanation generated only one diastereomer of the vinyl cyclopropyl alcohols (65–85% yield). Initially we were concerned that cyclopropanation of the electron rich trisubstituted olefins in entries 1–5 might be competitive with cyclopropanation of the allylic double bonds. Fortunately, this was not the case. For reasons that are not clear, the tandem asymmetric dienylation/diastereoselective cyclopropanation with benzaldehyde and 4-fluorobenzaldehyde did not result in formation of the desired product. These substrates exhibited low conversions and were complicated by formation of the bis(cyclopropane) and elimination byproducts. Similarly, problems were encountered in Scheme 1B with aromatic aldehydes.44
Table 3.
Scope of the One-pot Asymmetric Dienylation/Diastereoselective Cyclopropanation (Scheme 3).
| entry | product | dra | % ee (% y) | |
|---|---|---|---|---|
| 1 | 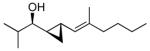 |
1b | >19:1 | 93 (75) |
| 2 | 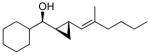 |
2b | >19:1 | 92 (80) |
| 3 | 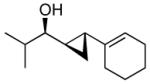 |
3b | >19:1 | 89 (80) |
| 4 | 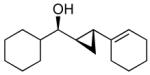 |
4b | >19:1 | 90 (85) |
| 5 | 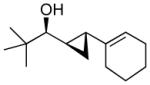 |
5b | >19:1 | 80 (65) |
| 6 | 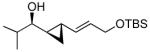 |
8b | >19:1 | 90 (71) |
| 7 | 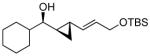 |
9b | >19:1 | 90 (79) |
| 8 | 10b | >19:1 | 76 (85) |
Determined by 1H NMR of the crude product.
3.2. Approaches Toward the Synthesis of anti-Cyclopropyl Alcohols
The ability of the alkoxy functionality in chiral allylic alkoxides to direct106 the cyclopropanation of neighboring double bonds with control of the incipient stereochemistry has been investigated and applied in synthesis.5 With acyclic (E)- and (Z)-allylic alcohols, the directed Simmons-Smith reaction is highly syn selective. Although the excellent directing ability of allylic alkoxides greatly facilitates synthesis of the syn cyclopropyl alcohols, efficient syntheses of the anti diastereomers remain challenging.107,108
An early approach to anti-cyclopropyl alcohols by Lautens involved reduction of the corresponding cyclopropyl ketones, which gave high anti-selectivity with trisubstituted and (Z)-disubstituted cyclopropyl ketones.109-111 Charette and coworkers found that enantioenriched allylic alcohols underwent cyclopropanation to generate the anti-cyclopropyl alcohols in the presence of stoichiometric enantioenriched tartrate-derived dioxaborolanes.112 A different strategy was developed by Charette and Lacasse that involved protection of allylic alcohols with bulky silyl groups followed by cyclopropanation (Table 4).107 The silyl group is believed to inhibit coordination of the zinc carbenoid to the allylic oxygen. Cyclopropanation is proposed to occur through a transition state that minimizes interaction between the silyl ether and carbenoid, affording the anti-stereoisomer, rather than through a directed pathway. Although this discovery represents a significant advance, the preparation of anti-cyclopropyl alcohols remains inefficient, involving the 1) synthesis of the allylic alcohol, 2) silyl ether formation, 3) cyclopropanation, and 4) deprotection, not to mention purifications after each step.
Table 4.
Synthesis of anti-Cyclopropyl Silyl Ethers by Charette and Lacasse.
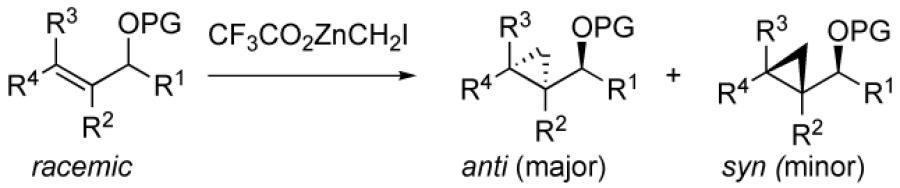 | |||||
|---|---|---|---|---|---|
| entry | SM | product | PG | dr | yield (%) |
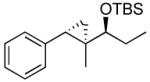
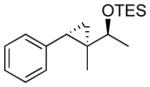
| 1 |  |
 |
TES | 97 : 3 | 87 |
| 2 | TBS | 97 : 3 | 85 | ||
| 3 | TIPS | 98 : 2 | 88 | ||
| 4 | 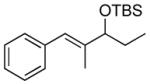 |
 |
97 : 3 | 88 | |
| 5 | 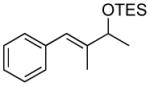 |
 |
>99:1 | 88 | |
3.2.1. Development of a One-pot Synthesis of anti-Cyclopropyl Alcohols
Our approach to anti-cyclopropyl alcohols blends our one-pot synthesis of syn-cyclopropyl alcohols (Scheme 1A) with Charette's synthesis of anti-cyclopropyl alcohols via silylated allylic alcohols.107
The key to success was development of an in situ silylation of the intermediate allylic zinc alkoxide that was compatible with the diastereoselective cyclopropanation. For this study, cinnamaldehyde and diethylzinc were employed with various silylating reagents (Table 5). On the basis of Charette's success with bulky silyl groups,107 we initiated our studies with TBDPS-Cl (tert-butyl diphenylsilyl chloride). Surprisingly, reaction of the zinc allylic alkoxide with 2 equiv TBDPS-Cl was very slow and only trace silylated products were observed (entry 1). We hypothesized that the reactivity of the zinc alkoxide product might be attenuated by formation of aggregates. To disrupt aggregation, we added 1.5 equiv triethylamine. In the presence of amine, conversion improved, but remained low (entry 2). Similar results were observed with TIPS-Cl (triisopropylsilyl chloride, entry 3). The smaller TBS-Cl (tert-butyl dimethylsilyl chloride) and TES-Cl (triethylsilyl chloride) exhibited greater reactivity toward the allylic zinc alkoxide, but full conversion to the silyl ether was not observed with 2 equiv of the silylating agent (entries 4 and 5). More reactive TIPS-OTf and TES-OTf resulted in complete consumption of the alkoxide. The reactions were not clean, however, due to formation of elimination byproducts and the silyl ethers were isolated in 50 and 35% yield, respectively (entries 6 and 7). Increasing the equivalents of TBS-Cl to 5 resulted in complete silylation. Unfortunately, the excess TBS-Cl caused difficulties in the subsequent cyclopropanation step. Finally we employed 1.5 equiv of TMS-Cl (trimethylsilyl chloride) with 1.5 equiv triethylamine (entry 9). Isolation of the silylated product did not accurately reflect the conversion, because TMS silyl ethers are very sensitive to cleavage of the Si-O bond. Nonetheless, we found that the silyl protection was complete by TLC in 18 h.
Table 5.
Optimization of the Asymmetric Addition/Silylation Procedure.
 | |||
|---|---|---|---|
| entry | silyl reagent | equiv | comment |
| 1 | TBDPS-Cl | 2.0 | very low conversiona |
| 2 | TBDPS-Cl | 2.0 | low conversion |
| 3 | TIPS-Cl | 2.0 | low conversion |
| 4 | TBS-Cl | 2.0 | incomplete |
| 5 | TES-Cl | 2.0 | incomplete |
| 6 | TIPS-OTf | 2.0 | 50% yield |
| 7 | TES-OTf | 2.0 | 35% yield |
| 8 | TBS-Cl | 5.0 | 75% yield |
| 9 | TMS-Cl | 1.5 | completeb |
No NEt3 added.
Isolation and purification resulted in partial loss of the TMS group.
The optimized asymmetric addition/silylation was then combined with the diastereoselective cyclopropanation as illustrated in Scheme 4. The cyclopropanation employing excess EtZnCH2I was sluggish. Use of Shi's more reactive CF3CH2OZnCH2I (5 equiv),113 however, generated the anti-cyclopropyl alcohols with high diastereoselectivity and yield in 24 h. The cyclopropanated silyl ether intermediates were desilylated by addition of 2 equiv of TBAF upon workup. Following Scheme 4, ethyl addition to cinnamaldehyde, α-methyl cinnamaldehyde, 3-methyl-2-butenal, and cyclohexene carboxaldehyde resulted in anti-cyclopropyl alcohol formation with high enantioselectivities (89–99%), good yields (67–82%) and excellent dr (>19:1, Table 6, entries 1–4). Methyl addition to α-methyl cinnamaldehyde followed by silylation, cyclopropanation and deprotection furnished the anti-cyclopropyl alcohol 15 in 80% yield with 95% ee and >19:1 dr (entry 5). The slightly lower dr of 16 (10:1) in entry 6 is likely a result of difficulties in generating the organozinc compound.114,115 The results in Table 6 demonstrate that anti-cyclopropyl alcohols can be prepared with high selectivities and good yields in an efficient one-pot procedure. The yields in Table 6 are only slightly lower than those reported by Charette in Table 4 for the diastereoselective cyclopropanation of racemic silyl ethers. To date, we have been unsuccessful employing the asymmetric vinylation of aldehydes44,50 (Scheme 1B) in tandem with the anti-cyclopropanation procedure. The enal alkylation/anti-cyclopropanation method of Scheme 4 is complementary to our syn-cyclopropanation chemistry shown in Scheme 1. In the next section we explore a method to couple generation of (Z)-allylic alkoxides with diastereoselective cyclopropanation.
Scheme 4.

Tandem Sequence to Afford anti-Cyclopropyl Alcohols with High Enantio- and Diastereoselectivity (Table 6).
Table 6.
Examples of anti-Cyclopropyl Alcohols Prepared in Scheme 4 in One-pot.
| entry | product | ee (%)a | drb | yield (%) | |
|---|---|---|---|---|---|
| 1 | 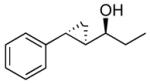 |
11 | 89 | >19:1 | 75 |
| 2 | 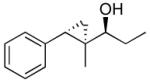 |
12 | 96 | >19:1 | 82 |
| 3 | 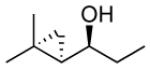 |
13 | 95 | >19:1 | 67 |
| 4 | 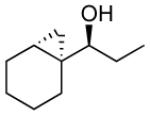 |
14 | 99 | >19:1 | 67 |
| 5 | 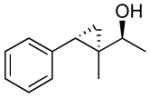 |
15 | 95 | >19:1 | 80 |
| 6 | 16 | 97 | ~ 10:1 | 60 |
Ee's determined by GC of HPLC.
Determined by 1H NMR analysis of the crude reaction mixture.
3.3. Generation of cis-Disubstituted Cyclopropyl Alcohols
Two approaches to cis-disubstituted cyclopropyl alcohols can be envisioned as illustrated in Scheme 5. They begin with asymmetric alkyl addition to (Z)-enals or asymmetric (Z)-vinylation to saturated aldehydes. The resulting (Z)-allylic alkoxide would then be subjected to diastereoselective syn-cyclopropanation. Both routes have challenges. (Z)-Enals can be difficult to prepare with high diastereopurity and they isomerize readily to the thermodynamically favored (E)-isomer. It is also difficult to prepare diastereopure (Z)-vinyl organometallic reagents. The latter method, however, is more versatile, because there are many more commercially available aldehydes compared to (Z)-enals.
Scheme 5.

Two Routes to cis-Disubstituted Cyclopropyl Alcohols.
As outlined in Scheme 1B, using Oppolzer's procedure85 we generated (E)-vinylzinc reagents as intermediates in the synthesis of trans-disubstituted cyclopropyl alcohols. This procedure, however, does not permit generation of diastereomeric cis-disubstituted cyclopropyl alcohols, which would be formed from (Z)-disubstituted vinylzinc reagents. To circumvent this deficiency, we recently developed a method to generate enantioenriched (Z)-allylic alcohols starting from 1-chloro-1-alkynes (Scheme 6).116 Initial hydroboration of 1-chloro-1-alkynes with dicyclohexylborane generated 1-chloro-1-alkenylboranes with excellent regioselectivity. It is known that nucleophiles react with 1-halo-1-alkenylboron derivatives, first adding to the open coordination site on boron followed by migration of the nucleophile117,118 or a boron alkyl to the vinylic position with inversion at the vinylic center.118-124 We used t-BuLi, which had been demonstrated to act as a hydride source in this process by Molander,118 in the formation of (Z)-vinylboranes. Vinylboranes are not very reactive toward carbonyl additions, however. On the basis of the work of Srebnik125 and Oppolzer85 we knew that (Z)-vinyl boranes would undergo boron to zinc transmetalation with dialkylzincs to generate more reactive (Z)-vinylzinc reagents. The increased nucleophilicity of vinylzinc reagents over their vinylborane counterparts enabled additions to aldehydes to proceed smoothly to generate (Z)-allylic alcohols.126 Using this method, a variety of racemic (Z)-allylic alcohols were prepared in high yields. Unfortunately, attempts toward enantioselective versions in the presence of (−)-MIB furnished only racemic products due to the rapid LiCl promoted background reaction. To suppress this background reaction, we inhibited the LiCl byproducts with tetraethylethylene diamine (TEEDA),124,127,128 which chelates to the lithium center and likely forms catalytically inactive 4-coordinate oligomeric [TEEDA•LiCl]n complexes with bridging chlorides.129,130 In the presence of the diamine inhibitor, enantioselectivities as high as 98% were recorded (Scheme 6, bottom left).116
Scheme 6.

Enantioselective Generation of (Z)-Allylic Alcohols in the Presence of TEEDA inhibitor.
To prepare enantio- and diastereoenriched cis-disubstituted cyclopropyl alcohols, the (Z)vinylation outlined in Scheme 6 was incorporated into the tandem (Z)-vinylation/cyclopropanation sequence in Scheme 7. Thus, hydroboration of the chloroalkyne and addition of tert-butyllithium generated the (Z)-vinyl borane. Transmetalation with diethylzinc was followed by addition of the diamine inhibitor (TEEDA), (−)-MIB, and the aldehyde. The resulting allylic alkoxide was then subjected to diastereoselective cyclopropanation with 5 equiv CF3CH2OZnCH2I under conditions similar to those used in Scheme 1B. After the usual workup, the desired syn-cis-disubstituted cyclopropyl alcohols were isolated in 42–70% yield with dr's >19:1 (Table 7). As shown in entries 1–3 of Table 7, chloroalkynes bearing TBDPS ethers underwent additions to saturated, aromatic and heteroaromatic aldehydes with enantioselectivities ≥90% and yields for the tandem reaction ranging from 52–65%. Likewise, 1-chloro substituted chloroalkynes were also good substrates, furnishing the cyclopropyl alcohols in 55–70% yield with 88–94% ee (entries 4–6). 1-Chloro phenyl acetylene and benzaldehyde were excellent substrates, affording the cis cyclopropyl alcohol in good yield and enantio- and diastereoselectivity (65% yield, 97% ee, dr>19:1). 2-Thiophenecarboxaldehyde proved more difficult and the tandem reaction exhibited low yield, but useful ee (92%) and dr (>19:1).
Scheme 7.

One-pot Synthesis of cis-Disubstituted Cyclopropyl Alcohols.
Table 7.
Substrate Scope of the Synthesis of cis-Disubstituted Cyclopropyl Alcohols.
| entry | product | ee (%)a | drb | yield (%) | |
|---|---|---|---|---|---|
| 1 | 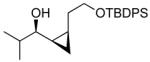 |
17 | 90 | >19:1 | 65 |
| 2 | 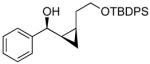 |
18 | 95 | >19:1 | 62 |
| 3 | 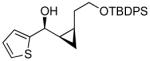 |
19 | 96 | >19:1 | 52 |
| 4 | 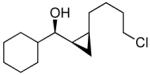 |
20 | 88 | >19:1 | 70 |
| 5 | 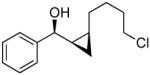 |
21 | 88 | >19:1 | 69 |
| 6 | 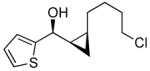 |
22 | 94 | >19:1 | 62 |
| 7 | 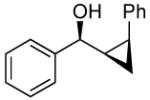 |
23 | 97 | >19:1 | 65 |
| 8 | 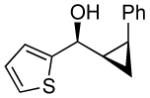 |
24 | 92 | >19:1 | 42 |
ee's determined by GC of HPLC
Determined by 1H NMR analysis of the crude reaction mixture
To confirm the relative stereochemistry of the cyclopropyl alcohols, 23 (Table 7, entry 7) was derivatized with (−)-camphanic acid chloride, the resulting ester crystallized by slow evaporation from a solution of hexanes and dichloromethane, and the structure determined by X-ray crystallography. Both the syn relationship between the hydroxyl and cyclopropane and cis geometry of the cyclopropane were found (Figure 2).
Figure 2.

X-ray structure of the camphanic ester of compound 23.
By using our method for the enantioselective (Z)-vinylation of aldehydes116 in tandem with the in situ cyclopropanation reaction, a variety of cis-disubstituted cyclopropyl alcohols (17–24) can be prepared in a one-pot procedure in respectable yields. This method circumvents the synthesis of thermodynamically unstable (Z)-enals and the isolation of (Z)-vinylzinc species. It is complementary to those cyclopropyl alcohol syntheses in Schemes 1, 3, and 4.
4. Summary and Outlook
Thousands of cyclopropane containing natural products and their derivatives have been described in the literature.2-4,9,14,65 Their interesting biological properties and rigid structures131 have made cyclopropanes a platform for development of new therapeutic agents. Nature's cyclopropanes are often enantioenriched and exhibit diverse substitution patterns and stereochemistries. Their efficient syntheses, therefore, require different approaches.
The cyclopropyl alcohols described herein all contain three stereogenic centers and possess distinct substitution patterns and stereochemical relationships. Their enantio- and diastereoselective syntheses have been designed to provide only the desired stereoisomer. The methods we have introduced for their syntheses are based on initial enantioselective C–C bond formations catalyzed by a (−)-MIB-based zinc catalyst. In the case of vinyl cyclopropyl alcohols, hydroboration of an enyne affords a dienyl borane. Boron to zinc transmetalation is followed by asymmetric aldehyde additions to form dienyl zinc alkoxides. Addition of EtZnCH2I results in an alkoxide-directed cyclopropanation of the allylic C=C double bond to afford vinyl cyclopropyl alcohols with high enantio- and diastereoselectivity. The zinc alkoxy group not only controls the chemoselectivity, it directs the cyclopropanation to afford the syn stereochemical relationship between the carbinol and cyclopropane.
To prepare the anti-diastereomers, the strong directing ability of the allylic alkoxide must be overridden. Based on work by Charette,107 we incorporated a silylation step into the tandem reaction to force cyclopropanation to occur on the opposite double bond face. Key to this procedure was addition of Et3N to facilitate silyl ether formation. Thus, asymmetric addition of an organozinc reagent to an enal was followed by silylation of the allylic zinc alkoxide to generate an allylic silyl ether. Cyclopropanation of the allylic silyl ether with CF3CH2OZnCH2I followed by TBAF workup provided the anti-cyclopropyl alcohols with excellent ee and dr. Selective formation of the syn- or anti-cyclopropyl alcohols is now readily achieved in one-pot.44 Previous methods to prepare these compounds required several synthetic steps and purifications.
We have previously employed Oppolzer's85 alkyne hydroboration/transmetalation to zinc and asymmetric addition to aldehydes to prepare cyclopropyl alcohols containing trans-disubstituted cyclopropanes (Scheme 1B).44 To access the cis-isomer, our asymmetric (Z)-vinylation of aldehydes116 was followed by cyclopropanation with CF3CH2OZnCH2I to provide the desired diastereomer with high enantio- and diastereoselectivities and moderate yields. The three methods introduced herein facilitate the synthesis of cyclopropyl alcohols with different stereochemical relationships. These compounds were not previously accessible in a synthetically efficient fashion. Given the rapid increase in molecular complexity with defined stereochemical outcomes, we anticipate that these methods will be useful in enantioselective synthesis.
Supplementary Material
Acknowledgment
We thank the NSF (CHE–065210 and 0848467) and NIH (National Institute of General Medical Sciences GM58101) for partial support of this work. We are also grateful to the NIH for the purchase of a Waters LCTOF- Xe Premier ESI mass spectrometer (grant number 1S10RR23444-1).
Footnotes
Supporting Information Available: Procedures, full characterization of new compounds, and structure of the camphanic ester of 23. This material is available free of charge via the Internet at http://pubs.acs.org.
5. References
- 1.Pietruszka J. Chem. Rev. 2003;103:1051–1070. doi: 10.1021/cr010027g. [DOI] [PubMed] [Google Scholar]
- 2.Wessjohann LA, Brandt W, Thiemann T. Chem. Rev. 2003;103:1625–1648. doi: 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]
- 3.Donaldson WA. Tetrahedron. 2001;57:8589–8627. [Google Scholar]
- 4.Salaun J. Top. Curr. Chem. 2000;207:1–67. [Google Scholar]
- 5.Pellissier H. Tetrahedron. 2008;64:7041–7095. [Google Scholar]
- 6.Liu HW, Walsh CT. In: The Chemistry of the Cyclopropyl Group. Rappoport Z, editor. John Wiley; New York, NY: 1997. p. 959. [Google Scholar]
- 7.Djerassi C, Doss GA. New J. Chem. 1990;14:713–19. [Google Scholar]
- 8.Lautens M, Klute W, Tam W. Chem. Rev. 1996;96:49–92. doi: 10.1021/cr950016l. [DOI] [PubMed] [Google Scholar]
- 9.Salaun J. Chem. Rev. 1989;89:1247–1270. [Google Scholar]
- 10.Reissig H-U, Zimmer R. Chem. Rev. 2003;103:1151–1196. doi: 10.1021/cr010016n. [DOI] [PubMed] [Google Scholar]
- 11.Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem. Rev. 2003;103:977–1050. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]
- 12.Charette AB, Beauchemin A. Org. React. 2001;58:1–415. [Google Scholar]
- 13.de Meijere A, editor. Small Ring Compounds in Organic Synthesis VI. Vol. 207. Springer; Berlin: 2000. [Google Scholar]
- 14.Wong HNC, Hon MY, Tse CW, Yip YC, Tanko JT, Hudlicky T. Chem. Rev. 1989;89:165–198. [Google Scholar]
- 15.Yu M, Pagenkopf BL. Tetrahedron. 2005;61:321–347. [Google Scholar]
- 16.Charette AB, Marcoux JF. Synlett. 1995:1197–1207. [Google Scholar]
- 17.Simmons HE, Smith RD. J. Am. Chem. Soc. 1959;81:4256–4264. [Google Scholar]
- 18.Rubin M, Rubina M, Gevorgyan V. Chem. Rev. 2007;107:3117–3179. doi: 10.1021/cr050988l. [DOI] [PubMed] [Google Scholar]
- 19.Kunz RK, MacMillan DWC. J. Am. Chem. Soc. 2005;127:3240–3241. doi: 10.1021/ja042774b. [DOI] [PubMed] [Google Scholar]
- 20.Xie H, Zu L, Li H, Wang J, Wang W. J. Am. Chem. Soc. 2007;129:10886–10894. doi: 10.1021/ja073262a. [DOI] [PubMed] [Google Scholar]
- 21.Rios R, Sunden H, Vesely J, Zhao GL, Dziedzic P, Cordova A. Adv. Syn. & Catal. 2007;349:1028–1032. [Google Scholar]
- 22.Papageorgiou CD, Dios M. A. C. d., Ley SV, Gaunt MJ. Angew. Chem., Int. Ed. 2004;43:4641–4644. doi: 10.1002/anie.200460234. [DOI] [PubMed] [Google Scholar]
- 23.Sun X-L, Tang Y. Acc. Chem. Res. 2008;41:937–948. doi: 10.1021/ar800108z. [DOI] [PubMed] [Google Scholar]
- 24.Charles DP, Steven VL, Matthew JG. Angew. Chem., Int. Ed. 2003;42:828–831. [Google Scholar]
- 25.Pellissier H. Tetrahedron. 2007;63:9267–9331. [Google Scholar]
- 26.Vicario JL, Badia D, Carrillo L. Synthesis-Stuttgart. 2007:2065–2092. [Google Scholar]
- 27.Rovis T, Evans DA. Prog. Inorg. Chem. 2001;50:1–150. [Google Scholar]
- 28.Davies HML, Antoulinakis EG. Org. React. 2001;57:1–326. [Google Scholar]
- 29.Doyle MP, McKervey MA. J. Chem. Soc., Chem. Commun. 1997:983–989. [Google Scholar]
- 30.Denmark SE, Christenson BL, O'Connor SP, Noriaki M. Pure Appl. Chem. 1996;68:23–27. [Google Scholar]
- 31.Zimmer LE, Charette AB. J. Am. Chem. Soc. 2009;131:15624–15626. doi: 10.1021/ja906033g. [DOI] [PubMed] [Google Scholar]
- 32.Goudreau SR, Charette AB. J. Am. Chem. Soc. 2009;131:15633–15635. doi: 10.1021/ja9074776. [DOI] [PubMed] [Google Scholar]
- 33.Long J, Yuan Y, Shi Y. J. Am. Chem. Soc. 2003;125:13632–13633. doi: 10.1021/ja030488e. [DOI] [PubMed] [Google Scholar]
- 34.Long J, Du H, Li K, Shi Y. Tetrahedron Lett. 2005;46:2737–2740. [Google Scholar]
- 35.Charette AB, Molinaro C, Brochu C. J. Am. Chem. Soc. 2001;123:12168–12175. doi: 10.1021/ja0108382. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi H, Yoshioka M, Ohno M, Kobayashi S. Tetrahedron Lett. 1992;33:2575–2578. [Google Scholar]
- 37.Denmark SE, O'Connor SP. J. Org. Chem. 1997;62:3390–3401. doi: 10.1021/jo9702397. [DOI] [PubMed] [Google Scholar]
- 38.Imai N, Sakamoto K, Takahashi H, Kobayashi S. Tetrahedron Lett. 1994;35:7045–7048. [Google Scholar]
- 39.Takahashi H, Yoshioka M, Shibasaki M, Ohno M, Imai N, Kobayashi S. Tetrahedron. 1995;51:12013–12026. [Google Scholar]
- 40.Voituriez A, Charette AB. Adv. Syn. & Catal. 2006;348:2363–2370. [Google Scholar]
- 41.Balsells J, Walsh PJ. J. Org. Chem. 2000;65:5005–5008. doi: 10.1021/jo991704y. [DOI] [PubMed] [Google Scholar]
- 42.Charette AB. In: In The Chemistry of Organozinc Compounds. Rappoport Z, Marek I, editors. John Wiley & Sons, Ltd; West Sussex: 2006. pp. 237–286. [Google Scholar]
- 43.For an exception see. Shitama H, Katsuki T. Angew. Chem., Int. Ed. 2008;47:2450–2453. doi: 10.1002/anie.200705641. [DOI] [PubMed] [Google Scholar]
- 44.Kim HY, Lurain AE, García-García P, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2005;127:13138–13139. doi: 10.1021/ja0539239. [DOI] [PubMed] [Google Scholar]
- 45.Nugent WA. J. Chem. Soc., Chem. Commun. 1999:1369–1370. [Google Scholar]
- 46.Chen YK, Jeon SJ, Walsh PJ, Nugent WA. Org. Synth. 2005;82:87–92. [Google Scholar]
- 47.Ratier M, Castaing M, Godet J-Y, Pereyre MJ. J. Chem. Res. Miniprint. 1978:2309–2318. [Google Scholar]
- 48.Charette AB, Lebel H. J. Org. Chem. 1995;60:2966–2967. [Google Scholar]
- 49.Charette AB, Mathieu S, Fournier J-F. Synlett. 2005:1779–1782. [Google Scholar]
- 50.Kim HY, Salvi L, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2009;131:954–962. doi: 10.1021/ja806989n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerwick WH. Chem. Rev. 2002;93:1807–1823. [Google Scholar]
- 52.Kumaraswamy G, Padmaja M. J. Org. Chem. 2008;73:5198–5201. doi: 10.1021/jo800631z. [DOI] [PubMed] [Google Scholar]
- 53.Baldwin JE. Chem. Rev. 2003;103:1197–1212. doi: 10.1021/cr010020z. [DOI] [PubMed] [Google Scholar]
- 54.Davies HML, Kong N, Churchill MR. J. Org. Chem. 1998;63:6586–6589. [Google Scholar]
- 55.Sebelius S, Olsson VJ, Wallner OA, Szabo KJ. J. Am. Chem. Soc. 2006;128:8150–8151. doi: 10.1021/ja062585o. [DOI] [PubMed] [Google Scholar]
- 56.Rozema MJ, Sidduri A, Knochel P. J. Org. Chem. 1992;57:1956–1958. [Google Scholar]
- 57.Langer F, Schwink L, Devasagayaraj A, Chavant P-Y, Knochel P. J. Org. Chem. 1996;61:8229–8243. doi: 10.1021/jo961129n. [DOI] [PubMed] [Google Scholar]
- 58.Wang SC, Tantillo DJ. J. Organomet. Chem. 2006;691:4386–4392. [Google Scholar]
- 59.Khusnutdinov RI, Dzhemilev UM. J. Organomet. Chem. 1994;471:1–18. [Google Scholar]
- 60.Faust R. Angew. Chem., Int. Ed. 2001;40:2251–2253. doi: 10.1002/1521-3773(20010618)40:12<2251::aid-anie2251>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 61.Sonawane HR, Bellur NS, Kulkarni DG, Ahuja JR. Synlett. 1993;1993:875–884. [Google Scholar]
- 62.Connor DT, Greenough RC, Strandtmann MV. J. Org. Chem. 1977;42:3664–3669. doi: 10.1021/jo00443a006. [DOI] [PubMed] [Google Scholar]
- 63.Henrick CA. In: Pyrethroids in Agrochemicals from Natural Products. Godfrey CRA, editor. Marcel Dekker; New York: 1995. pp. 147–213. [Google Scholar]
- 64.Yoshida M, Ezaki M, Hashimoto M, Yamashita M, Shigematsu N, Okuhara M, Kohsaka M, Horikoshi K. J. Antibiot. 1990;43:748–754. doi: 10.7164/antibiotics.43.748. [DOI] [PubMed] [Google Scholar]
- 65.Salaun J, Baird MS. Curr. Med. Chem. 1995;2:511–542. [Google Scholar]
- 66.Berberich SM, Cherney RJ, Colucci J, Courillon C, Geraci LS, Kirkland TA, Marx MA, Schneider MF, Martin SF. Tetrahedron. 2003;59:6819–6832. [Google Scholar]
- 67.Doyle MP. Chem. Rev. 1986;86:919–939. [Google Scholar]
- 68.Beumer R, Reiser O. Tetrahedron. 2001;57:6497–6503. [Google Scholar]
- 69.Fox ME, Li C, Marino JJP, Overman LE. J. Am. Chem. Soc. 1999;121:5467–5480. [Google Scholar]
- 70.Corey EJ, Gant TG. Tetrahedron Lett. 1994;35:5373–5376. [Google Scholar]
- 71.Murelli RP, Catal·n S, Gannon MP, Snapper ML. Tetrahedron Lett. 2008;49:5714–5717. [Google Scholar]
- 72.Wender PA, Gamber GG, Williams TJ. In: In Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Wiley; Weinheim: 2005. p. 263. [Google Scholar]
- 73.Wegner HA, de Meijere A, Wender PA. J. Am. Chem. Soc. 2005;127:6530–6531. doi: 10.1021/ja043671w. [DOI] [PubMed] [Google Scholar]
- 74.Kurahashi T, de Meijere A. Synlett. 2005:2619–2622. [Google Scholar]
- 75.Trost BM, Shen HC, Horne DB, Toste FD, Steinmetz BG, Koradin C. Chem. Eur. J. 2005;11:2577–2590. doi: 10.1002/chem.200401065. [DOI] [PubMed] [Google Scholar]
- 76.Trost BM, Yasukata T. J. Am. Chem. Soc. 2001;123:7162–7163. doi: 10.1021/ja010504c. [DOI] [PubMed] [Google Scholar]
- 77.Zheng J-C, Liao W-W, Tang Y, Sun X-L, Dai L-X. J. Am. Chem. Soc. 2005;127:12222–12223. doi: 10.1021/ja052228y. [DOI] [PubMed] [Google Scholar]
- 78.Deng X-M, Cai P, Ye S, Sun X-L, Liao W-W, Li K, Tang Y, Wu Y-D, Dai L-X. J. Am. Chem. Soc. 2006;128:9730–9740. doi: 10.1021/ja056751o. [DOI] [PubMed] [Google Scholar]
- 79.Davies HML, Bruzinski PR, Lake D, Kong N, Fall M. J. Am. Chem. Soc. 1996;118:6897–6907. [Google Scholar]
- 80.Davies HML, Panaro SA. Tetrahedron Lett. 1999;40:5287–5290. [Google Scholar]
- 81.Lowenthal RE, Masamune S. Tetrahedron Lett. 1991;32:7373–7376. [Google Scholar]
- 82.Barrett AGM, Tustin GJ. J. Chem. Soc., Chem. Commun. 1995:355–356. [Google Scholar]
- 83.Ukaji Y, Nishimura M, Fujisawa T. Chem. Lett. 1992:61–64. [Google Scholar]
- 84.Charette AB, Juteau H, Lebel H, Molinaro C. J. Am. Chem. Soc. 1998;120:11943–11952. [Google Scholar]
- 85.Oppolzer W, Radinov RN. Helv. Chim. Acta. 1992;75:170–173. [Google Scholar]
- 86.Oppolzer W, Radinov RN, El-Sayed E. J. Org. Chem. 2001;66:4766–4770. doi: 10.1021/jo000463n. [DOI] [PubMed] [Google Scholar]
- 87.Hussain MH, Walsh PJ. Acc. Chem. Res. 2008;41:883–893. doi: 10.1021/ar800006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rowley Kelly A, Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2005;127:14668–14674. doi: 10.1021/ja051291k. [DOI] [PubMed] [Google Scholar]
- 89.Lurain AE, Carroll PJ, Walsh PJ. J. Org. Chem. 2005;70:1262–1268. doi: 10.1021/jo048345d. [DOI] [PubMed] [Google Scholar]
- 90.Jeon S-J, Chen YK, Walsh PJ. Org. Lett. 2005;7:1729–1732. doi: 10.1021/ol050255n. [DOI] [PubMed] [Google Scholar]
- 91.Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J. Am. Chem. Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735. [DOI] [PubMed] [Google Scholar]
- 92.Mahandru GM, Liu G, Montgomery J. J. Am. Chem. Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]
- 93.Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448–16449. doi: 10.1021/ja0673027. [DOI] [PubMed] [Google Scholar]
- 94.Miller KM, Jamison TF. Org. Lett. 2005;7:3077–3080. doi: 10.1021/ol051075g. [DOI] [PubMed] [Google Scholar]
- 95.Kong JR, Krische MJ. J. Am. Chem. Soc. 2006;128:16040–16041. doi: 10.1021/ja0664786. [DOI] [PubMed] [Google Scholar]
- 96.Yu C-M, Lee S-J, Jeon M. j. C. S.-Perkin Trans. 1. 1999:3557–3558. [Google Scholar]
- 97.Yu C-M, Yoon S-K, Lee S-J, Lee J-Y, Yoon S-K, Kim SS. Chem. Commun. 1998:2749–2750. [Google Scholar]
- 98.Li H, Walsh PJ. J. Am. Chem. Soc. 2004;126:6538–6539. doi: 10.1021/ja049206g. [DOI] [PubMed] [Google Scholar]
- 99.Li H, Walsh PJ. J. Am. Chem. Soc. 2005;127:8355–8361. doi: 10.1021/ja0425740. [DOI] [PubMed] [Google Scholar]
- 100.Anaya de Parrodi C, Walsh PJ. Synlett. 2004:2417–2420. [Google Scholar]
- 101.Wipf P, Ribe S. J. Org. Chem. 1998;63:6454–6455. [Google Scholar]
- 102.Charette AB, Marcoux JF. J. Am. Chem. Soc. 1996;118:4539–4549. [Google Scholar]
- 103.Denmark SE, Edwards JP. J. Org. Chem. 1991;56:6974–6981. [Google Scholar]
- 104.Furukawa J, Kawabata K, Nishimura J. Tetrahedron Lett. 1966;28:3353–3354. [Google Scholar]
- 105.Furukawa J, Kawabata K, Nishimura J. Tetrahedron. 1968;24:53–58. [Google Scholar]
- 106.Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307–1370. [Google Scholar]
- 107.Charette AB, Lacasse M-C. Org. Lett. 2002;4:3351–3354. doi: 10.1021/ol0264051. [DOI] [PubMed] [Google Scholar]
- 108.Zohar E, Marek I. Org. Lett. 2004;6:341–343. doi: 10.1021/ol036143i. [DOI] [PubMed] [Google Scholar]
- 109.Delanghe PHM, Lautens M. Tetrahedron Lett. 1994;35:9513–9516. [Google Scholar]
- 110.Lautens M, Delanghe PHM. J. Org. Chem. 1995;60:2474–2487. [Google Scholar]
- 111.Kazuta Y, Abe H, Yamamoto T, Matsuda A, Shuto S. J. Org. Chem. 2003;68:3511–3521. doi: 10.1021/jo030019v. [DOI] [PubMed] [Google Scholar]
- 112.Charette AB, Lebel H, Gagnon A. Tetrahedron. 1999;55:8845–8856. [Google Scholar]
- 113.Yang ZQ, Lorenz JC, Shi Y. Tetrahedron Lett. 1998;39:8621–8624. [Google Scholar]
- 114.Knochel P, Vettel S, Eisenberg C. Appl. Organomet.Chem. 1995;9:175–188. [Google Scholar]
- 115.Knochel P, Singer RD. Chem. Rev. 1993;93:2117–2188. [Google Scholar]
- 116.Salvi L, Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2007;129:16119–16125. doi: 10.1021/ja0762285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Corey EJ, Albonico SM, Koelliker U, Schaaf TK, Varma RK. J. Am. Chem. Soc. 1971;93:1491–1493. doi: 10.1021/ja00735a033. [DOI] [PubMed] [Google Scholar]
- 118.Campbell JBJ, Molander GA. J. Organomet. Chem. 1978;156:71–79. [Google Scholar]
- 119.Zweifel G, Arzoumanian H. J. Am. Chem. Soc. 1967;89:5086–5088. [Google Scholar]
- 120.Negishi E, Yoshida T. J. Chem. Soc., Chem. Commun. 1973:606–607. doi: 10.1021/ja00801a056. [DOI] [PubMed] [Google Scholar]
- 121.Negishi E, Williams RM, Lew G, Yoshida T. J. Organomet. Chem. 1975;92:C4–C6. [Google Scholar]
- 122.Brown HC, Imai T, Bhat NG. J. Org. Chem. 1986;51:5277–5282. [Google Scholar]
- 123.Chen YK, Walsh PJ. J. Am. Chem. Soc. 2004;126:3702–3703. doi: 10.1021/ja0396145. [DOI] [PubMed] [Google Scholar]
- 124.Kerrigan MH, Jeon S-J, Chen Y, Salvi L, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2009;131:8434–8445. doi: 10.1021/ja809821x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Srebnik M. Tetrahedron Lett. 1991;32:2449–2452. [Google Scholar]
- 126.Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2006;128:9618–9619. doi: 10.1021/ja061973n. [DOI] [PubMed] [Google Scholar]
- 127.Kim JG, Walsh PJ. Angew. Chem., Int. Ed. 2006;45:4175–4178. doi: 10.1002/anie.200600741. [DOI] [PubMed] [Google Scholar]
- 128.Salvi L, Kim JG, Walsh PJ. J. Am. Chem. Soc. 2009;131:12483–12493. doi: 10.1021/ja9046747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chitsaz S, Pauls J, Neumuller B. Z. Naturforsch., B. 2001;56:245–248. [Google Scholar]
- 130.Hoffmann D, Dorigo A, Schleyer P. v. R., Reif H, Stalke D, Sheldrick GM, Weiss E, Geissler M. Inorg. Chem. 1995;34:262–269. [Google Scholar]
- 131.Reichelt A, Martin SF. Acc. Chem. Res. 2006;39:433–442. doi: 10.1021/ar030255s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.