

Abstract
Acyltransferase (AT) domains of multimodular polyketide synthases are the primary gatekeepers for stepwise incorporation of building blocks into a growing polyketide chain. Each AT domain has two substrates, an α-carboxylated CoA thioester (e.g. malonyl-CoA or methylmalonyl-CoA) and an acyl carrier protein (ACP). Whereas the acyl-CoA specificity of AT domains has been extensively investigated, little is known about their ACP specificity. Guided by recent high-resolution structural insights, we have systematically probed the protein-protein interactions between AT domains, ACP domains and the linkers that flank AT domains. Representative AT domains of the 6-deoxyerythronolide B synthase (DEBS) have greater than 10-fold specificity for their cognate ACP substrates as compared to other ACP domains from the same synthase. Both the flanking (N- and C-terminal) linkers of an AT domain contributed to the efficiency and specificity of transacylation. As a frame of reference, the activity and specificity of a stand-alone AT domain from the “AT-less” disorazole synthase (DSZS) were also quantified. The activity (kcat/KM) of this AT was >250-fold higher than the corresponding values for DEBS AT domains. Although the AT from DSZS discriminated modestly against ACP domains from DEBS, it exhibited >40-fold higher activity in trans in the presence of these heterologous substrates than their natural AT domains. Our results highlight the opportunity for regioselective modification of a polyketide backbone by in trans complementation of inactivated AT domains. They also reinforce the need for more careful consideration of protein-protein interactions in the engineering of these assembly line enzymes.
Polyketide synthases (PKSs) are multifunctional enzymes responsible for the production of polyketide natural products, many of which have important medicinal applications [1-6]. Modular polyketide synthases, such as the 6-deoxyerythronolide B synthase (DEBS), are organized into modules comprised of multiple catalytic domains. To assemble short acyl-CoA precursors into complex polyketide products, each module contains minimally three functional domains: a β-ketosynthase (KS), an acyltransferase (AT), and an acyl carrier protein (ACP). The KS domain receives the growing polyketide chain from the upstream module and subsequently catalyzes formation of the C-C bond between this substrate and an ACP-bound extender unit that is selected by the AT domain.
Many biosynthetic engineering strategies for the modification of polyketides are predicated on the apparent modularity of these multimodular assembly lines. For example, two different modular approaches have been proposed for the incorporation of unnatural extender units into the polyketide backbone. In one approach, the AT domain of a target module is replaced by a naturally occurring homolog with a distinct extender unit specificity [7-9]. An alternative approach involves complementation of a module harboring a site-specifically inactivated AT with a stand-alone AT protein having the desired extender unit specificity [10]. In practice, the efficiency of both approaches rests upon the extent to which the protein-protein interactions necessary for structural integrity and catalytic activity of the engineered module are preserved as a result of introducing a heterologous AT. Here we attempt to pinpoint these protein-protein interactions and to quantify their contributions to acyl transfer kinetics.
Our investigations into specific protein-protein interactions involving the AT domain took advantage of the development of a multidomain [KS][AT] construct harboring catalytically active KS and AT domains as well as three flanking “linker” sequences [11,12], a catalytically active AT construct that includes flanking linkers at both the N- and C-termini [13], and an active ACP construct without any linkers [12]. The X-ray structures of prototypical [KS][AT] constructs [14, 15] and a solution NMR structure of a prototypical ACP from DEBS [16] have been solved (Figure 1). For comparison purposes, we have also studied a representative stand-alone AT protein from an “AT-less” multimodular PKS, the disorazole synthase (DSZS) [17, 18]. The DSZS and homologous synthases are comprised of modules lacking dedicated AT domains; instead, extender units are transacylated onto all the ACP domains of these synthases by a single stand-alone AT protein [19, 20]. Our results reveal that AT-ACP interactions are both specific and variable, and therefore warrant more careful consideration in designing efficient strategies for engineering polyketide biosynthesis.
Figure 1.
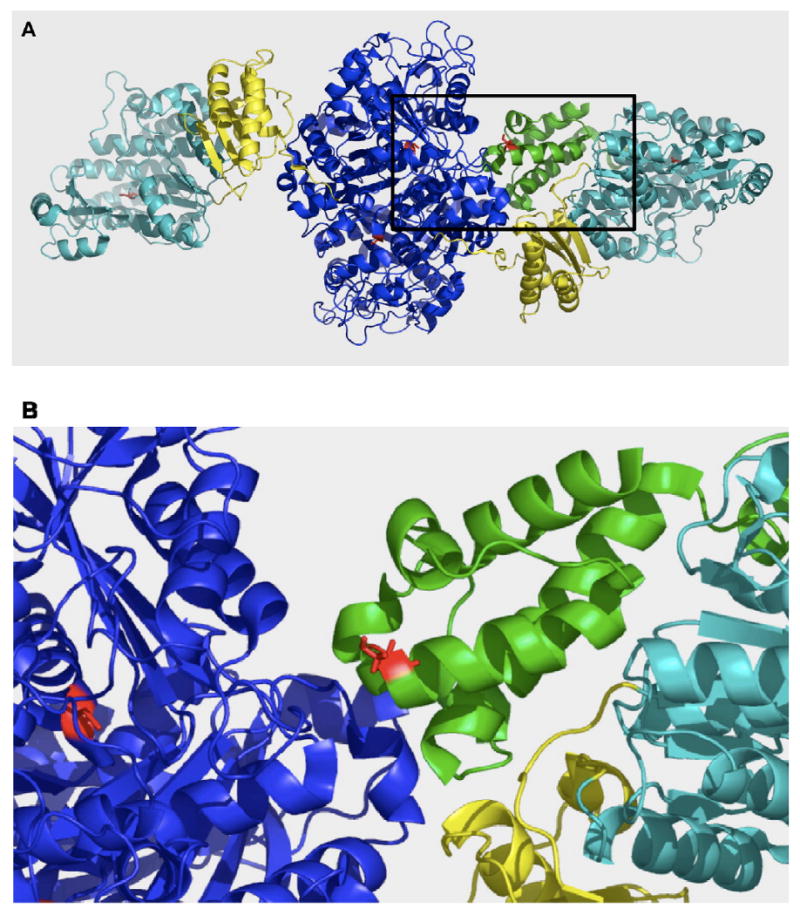
PatchDock model for the NMR solution structure of the ACP2 domain of DEBS (PDB ID 2JU2, [16]) docked onto the X-ray crystal structure of the [KS3][AT3] fragment of DEBS (PDB ID 2QO3, [15]). The [KS][AT] protein forms a homodimer. KS domain, AT domain and the linkers are shown in blue, cyan and yellow respectively. ACP domain is shown in green. The active site residues of the KS, AT and ACP domains are highlighted red. (B) is the enlarged view of boxed area in (A).
Experimental Procedures (Methods and Materials)
Reagents and Chemicals
DL-[2-Methyl-14C]-methylmalonyl-CoA and [14C]-malonyl-CoA were from American Radiolabeled Chemicals. All other chemicals were from Sigma. SDS PAGE-gradient gels were from BioRad and Invitrogen. Ni-NTA affinity resin was from Qiagen. HiTrap-Q anion exchange column was from Amersham Pharmacia.
Construction and expression of discrete AT domains from DEBS and DSZS
All AT domains from DEBS were expressed as stand-alone proteins with their KS-to-AT and post AT linkers [13]. The DNA sequence encoding DEBS AT1 was amplified by PCR as an NdeI-EcoRI fragment using primers 5′- AAAAAACATATGCAGGTCGTCGAAGGCGAGCGG –3′and 5′-TTTTGAATTCACGCGGTAGCGCAGCGCGGAAACC -3′. This NdeI-EcoRI fragment was cloned into the pET28 expression vector to yield plasmid pAYC129. The DNA sequence encoding DEBS AT2 was constructed similarly with primers 5′-AAAACATATGGAACCGGAGCCCGTGCCGCAACC-3′and 5′- TTTTGAATTCACCCGGTAGAACCAGCCGTCGAGC -3′ to yield plasmid pAYC130. The DNA sequence encoding DEBS AT4 was amplified similarly with primers 5′-AAAAAACATATGGCAGAGCAGGAGGCCGCCCGC-3′and 5′-TTTTGAATTCACCTCCGCGCCGCGCACGCCCAGC -3′to yield plasmid pAYC131. The DNA sequence encoding DEBS AT5 was amplified similarly with primers 5′-AAAAACATATGGAGGCCGACGAGCCCGAGCCGG-3′and 5′-TTTTGAATTCACCTGGTAGCGCCAGTCGTCGTCC-3′ to yield plasmid pAYC132. The DNA sequence encoding DEBS AT6 was constructed similarly with primers 5′-AAAACATATGGAGCCCGAGCCGCTGCCGGAACC-3′and 5′-TTTTGAATTCACGCGGTAGCGGCTGTCGGCGAGC-3′ to yield plasmid pAYC133. All five plasmids were introduced into E. coli BL21(DE3) via transformation. Proteins were expressed and purified using the previously described protocol [11]. The expression and purification of DEBS AT3 with the post-AT linker (pAYC23) has been previously described [13].
Two versions of AT gene from DSZS were constructed. The DNA sequence encoding DSZS AT without the post AT linker (denoted DSZS ATS) was amplified by PCR as an NdeI-EcoRI fragment using primers 5′-AAAAACATATGAAAGCATACATGTTTCCCGGGC–3′and 5′-TTTTTGAATTCGCGACGACGAGGGGCTGGGCG-3′. Cosmid pKOS254-190.1 was used as the template [17]. This NdeI-EcoRI fragment was cloned into the pET21 expression vector to yield plasmid pFW3. The DNA sequence encoding DSZS AT with the post AT linker (denoted DSZS ATL) was constructed similarly with primers 5′- AAAAACATATGAAAGCATACATGTTTCCCGGGC-3′and 5′- TTTTTGAATTCGCCGCGGCGCTGCCCAGCACC -3′ to yield plasmid pFW5. The plasmids were transformed into E. coli BL21(DE3) for protein expression. Proteins were expressed and purified using the previously described protocol [11].
DNA sequences encoding various AT6 constructs (AT6AT, AT6PAT, AT6KSAT) were amplified from pAYC133 as NdeI-EcoRI fragments and cloned into pET21 expression vector. The DNA sequence encoding core AT6 domain, excluding the C-terminal helix from the AT domain and flanking linkers, (denoted AT6AT) was constructed with primers 5′-AAAAACATATGGGCGTGGTTTTCGTCTTCCCAGGTC-3′ and 5′-TTTTTGAATTCGCCCCGATGGCCACGGCGTCCGC-3′ to yield pFW59. The DNA sequence encoding core AT6 domain with C-terminal flanking (post AT) linker (denoted AT6PAT) was constructed similarly with primers 5′-AAAAACATATGGGCGTGGTTTTCGTCTTCCCAGGTC-3′ and 5′-TTTTTGAATTCGCGCGGTAGCGGCTGTCGGCGAG-3′ to yield pFW60. The DNA sequence encoding core AT6 domain with N-terminal flanking KS-to-AT linker, excluding the C-terminal helix and post AT linker, (denoted AT6KSAT) was constructed with primers 5′-AAAAACATATGGAGCCCGAGCCGCTGCCGGAA-3′ and 5′-TTTTTGAATTCGCCCCGATGGCCACGGCGTCCGC-3′ to yield plasmid pFW61.
DNA sequences encoding the rest of the AT6 constructs (AT6small and AT6small,PAT) were amplified from pAYC133 as NdeI-EcoRI fragments and cloned into pET28 expression vector. The DNA sequence encoding core AT6 domain with proximal 15 residues from the KS-to-AT linker and less the post AT linker and its C-terminal helix (denoted as ATsmall) was constructed with primers 5′-AAAAACATATGGGCGTTGCGGCTCCCGGTGCCA-3′ and 5′-TTTTTGAATTCGCCCCGATGGCCACGGCGTCCGC-3′ to yield plasmid pFW75. The DNA sequence encoding AT6 along with 15 residues from the KS-to-AT linker and the post AT linker (denoted as ATsmall, PAT) was constructed with 5′-AAAAACATATGGGCGTTGCGGCTCCCGGTGCCA-3′ and 5′-TTTTTGAATTCGCGCGGTAGCGGCTGTCGGCGAG-3′ to yield plasmid pFW76. The plasmids were transformed into E. coli BL21(DE3) for protein expression. Proteins were expressed and purified using the previously described protocol [11].
Construction and expression of DEBS [KS][AT] didomains
The expression and purification of [KS3][AT3] (pAYC02) and [KS6][AT6] (pAYC11) didomains from DEBS have been previously described [11].
Construction and expression of AT° mutants of DEBS module 3+TE and module 6+TE
The construction of plasmids encoding module 3 (pRSG34) and module 6 (pRSG54) with thioesterase (TE) domain covalently attached has been previously described [21]. Module 3/AT°+TE: To inactivate the acyltransferase domain of module 3+TE (pRSG34), the AT active site Ser was mutated into Ala using primers 5′- GCGGTCGTGGGGCACGCGCAGGGCGAGATCG -3′and 5′-CGATCTCGCCCTGCGCGTGCCCCACGACCGC -3′ to yield plasmid pAYC136. Module 6/AT°+TE: Similarly, primers 5′- GCCGTCATCGGCCATGCGCAGGGCGAGATCG -3′and 5′-CGATCTCGCCCTGCGCATGGCCGATGACGGC -3′ were used to inactivate the AT domain of module 6+TE (pRSG54) to yield plasmid pAYC138. The plasmids, along with pRSG34 and pRSG54, were transformed into E. coli BAP1 [22] to produce the desired holo-proteins with pantetheinylated ACP domains [23]. Proteins were expressed and purified using the previously described protocol [11].
Construction and expression of discrete DEBS and DSZS acyl carrier proteins
A N-terminal hexahistidine tagged DEBS ACP6 was constructed for ACP6 protein expression. ACP6 sequence was amplified from pRSG54 with primers 5′-AAAAACATATGGCGGCCCCGGCGCGGGAGATG-3′ and 5′-TTTTTGAATTCTCAGAGCTGCTGTCCTATGTGGTC-3′. The resulting NdeI-EcoRI fragment was then inserted into a pET28 expression vector to yield pFW55.
Expression plasmid for DEBS ACP3 was constructed from a previously described ACP3 expressing construct, pVYA5 [12]. An XbaI-NdeI cassette, containing a hexahistidine-tag upstream of a flag-tag, was prepared by thermally annealing primer 5′ – AAAAAATCTAGAAATAATTTTGTTTAACTTTAACTTTAAGAAGGAGATATACCATGCCGCA TCATCATCATCATCACAGCAGCGACTACAAAGACGATGACGACAAGCTGCATATGAAAAA A-3′ and its complementary primer for 5 minutes at 95°C. This cassette was then digested and inserted into pVYA5, replacing the corresponding XbaI-NdeI fragment to yield pSHIV9. The original XbaI-NdeI fragment in pVYA5 contains only an N-terminal hexahistidine-tag.
The sequence encoding DSZS ACP1 was amplified from cosmid pKOS254-190.4 [17], using primers 5′-AAAAACATATGGCGCCTGCAGGGGCAGGACAG-3′ and 5′-TTTTTGAATTCGCTGCCGACCTCGCGGGGACGCG-3′. The NdeI-EcoRI fragment was then inserted into pET21 expression vector to yield pFW50.
The plasmids were transformed into E. coli BAP1 [22] to produce the desired holo-proteins with pantetheinylated ACP domains [23]. Proteins were expressed and purified using the previously described protocol [11].
Limited Proteolysis of stand-alone DEBS AT6
Stand-alone DEBS AT6 with both linkers (30 μM, 100mM phosphate at pH 7.2) was incubated with varying concentrations of trypsin with 2.5mM TCEP at room temperature for 30 minutes. Trypsin (4.2-42 μM in ddH2O) was added to make up final molar Trypsin: AT protein ratios of 1:5, 1:15, 1:50 and 1:100. The total reaction volume is 10 μL. The reaction was quenched in 5 μL of SDS-PAGE sample buffer. The proteins were separated on a SDS-PAGE gel and the protein of interest was eluted from the gel and subjected to in-gel trypsin digestion and LC-MS/MS analysis.
In-gel trypsin digestion and LC-MS/MS analysis
The excised gel band from above was cut into 1×1 mm pieces before being reduced with 50 mM DTT and alkylated with 100 mM acrylamide. The gel was then destained with 50 mM 1:1 acetonitrile: ammonium bicarbonate solution. This was dried on a Speedvac and reconstituted in 12 ng/μL of trypsin, in the presence of protease Max. The mixture was incubated at 50°C for 1 hour before being spun down at 12 g for 30 seconds. Peptides were extracted and dried using a speedvac. This was reconstituted in 10 μL of 2% acetonitrile/water with 0.1% formic acid.
The sample was then injected onto Eksigent NanoLC-2D capillary HPLC with a self-packed C18 reverse phase column. MS spectra were collected with an LCQ Deca XP Plus (Thermo Scientific) that was set to data dependent acquisition mode to analyze the top 3 abundant peaks. Data were analyzed using SORCERER processor (SageN Research) with Swiss-PROT database. An 8-node cluster for accelerated Sequest database searching was used.
Acylation of trypsin-digested fragments of stand-alone AT6
Stand-alone DEBS AT6 (30 μM, 100mM phosphate at pH 7.2) was digested, as described above, at molar Trypsin: AT protein ratios of 1:5, 1:15, 1:50 and 1:100 with 2.5mM TCEP at room temperature. No trypsin was added to the control reaction. The mixture was incubated with 200 μM [14C]-methylmalonyl-CoA after 30 minutes of trypsin digestion. The total volume of the reaction mixture is 10 μL. The reaction was quenched after 1 minute of [14C]-methylmalonyl CoA addition with 5 μL of SDS-PAGE sample buffer. The samples were loaded on a SDS-PAGE gel. The gel was dried using a BioRad gel-drying system and analyzed using a Packard phosphorimager.
Acylation and transacylation of module 3+TE, module 6+TE mutants and stand-alone ACPs in the presence of discrete AT proteins
For kinetic assays, the reaction volume corresponding to each time point was 10 μL. All ACPs used were in their holo-form.
Transacylation of DEBS ACP6 in the presence of AT6 constructs
AT6 constructs (20 μM, in 100 mM phosphate at pH 7.2) were incubated at room temperature with 2.5 mM TCEP and 200 μM [14C]-methylmalonyl-CoA, with discrete DEBS ACP6 protein (80 μM). 9 ⎧L of the mixture was removed at 1 and 5 minutes time points and quenched with 5 ⎧L SDS-PAGE loading buffer and loaded on a SDS-PAGE gel. The gel was dried using a BioRad gel-drying system and analyzed using a phosphorimager.
Acylation and transacylation of module 3+TE and module 6+TE mutants in the presence of discrete AT proteins
Wild-type or mutant modules (30 μM, in 100 mM phosphate at pH 7.2) were incubated for 10 minutes on ice with 2.5 mM TCEP and 30 μM [14C]-methylmalonyl-CoA (for DEBS ATs) or [14C]-malonyl-CoA (for DSZS ATs), alone or in conjunction with a discrete AT protein (30 μM). Samples were quenched with 5 ⎧L SDS-PAGE loading buffer and loaded on a SDS-PAGE gel. Samples were processed as described above.
Transacylation rates of modular ACPs by discrete AT proteins
Module 3/AT°+TE (20-100 ⎧M, in 100mM phosphate at pH 7.2) were incubated at room temperature with 0.03 ⎧M discrete DSZS ATS, 2.5mM TCEP and 200 ⎧M [14C]-malonyl-CoA. 9 ⎧L of the mixture was removed at intervals of 1 minute and quenched with 5 ⎧L SDS-PAGE loading buffer. The samples were loaded on a SDS-PAGE gel and processed as described above.
Transacylation rates of discrete ACPs by DSZS ATS
Discrete ACPs, (20-150 ⎧M, in 100mM phosphate, pH 7.2) were incubated on room temperature with DSZS ATS (0.003 ⎧M for cognate DSZS ACP1 transacylation, 0.03 ⎧M for transacylation of DEBS ACPs), 2.5mM TCEP and 200 ⎧M [14C]-malonyl-CoA. 9 ⎧L of the mixture was removed at 1-minute intervals and quenched with 5 ⎧L SDS-PAGE loading buffer and loaded on a SDS-PAGE gel. Samples were processed as described above.
Transacylation rates of discrete ACPs by DEBS ATs and [KS][AT]s
Discrete ACPs, (20-100 ⎧M, in 100mM phosphate, pH 7.2) were incubated on room temperature with discrete DEBS ATs (and [KS][AT]s), 2.5mM TCEP and 200 ⎧M [14C]-methylmalonyl-CoA. Depending on the ACP – AT combination used, 0.5-10 ⎧M DEBS AT (or [KS][AT]) was used for each series of tranacylation assays. 9 ⎧L of the mixture was removed at intervals of 2 minutes and quenched with 5 ⎧L SDS-PAGE loading buffer and loaded on a SDS-PAGE gel. Samples were processed as described above.
Transacylation assays for kcat/KM calculations (Table 1) were repeated at least three independent times.
Table 1.
Apparent kcat/KM values for transacylation of ACPs by alternative AT constructsa.
| AT | ACP | kcat/KM (M s)-1 |
|---|---|---|
| [KS3][AT3] | ACP3 | 390 ± 50 |
| ACP6 | 21 ± 8 | |
| [KS6][AT6] | ACP3 | 6 ± 1 |
| ACP6 | 86 ± 14 | |
| AT6 | ACP3 | 3 ± 0.3 |
| ACP6 | 69 ± 7 | |
| AT6small, PAT | ACP3 | 4 ± 1 |
| ACP6 | 19 ± 3 | |
| DSZS ATS | DSZS ACP1 | (87 ± 13) × 103 |
| ACP3 | (14 ± 3) × 103 | |
| Module 3/AT°+TE | (9 ± 1) × 103 | |
ATs and ACPs are from DEBS, unless otherwise indicated
Docking models of DEBS AT and ACP
Homology models for ACP3, ACP6 and AT6 from DEBS were derived using the I-TASSER server [24-26].
In silico docking experiments involving AT (or [KS][AT]) and ACP proteins were performed using PatchDock [27, 28] and then further refined and re-ranked with FireDock [29, 30]. For PatchDock simulations, ATs and ACPs were set as receptor and ligand respectively under default complex type settings.To simulate ACP docking on AT, a maximum distance constraint of 25 Å was placed between AT and ACP active site Ser residues. This was to ensure that the catalytic AT Ser residue was not beyond the reach of the approximately 18 Å-long panethetinylated arm of the holo-ACP.
Results
Design and production of stand-alone AT constructs from DEBS
Analysis of the X-ray structures of [KS3][AT3] and [KS5][AT5] from DEBS led to the design of a prototypical stand-alone AT from module 3 of DEBS [13]. From an initial analysis of two AT3 constructs, both of which contain the N-terminal KS-to-AT linker with or without the C-terminal post AT linker, it was observed that the absence of the post AT linker negatively affects KS activity but not AT activity [13]. In this study we constructed and analyzed a panel of stand-alone AT constructs from module 3 as well as other DEBS modules. As shown in Figure 2, these constructs included the KS-to-AT linker, the post AT linker, neither linker, or both linkers.
Figure 2.
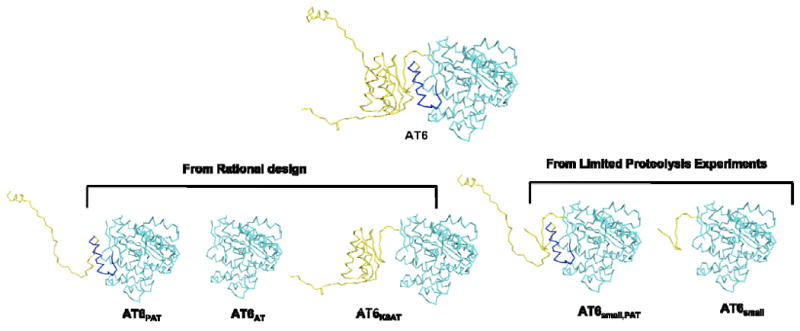
Ribbon models of truncated AT6 constructs from rational design and limited proteolysis experiments. The model for AT6 was generated using I-TASSER server [24-26]. The AT domain, C-terminal helix, and both linkers are shown in cyan, dark blue and yellow respectively, and are all present in construct “AT6”.
AT constructs harboring both the KS-to-AT linker and the post AT linker from DEBS modules 3, 4, 5, and 6 (denoted as AT3, AT4, AT5, and AT6, respectively) were expressed in E. coli as soluble proteins at approximately 3 mg, 10 mg, 15 mg, and 50 mg purified protein per liter culture, respectively. No soluble protein was obtained for the AT domains from modules 1 or 2. To understand the contributions of the N- and C-terminal linkers to AT activity and specificity, we also attempted to produce truncated derivatives of AT6 that lacked either or both linkers (Figure 2). Our initial attempt at determining linker boundaries for these truncated constructs entailed rational design based on our knowledge of previously solved crystal structures of [KS][AT] [14, 15]. Because the AT domain harbors a conserved C-terminal helix that interacts with the KS-to-AT linker, some of our constructs also lacked this helix. AT6 constructs without the KS-to-AT linker (denoted AT6PAT) and without both linkers (denoted as AT6AT) were produced with a yield of 30 and 7 mg per liter culture respectively. Soluble proteins were not obtained for AT6 construct without the post AT linker and C-terminal helix (denoted as AT6KSAT).
A truncated acyl transferase construct developed through limited proteolysis
Analysis of the above constructs (detailed below) revealed that some proteins had comparable activity to the reference stand-alone AT constructs, whereas the activity of others was substantially attenuated. Together, these results illustrated the limits of structure- and sequence-based engineering of stand-alone enzymes that are ordinarily part of larger multi-domain proteins. We therefore turned to limited proteolysis as an alternate strategy for identifying an active truncated derivative of AT6, which included the KS-to-AT linker and post AT linker (Figure 2). After treatment of the AT6 protein with varying amounts of trypsin, [14C]-methylmalonyl-CoA was added to the partially digested mixture in order to identify fragments competent for self-acylation. The corresponding protein bands from an SDS-PAGE gel were extracted and subjected to LC-MS/MS analysis in order to identify the relevant N- and C-termini (Figure 3). In particular, the sequence of the 40 kDa proteolytic fragment was analyzed. LC-MS/MS generated peptide sequences (>95% confidence) were used to determine its boundaries. Overall, 67% coverage of the predicted amino acid sequence of this fragment (predicted MW 39,422 Da) was obtained. More importantly, continuous coverage of the N- and C-termini was obtained over 37 and 59 residues, respectively. Whereas unambiguous border identification is not possible by LC-MS/MS, these results motivated us to test the properties of the predicted construct, which starts at the amino acid sequence GVAAPGATTGTA (denoted as AT6small, PAT, Figure 2). The construct was expressed as a soluble protein and purified in a yield of 6 mg per liter culture. As seen in Figure 4, robust self-acylation of this protein was observed in the presence of [14C]-methylmalonyl-CoA. The corresponding protein with the same N-terminus but lacking the C-terminal helix was obtained as a soluble protein (AT6small), but was only weakly acylated by [14C]-methylmalonyl-CoA.
Figure 3.
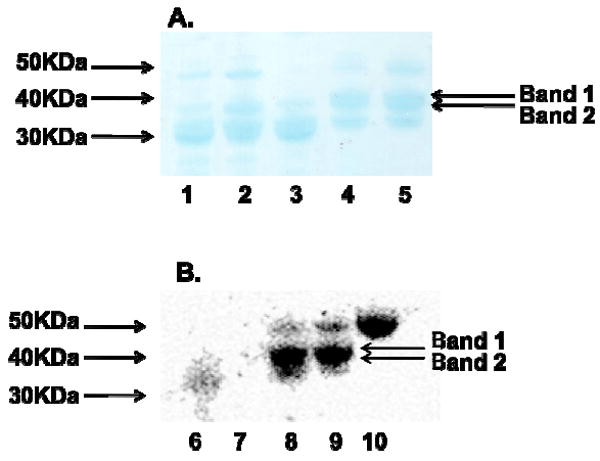
Limited Proteolysis of AT6 and identification of active truncated variants. (Panel A) Partial trypsin digestion of DEBS AT6 (30 minutes at room temperature). AT6 was incubated with molar ratios of trypsin: AT6 ranging from 1:5 (Lane 1), 1:15 (Lane 2), 1:15 (Lane 3, incubated for 1 hour), 1:50 (Lane 4) to 1:100 (Lane 5). (Panel B) Radio-SDS-PAGE analysis of trypsin digested AT6 fragments incubated with [14C]-methylmalonyl CoA. AT6 was incubated with molar ratios of trypsin: AT6 of 1:5 (Lane 6), 1:15 (Lane 7), 1:50 (Lane 8), 1:100 (Lane 9). AT6 with no trypsin added was used as a control (Lane 10).
Figure 4.

Radio-SDS-PAGE analysis of transacylation and acylation activities of truncated DEBS AT6 constructs with DEBS ACP6. [14C]-methylmalonyl-CoA was incubated for 1 minute (Panel A) and 5 minutes (Panel B) with AT6AT (Lane 1), AT6PAT (Lane 2), AT6small (Lane 3), AT6small,PAT (Lane 4) and AT6 (Lane 5) with DEBS ACP6.
Acylation and transacylation activities of alternative stand-alone AT constructs
A functional AT construct from DEBS should be able to self-acylate as well as to transacylate its cognate ACP domain, while also retaining specificity for methylmalonyl-CoA. To investigate these properties of the alternative forms of AT6 and as well as the truncated AT6 derivatives that were obtained via limited proteolysis (Figure 2), assays with [14C]-methylmalonyl-CoA and ACP6 were performed. Radio-SDS-PAGE (Figure 4) revealed that AT6AT and AT6small self-labeled more weakly than AT6. Similarly, AT6AT and AT6small were unable to transfer the extender unit onto the ACP (Figure 4). AT6pat, AT6small,PAT and AT6 showed a comparable ability to acylate the ACP.
Expression and purification of recombinant DSZS AT
The disorazole synthase (DSZS) is a member of an “AT-less” Type I polyketide synthase with a standalone AT that transacylates extender units onto all ACP domains of this PKS. This AT is part of a larger (91 kD) protein that also includes a C-terminal putative oxidoreductase domain separated by a linker of unknown function [17]. Based on sequence alignments with known acyltransferases from structurally characterized bacterial fatty acid synthases [31], two versions of the disorazole AT were engineered, one with the ca. 100 residue linker (DSZS ATl) and the other without this linker (DSZS ATS). Both constructs were expressed as soluble proteins at yields of 80-110 mg purified protein per liter culture.
Expression, purification and characterization of ACP proteins
To measure transacylation rate of an ACP by an AT, the kinetics of radiolabel transfer from [14C]-malonyl-CoA (for DSZS AT) or [14C]-methylmalonyl-CoA onto each ACP was quantified. The ACP3 and ACP6 domains from DEBS were expressed using similar constructs as described before [12]. DSZS is predicted to have eight distinct ACP domains, one in each module except for module 7, which has two ACPs [17]. Based on sequence alignments with Type I and II PKS acyl carrier proteins, a stand-alone acyl carrier protein (DSZS ACP1) was constructed from module 1 of DSZS. This construct was expressed in E. coli BAP1 [22], and purified as soluble holo-ACP at a yield of 1 mg per liter culture.
Expression and purification of AT° mutants of DEBS module 3+TE and module 6+TE
To attempt complementation of intact modules by stand-alone AT domains, mutants of DEBS modules 3 and 6 with inactive AT domains were engineered (denoted module 3/AT°+TE and module 6/AT°+TE, respectively). In each case AT inactivation was achieved via a Ser→Ala mutation at the AT active site. Both mutants were produced as soluble proteins at approximately 50 mg purified protein per liter culture.
ACP specificity of acyltransferases
In an intact DEBS module, an AT domain catalyzes acyl transfer from methylmalonyl-CoA to the ACP domain within the same module via a covalent acyl-AT intermediate. As an initial assessment of whether DEBS AT domains are specific for their natural ACP partners, the AT domains from modules 3-6 of DEBS (with intact KS-to-AT and post AT linker) were individually incubated with DEBS module 3/AT°+TE or module 6/AT°+TE derivatives of DEBS in the presence of [14C]-methylmalonyl-CoA. As seen in Figure 5, although the AT proteins were comparably labeled by the methylmalonyl extender unit, different combinations of modules and stand-alone AT proteins showed different degrees of AT-to-ACP transacylation capability. Moreover, the transacylation preferences for the two modules were distinct from one another.
Figure 5.
Radio SDS-PAGE analysis of acylation and transacylation activities of stand-alone AT proteins from DEBS using AT° mutants of modules 3 and 6 as ACP substrates. [14C]-methylmalonyl-CoA was incubated with module 3/AT°+TE (panel A, lanes 1-4) or module 6/AT°+TE (panel B, lanes 1-4) in the presence of alternative acyltransferases. In each panel: Lane 1: DEBS AT3. Lane 2: DEBS AT4. Lane 3: DEBS AT5. Lane 4: DEBS AT6. Lane 5: The corresponding wild-type module with an active AT (control).
We therefore quantified the specificity of different AT constructs from DEBS modules 3 and 6 for ACP3 and ACP6. A representative transacylation rate versus [ACP] data set is plotted in Figure 6, the slope of which yielded an apparent kcat/KM value. The results of all such experiments are summarized in Table 1. For both the [KS][AT] and the AT6 constructs, a 10-20 fold preference is observed for the cognate ACP. Moreover, the rate constants are comparable in magnitude for the [KS6][AT6] and the AT6 constructs. In contrast, both the activity and specificity of the smaller AT construct AT6small,PAT were modestly reduced. The latter construct was chosen for kinetic analysis, because it was the most active of the truncated AT6 derivatives analyzed in this study.
Figure 6.
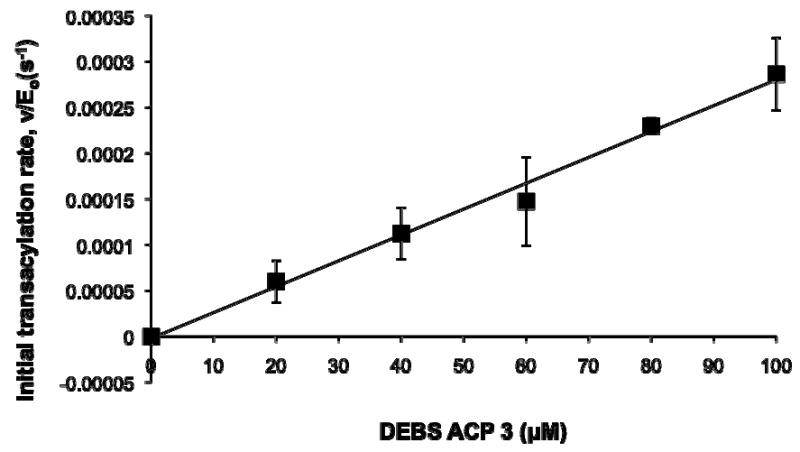
A typical transacylation rate (s-1) vs DEBS [ACP] (μM) plot. 20-100 μM DEBS ACP3 was incubated with 10 μM DEBS AT6. [14C]-methylmalonyl-CoA was held constant at 200 μM. Initial rates were determined at room temperature at least three independent times. kcat/KM (μM s)-1 was obtained through linear fitting of the data.
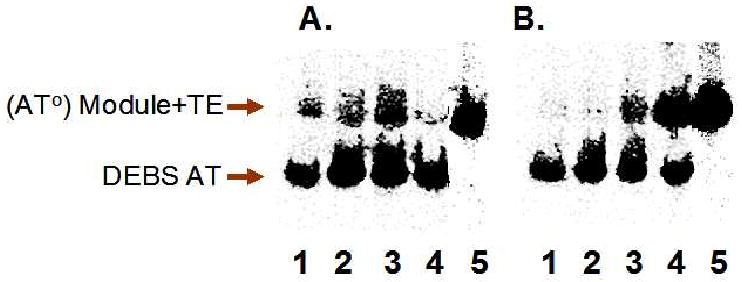
We also performed similar measurements with the AT protein from the DSZS. To test the acylation and transacylation activities of DSZS ATS and DSZS ATL, each protein was incubated with [14C]-malonyl-CoA in the presence of the AT° mutants of DEBS module 3+TE or module 6+TE. As seen in Figure 7, both enzymes catalyzed self-acylation and transacylation to the ACP domain with comparable efficiency. Thus, the 100-residue post AT linker did not appear to have a significant effect on either component of DSZS AT activity. When [14C]-methylmalonyl-CoA was used instead of [14C]-malonyl-CoA, no acyl transfer to either of the two DEBS modules was detected, although some labeling of the DSZS AT proteins could be observed (data not shown). Most remarkably, the activity of DSZS ATS was considerably higher against all ACPs evaluated than even that of the cognate AT (Table 1). Thus, although DSZS AT has higher specificity for its own substrates, it is still able to efficiently charge heterologous ACP domains.
Figure 7.
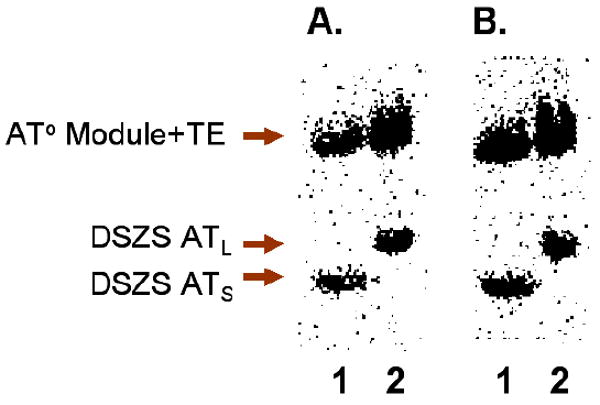
Radio SDS-PAGE analysis of DSZS AT activity. [14C]-malonyl-CoA was incubated with DEBS module 3/AT°+TE (panel A) or module 6/AT°+TE (panel B) in the presence of recombinant DSZS AT constructs. The discrete AT labeled the ACP domain of each module. Lane 1: DSZS ATS (DSZS AT without post-AT linker). Lane 2: DSZS ATL (DSZS AT with post AT linker).
It should be noted that, in all of the above experiments, AT-ACP specificity was quantified by measuring and comparing the overall specificity constant (kcat/KM) of the transacylation reaction. The observed differences in this parameter should not be mistaken for inherent AT-ACP affinity, which would be more appropriately investigated via protein binding experiments such as isothermal titration calorimetry.
Discussion
In a multimodular PKS, the starter unit is elongated by decarboxylative condensation with extender units as it progresses through the enzymatic assembly line. These extender units are selected by acyltransferase (AT) domains that acts as gatekeepers in each PKS module. The importance of the AT to the stringent incorporation of specific extender unit in the synthesis of polyketide building blocks makes it vital that the mechanism and structure of these domains be well-elucidated in order to develop efficient strategies for the regiospecific engineering of extender unit incorporation in polyketide biosynthesis. Here we have quantified the ACP specificity of representative AT domains derived from multimodular PKSs, and have examined the role of flanking linkers on both AT activity and specificity.
A key finding of our study is that, whereas stand-alone AT constructs harboring intact N- and C-terminal linkers have properties comparable to the corresponding AT domains that are part of larger multidomain systems, deletion of either linker has a deleterious effect on both the activity and specificity of the mutant AT. The most active of the truncated constructs consisted of core AT domain flanked by the proximal 15 C-terminal residues of the KS-to-AT linker and the post AT linker (AT6small, PAT). Nonetheless, the activity of even that truncated AT was measurably attenuated. It thus appears that the intact linkers play an important role in ACP recognition and/or catalysis of acyl transfer. The mechanistic basis for such an effect remains to be elucidated.
Our investigations into the ACP specificity of AT domains focused on DEBS ACP3 and ACP6 as substrates, because these two ACPs are anticipated to have the most different electrostatic surfaces among the six DEBS ACPs [16]. Similarities in kcat/KM values between [KS][AT] didomain and stand-alone AT constructs (Table 1) suggested that the presence of the KS domain does not significantly influence AT-ACP recognition. This situation is radically different from KS-ACP recognition, which is influenced by the presence of both the AT domain and the post AT linker [13]. We note that the above analysis of AT-ACP specificity is based on in trans analysis of enzyme activity that ordinarily functions in cis. Earlier studies on nonribosomal peptide synthetases [32] and polyketide synthases [7] have observed minimal effects on the catalytic activities of heterologous adenylation and AT domains, respectively, when introduced in an in cis configuration. Thus, the actual consequence of sub-optimal AT-ACP interactions on the turnover number of the overall assembly line remains to be elucidated.
Protein-protein docking simulations were performed with PatchDock [27, 28] using the known structures of DEBS [KS3][AT3] (PDB ID 2QO3) and ACP2 (PDB ID 2JU2) as well as a homology model of ACP3 (generated via the I-TASSER server [24-26]). Several of the lowest energy docking models emerging from this exercise placed the ACP on the side of the AT protein that opens to the gorge leading to the AT active site serine. A representative AT3-ACP3 docking model is shown in Figure 8. In this model, helix I, helix II and loop II regions of the ACP are docked onto the beta strands of the KS-to-AT linker, the smaller subdomain of AT3, and the C-terminal helix of the AT. All other docking models generated by PatchDock simulations exhibited similar orientations between the AT and the ACP. The distance between the active sites was 23 Å, consistent with the interaction of the 18 Å phosphopantetheinyl arms with the AT active site Ser.
Figure 8.
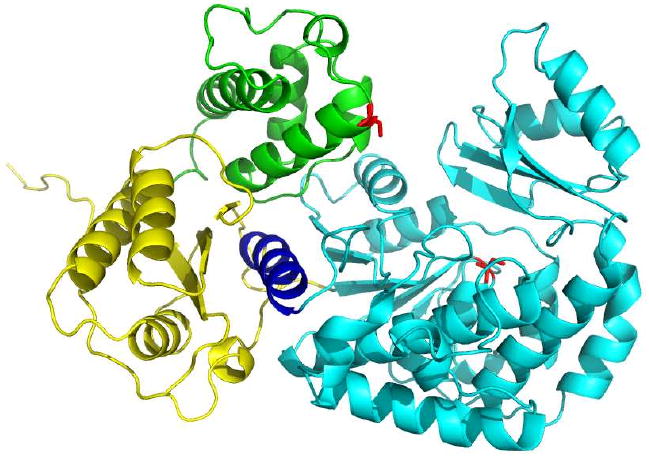
PatchDock docking model for AT3 with linkers (truncated from PDB ID 2QO3, [15]) and DEBS ACP3 (homology model generated by I-TASSER). AT domain, linkers, C-terminal helix and ACP domain are shown in cyan, yellow, dark blue and green respectively. Active site Ser residues are shown as red.
This model for AT-ACP interaction may explain the observed specificity of AT3 and AT6 for their cognate ACPs (Table 1). The proposed AT-ACP interactions involves docking of the ACP onto the KS-to-AT linker and C-terminal helix of the AT. The sequences of both regions are highly variable amongst the DEBS modules, and highlight the importance of considering this inter-domain interface in future biosynthetic engineering strategies for altering extender unit specificity of PKSs. Indeed, earlier work has already demonstrated that mutations in the C-terminal helix can influence AT substrate specificity [33].
Perhaps not surprisingly, the DSZS AT has a markedly higher (250-1000 fold) transacylation efficiency for its cognate DSZS ACP1 in comparison to the maximum activity of stand-alone AT domains from DEBS. The evolution of this high in trans AT activity is presumably facilitated by the intermodular nature of DSZS AT's mode of action on its cognate ACP substrates. It is nonetheless impressive that the rate at which DSZS AT catalyzes acyl transfer onto heterologous ACP domains is only modestly lower (10-fold) than acyl transfer to its cognate ACP domains. This observation is in stark contrast to an estimated ∼5000 fold difference in the rate at which the malonyl CoA: acyl carrier protein transacylase from the fatty acid synthase acylates its own ACP as compared to DEBS ACP2, and suggests that the DSZS acyltransferase may be a superior candidate for in trans complementation of engineered PKSs [10]. This hypothesis is currently being tested in our laboratories.
In conclusion, this study highlights the importance of AT-ACP specificity in multimodular PKSs. Such interactions, which were overlooked in virtually all earlier attempts to engineer PKSs via domain substitution, must clearly be carefully considered in the design of structurally stable and catalytically active hybrid PKSs. Our results also suggest improved boundaries for AT domain substitution that include the flanking linker regions.
Acknowledgments
We thank the Vincent Coates Foundation Mass Spectrometry Laboratory at Stanford University (SUMS). We are also grateful to Chris Adams at SUMS for help with LC-MS/MS analysis. We thank Shiven Kapur for plasmid pSHIV9. This work was supported by grants from the NIH (GM087934 to C.K. and GM022172 to D.E.C.), by a Stanford Graduate Fellowship to A.Y.C., and by a National Science Scholarship from the Agency of Science, Technology and Research (A*STAR), Singapore, to F.T.W.
Abbreviations
- AT
Acyltransferase
- CoA
Coenzyme A
- ACP
Acyl Carrier Protein
- DEBS
6-Deoxyerythronolide B Synthase
- DSZS
Disorazole Synthase
- KS
β-Ketosynthase
- PKS
Polyketide Synthase
- TE
Thioesterase
- DTT
Dithiothreitol
- LC-MS/MS
Liquid Chromatography Tandem Mass Spectrometry
- TCEP
Tris(2-carboxyethyl)phosphine
- SDS-PAGE
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis
Footnotes
This research was supported by grants from the NIH (GM087934 to C.K. and GM022172 to D.E.C.).
References
- 1.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Structure and Mechanism of the 6-Deoxyerythronolide B Synthase. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA, Walsh CT. Assembly-Line Enzymology for Polyketide and Nonribosomal Peptide Antibiotics: Logic, Machinery, and Mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 3.Hill AM. The Biosynthesis, Molecular Genetics and Enzymology of the Polyketide-Derived Metabolites. Nat Prod Rep. 2006;23:256–320. doi: 10.1039/b301028g. [DOI] [PubMed] [Google Scholar]
- 4.Weissman KJ, Leadlay PF. Combinatorial Biosynthesis of Reduced Polyketides. Nature Reviews Microbiology. 2005;3:925. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- 5.Wenzel SC, Müller R. Formation of Novel Secondary Metabolites by Bacterial Multimodular Assembly Lines: Deviations from Textbook Biosynthetic Logic. Curr Opin Chem Biol. 2005;9:447. doi: 10.1016/j.cbpa.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 6.McDaniel R, Welch M, Hutchinson CR. Genetic Approaches to Polyketide Antibiotics. 1. Chem Rev. 2005;105:543. doi: 10.1021/cr0301189. [DOI] [PubMed] [Google Scholar]
- 7.Hans M, Hornung A, Dziarnowski A, Cane DE, Khosla C. Mechanistic Analysis of Acyl Transferase Domain Exchange in Polyketide Synthase Modules. J Am Chem Soc. 2003;125:5366–5374. doi: 10.1021/ja029539i. [DOI] [PubMed] [Google Scholar]
- 8.Ruan X, Pereda A, Stassi DL, Zeidner D, Summers RG, Jackson M, Shivakumar A, Kakavas S, Staver M, Donadio S, Katz L. Acyltransferase Domain Substitutions in Erythromycin Polyketide Synthase Yield Novel Erythromycin Derivatives. J Bacteriol. 1997;179:6416–6425. doi: 10.1128/jb.179.20.6416-6425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oliynyk M, Brown MJB, Cortés J, Staunton J, Leadlay PF. A Hybrid Modular Polyketide Synthase obtained by Domain Swapping. Chem Biol. 1996;3:833–839. doi: 10.1016/s1074-5521(96)90069-1. [DOI] [PubMed] [Google Scholar]
- 10.Kumar P, Koppisch AT, Cane DE, Khosla C. Enhancing the Modularity of the Modular Polyketide Synthases: Transacylation in Modular Polyketide Synthases Catalyzed by Malonyl-CoA:ACP Transacylase. J Am Chem Soc. 2003;125:14307–14312. doi: 10.1021/ja037429l. [DOI] [PubMed] [Google Scholar]
- 11.Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. Extender Unit and Acyl Carrier Protein Specificity of Ketosynthase Domains of the 6-Deoxyerythronolide B Synthase. J Am Chem Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim CY, Alekseyev VY, Chen AY, Tang Y, Cane DE, Khosla C. Reconstituting Modular Activity from Separated Domains of 6-Deoxyerythronolide B Synthase. Biochemistry. 2004;43:13892–13898. doi: 10.1021/bi048418n. [DOI] [PubMed] [Google Scholar]
- 13.Chen AY, Cane DE, Khosla C. Structure-Based Dissociation of a Type I Polyketide Synthase Module. Chemistry & Biology. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C. The 2.7-Å Crystal Structure of a 194-kDa Homodimeric Fragment of the 6-Deoxyerythronolide B Synthase. Proc Natl Acad Sci USA. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang Y, Chen AY, Kim CY, Cane DE, Khosla C. Structural and Mechanistic Analysis of Protein Interactions in Module 3 of the 6-Deoxyerythronolide B Synthase. Chemistry & Biology. 2007;14:931–943. doi: 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alekseyev VY, Liu CW, Cane DE, Puglisi JD, Khosla C. Solution Structure and Proposed Domain Domain Recognition Interface of an Acyl Carrier Protein Domain from a Modular Polyketide Synthase. Protein Sci. 2007;16:2093–107. doi: 10.1110/ps.073011407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carvalho R, Reid R, Viswanathan N, Gramajo H, Julien B. The Biosynthetic Genes for Disorazoles, Potent Cytotoxic Compounds that Disrupt Microtubule Formation. Gene. 2005;359:91–98. doi: 10.1016/j.gene.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Kopp M, Irschik H, Pradella S, Rolf Müller R. Production of the Tubulin Destabilizer Disorazol in Sorangium Cellulosum: Biosynthetic Machinery and Regulatory Genes. ChemBioChem. 2005;6:1277–1286. doi: 10.1002/cbic.200400459. [DOI] [PubMed] [Google Scholar]
- 19.Cheng Y, Tang G, Shen B. Type I Polyketide Synthase Requiring a Discrete Acyltransferase for Polyketide Biosynthesis. Proc Natl Acad Sci USA. 2003;100:3149–3154. doi: 10.1073/pnas.0537286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen T, Ishida K, Jenke-Kodama H, Dittmann E, Gurgui C, Hochmuth T, Taudien S, Platzer M, Hertweck C, Piel J. Exploiting the Mosaic Structure of Trans-Acyltransferase Polyketide Synthases for Natural Product Discovery and Pathway Dissection. Nat Biotech. 2008;26:225–233. doi: 10.1038/nbt1379. [DOI] [PubMed] [Google Scholar]
- 21.Gokhale RS, Tsuji SY, Cane DE, Khosla C. Dissecting and Exploiting Intermodular Communication in Polyketide Synthases. Science. 1999;284:482–485. doi: 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]
- 22.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Biosynthesis of Complex Polyketides in a Metabolically Engineered Strain of E. Coli. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 23.Walsh CT, Gehring AM, Weinreb PH, Quadri LE, Flugel RS. Post-Translational Modification of Polyketide and Nonribosomal Peptide Synthases. Curr Opin Chem Biol. 1997;1:309–315. doi: 10.1016/s1367-5931(97)80067-1. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y. Template-Based Modeling and Free Modeling by I-TASSER in CASP7. Proteins. 2007;69:108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- 26.Wu ST, Skolnick J, Zhang Y. Ab Initio Modeling of Small Proteins by Iterative TASSER Simulations. BMC Biology. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duhovny D, Nussinov R, Wolfson HJ. Efficient Unbound Docking of Rigid Molecules. Lecture Notes in Computer Science. 2002;2452:185–200. [Google Scholar]
- 28.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucl Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrusier N, Nussinov R, Wolfson HJ. FireDock: Fast Interaction Refinement in Molecular Docking. Proteins: Structure, Function, and Bioinformatics. 2007;69:139–159. doi: 10.1002/prot.21495. [DOI] [PubMed] [Google Scholar]
- 30.Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ. FireDock: A Web Server for Fast Interaction Refinement in Molecular Docking. Nucl Acids Res. 2008;36:W229–232. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenke-Kodama H, Sandmann A, Muller R, Dittmann E. Evolutionary Implications of Bacterial Polyketide Synthases. Mol Biol Evol. 2005;22:2027–2039. doi: 10.1093/molbev/msi193. [DOI] [PubMed] [Google Scholar]
- 32.Linne U, Stein DB, Mootz HD, Marahiel MA. Systematic and Quantitative Analysis of Protein-Protein Recognition between Nonribosomal Peptide Synthetases Investigated in the Tyrocidine Biosynthetic Template. Biochemistry. 2003;42:5114–5124. doi: 10.1021/bi034223o. [DOI] [PubMed] [Google Scholar]
- 33.Lau J, Fu H, Cane DE, Khosla C. Dissecting the Role of Acyltransferase Domains of Modular Polyketide Synthases in the Choice and Stereochemical Fate of Extender Units. Biochemistry. 1999;38:1643–1651. doi: 10.1021/bi9820311. [DOI] [PubMed] [Google Scholar]