

Abstract
Background
There is evidence that disaccharide sucrose produce a greater increase in serum fructose and triglycerides (TGs) than the effect produced by their equivalent monosaccharides, suggesting that long-term exposure to sucrose or fructose + glucose could potentially result in different effects.
Aim of the study
We studied the chronic effects of a combination of free fructose and glucose relative to sucrose on rat liver.
Methods
Rats were fed either a combination of 30% fructose and 30% glucose (FG) or 60% sucrose (S). Control rats were fed normal rat chow (C). All rats were pair fed and were followed for 4 months. After killing, blood chemistries and liver tissue were examined.
Results
Both FG-fed- and S-fed rats developed early features of metabolic syndrome when compared with C. In addition, both diets induced hepatic alterations, including variable increases in hepatic TG accumulation and fatty liver, an increase in uric acid content in the liver, as well as an increase in hepatic levels of monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor-alpha (TNF-α) measured in liver homogenates.
Conclusions
Diets containing 30% of fructose either as free fructose and glucose, or as sucrose, induce metabolic syndrome, intrahepatic accumulation of uric acid and TGs, increased MCP-1 and TNF-α as well as fatty liver in rats. It will be relevant to determine clinically whether pharmacological reduction in uric acid levels might have a therapeutic advantage in the treatment of non-alcoholic fatty liver disease.
Keywords: Non-alcoholic steatosis, Metabolic syndrome, Sucrose, Fructose
Introduction
Fructose intake and/or sugar-sweetened beverages have recently received attention as a potential risk factor for metabolic syndrome and diabetes [15, 16, 33, 38]. The intake of fructose, as sucrose (disaccharide consisting of glucose and fructose) or as high-fructose corn syrup (HFCS), has increased markedly in the last 2 decades in association with increasing rates of obesity, diabetes, hypertension, and kidney disease [16]. Moreover, experimental evidence has shown that fructose can induce metabolic syndrome in animals, whereas this is not observed in controls fed equal amounts of glucose [28]. Studies in humans have also shown that high doses of fructose can result in insulin resistance, postprandial hypertriglyceridemia, intra-abdominal fat accumulation, and elevated blood pressure [3, 5, 13, 14, 40, 41, 43]. In this regard, Swarbrick et al. showed that prolonged fructose consumption can increase fasting glucose, postprandial triglycerides (TGs), and apolipoprotein-B in overweight women [42].
The mechanism by which fructose causes metabolic syndrome is due to its unique metabolism which facilitates the synthesis of TGs and uric acid (reviewed in ref. [15, 29]). Furthermore, the incubation of human endothelial cells, renal tubular cells and hepatocytes with postprandial concentrations of fructose induced the overexpression and increased activity of fructokinase, reduced intracellular ATP, increased intracellular uric acid levels and activated proinflammatory and pro-oxidative responses [7, 12, 32]. Of note is that allopurinol blocked fructose-induced monocyte chemoattractant protein-1 (MCP-1) production in renal tubular cells and stimulation with 15 mg/dl uric acid mimicked the effect of fructose, suggesting that xanthine oxidase pathway might mediate the proinflammatory effects of fructose [7].
Long-term fructose intake is also associated with nonalcoholic fatty liver disease (NAFLD) which is another manifestation of the metabolic syndrome [32]. Fructose rapidly increases hepatic TG synthesis and deposition, and can cause fatty liver in rats [2]. Chronic sucrose ingestion has also been reported to raise TG and liver transaminase enzymes in humans [34]. In one study, the amount of fructose ingested from soft drinks was over twofold higher in subjects with NAFLD than that in the control group (89 vs. 42 g/day) [32]. These individuals also displayed twofold higher levels of fructokinase mRNA, consistent with a history of increased fructose intake [32].
The hallmark feature of the pathogenesis of NAFLD, both histologically and metabolically, is the accumulation of TGs in the liver [10]. Regulation of fatty acid metabolism is highly different in fasting and fed states. Recent studies showed that 59% of TG within hepatocytes arose from the plasma-free fatty acid pool in the fed state [10]. Interestingly, three studies have shown that postprandial hypertriglyceridemia correlates with fat liver content [25, 30, 35]. In one study of patients with type-II diabetes mellitus and normal fasting TG, the increase in postprandial TG levels was directly predicted by the amount of hepatic fat [30]. In this regard, the administration of fructose-sweetened beverages providing 25% of the daily caloric intake for 10 weeks was shown to increase hepatic de novo lipogenesis and reduce postprandial lipoprotein lipase activity suggesting that both mechanisms may contribute to fructose-induced postprandial hypertriglyceridemia [41].
If fructose is a mediator of metabolic syndrome, a key question is whether sucrose is different from their individual components, fructose and glucose, in its metabolic effects. Sucrose is degraded by intestinal sucrase (EC 3.2.1.26) prior to the release of fructose for absorption. Acute studies have shown that serum fructose and TGs increase more after a sucrose drink than when an equivalent amount of a mixture of fructose and glucose is administered to healthy individuals (disaccharide effect) [22, 44]. Moreover, there are a few short-term studies that have compared the effects of sucrose with HFCS, which contains roughly the same amount of fructose and glucose as free sugars. In general, those studies have not found significantly different effects on appetite and satiety between both sweeteners [26, 27]. Interestingly, in male humans, a short term study showed that consuming HFCS- and sucrose-sweetened beverages increased postprandial TGs concentrations to the same extent as fructose alone [39]. However, it is unknown whether the effects of sucrose (disaccharide) vs. free monosaccharides (fructose + glucose) on liver adaptation differ in a chronic setting.
We, therefore, studied the long-term effects of a combination of free fructose and free glucose compared with equivalent amounts of fructose and glucose supplied as sucrose on the development of fatty liver in rats.
Methods
Animal protocol
All animal protocols were approved by the University of Florida Institutional Animal Care and Use Committee. Twelve 150 g male Sprague-Dawley rats (Charles Rivers Laboratories, Wilmington, MA, USA) were divided in two groups and received a diet of either 60% sucrose (S) or 30% fructose + 30% glucose (FG); in addition, six 150 g male Sprague-Dawley rats were used as normal controls and received standard rat chow (C). Details of experimental diets used in this study are given in Table 1. The animals were pair fed to ensure equal food intake. Weights were obtained weekly. Animals were maintained in temperature- and humidity-controlled specific pathogen-free conditions on a 12:12-h light–dark cycle. Postprandial serum samples were obtained from the tail vein at 8 weeks; at 16 weeks, the animals were anesthetized with isoflurane, and killed and blood obtained by cardiac puncture. Total abdominal fat from both mesenteric and retroperitoneal areas was excised and weighed. Liver tissue was retrieved for histology and molecular biology studies.
Table 1.
Relevant nutrient content of the rodent experimental diets
| Constituent | Normal diet (n = 6) |
Sucrose diet (n = 6) |
Fructose + glucose diet (n = 6) |
|---|---|---|---|
| Energy (kcal/g) | 3.6 | 3.6 | 3.6 |
| Starch (%) | 45.6 | – | – |
| Maltodextrin (%) | 20 | – | – |
| Sucrose (%) | – | 60.0 | – |
| Fructose (%) | – | – | 30.0 |
| Glucose (%) | – | – | 32.9 |
| Protein (%) | 20.7 | 20.7 | 20.7 |
| Fat (%) | 5.0 | 5.0 | 5.0 |
| Vitamins (%) | 1.0 | 1.0 | 1.0 |
Control diet (Cat. No. 05075, Harlan Teklad, Madison, WI, USA), sucrose diet (Cat. No. TD 06685. Harlan Teklad, Madison, WI, USA); fructose + glucose diet (Cat. No. TD 06684, Harlan Teklad, Madison, WI, USA); Nutrient composition presented as % of weight
Serum glucose, TGs, total cholesterol, and uric acid levels were measured using the VetAce autoanalyzer (Alpha Wasserman, West Caldwell, NJ, USA). Rat serum insulin, MCP-1 and tumor necrosis factor (TNF) were measured by ELISA using commercial kits (Insulin, Crystal Chem, Chicago, IL, USA; MCP-1 and TNF, BD Biosciences Pharmingen, San Diego, CA, USA).
Liver histology
Tissue was fixed in methyl-Carnoy's solution, dehydrated in alcohols, and embedded in paraffin (Sigma-Aldrich, St Louis, MO, USA), and 4-μm sections were stained with periodic-acid Schiff (PAS) stain. The histologic analysis was performed blinded regarding treatment groups. Steatosis attributable to NAFLD is typically macrovesicular rather than microvesicular [6]. In the present study, macrovesicular steatosis was defined as a single vacuole that is larger than the nucleus and usually displaces it to the periphery of the cell [31]. The scoring grade as the percentage of macrovesicles on PAS-stained liver tissue (20×) was set as follows; <25% = 0, 25% = 1+, 50% = 2+, 75% = 3+, and 100% = 4+. In addition, indirect immunoperoxidase for macrophages and for CD8-positive lymphocytes was performed using buffered paraformaldehyde-fixed tissue that had been frozen in liquid nitrogen using ED-1 and antiCD8 antibodies from BD Pharmingen (San Diego, CA, USA) as previously described. Twenty fields (1,100 μm × 1,400 μm) of each biopsy were measured at 200× magnification, and the mean number of macrophages per field was calculated. We also stained type-III collagen in methyl-Carnoy's fixed tissue with a goat anti-collagen III antibody (Southern Biotech, Birmingham, AL, USA) as a measure of fibrosis and determined the percent area of collagen deposition using an Axioplan 2 imaging microscope (Carl Zeiss, Munich, Germany) with Zeiss AutoMeasure software (Axiovision 4.1).
Measurements of liver tissue homogenates
Liver tissue homogenates were prepared by mechanical homogenization after the addition of fresh tissue to RIPA buffer (Sigma-Aldrich) to which Compete (Roche, Mannheim, Germany) protease inhibitors were added. Samples were incubated at 4°C for 1 h and then centrifuged at 4°C at 16,000g for 60 min. The supernatant was removed, and the protein concentration was standardized at 8 mg/ml after total protein measurement (Pierce, Rockford, IL, USA). Rat MCP-1 and TNF-alpha (TNF-α) protein was measured in the homogenates by ELISA with a kit (BD Biosciences Pharmingen, San Diego, CA, USA) according to the manufacturer's directions. Both the TG and uric acid levels in the liver tissue homogenates were determined using the VetAce autoanalyzer (Alpha Wasserman, West Caldwell, NJ, USA).
Statistical analysis
Statistical analysis was performed using Prism 5 for Windows (Graph Pad Software, Inc., San Diego, CA, USA). Values are expressed as mean ± SD. All studied parameters were analyzed by one-way ANOVA. When the ANOVA P value was <0.05, post-test comparisons were made using a Bonferroni multiple-comparison test. Correlation analysis was used to measure the degree to which two variables were related.
Results
General characteristics and systemic chemistries
The animals consumed a similar amount of calories per day and at the end of the experiment they had comparable body weights. These findings suggested that experimental diets were well tolerated. Data of total caloric intake, body weight, as well as abdominal, retroperitoneal and total fat are shown in Table 2. There were no differences among groups; however, both S- and FG-fed rats tended to have greater intra-abdominal, retroperitoneal and total fat compared with rats on the control diet (Table 2).
Table 2.
Mean caloric intake, and body weight (BW), abdominal, retroperitoneal and total fat (per 100 g of BW) after 4 months of a control, sucrose, or fructose–glucose diet
| Daily intake (kcal) | BW (g) | Abdominal fat (g) | Retroperitoneal fat (g) | Total fat (g) | |
|---|---|---|---|---|---|
| C (n = 6) | 202 ± 24 | 634 ± 63 | 1.53 ± 0.62 | 3.1 ± 0.79 | 4.6 ± 1.35 |
| S (n = 6) | 201 ± 24 | 662 ± 48 | 2.37 ± 0.41 | 4.2 ± 0.92 | 6.58 ± 1.23 |
| FG (n = 6) | 203 ± 6 | 647 ± 53 | 2.33 ± 0.70 | 4.2 ± 0.7 | 6.48 ± 1.36 |
C control diet, S sucrose diet, FG fructose + glucose diet
The effect of the various diets on postprandial blood chemistries obtained at 8 and 16 weeks are shown in Table 3. After 8 weeks, both experimental diets induced a significant increase in serum TGs and cholesterol relative to the control diet. Particularly, the F + G diet induced a 2.6 increase in serum TGs which reached statistical significance. Neither plasma glucose nor uric acid levels were different at this time point. At 16 weeks (Table 3), we observed a similar increase in TGs and cholesterol with the S and F + G diets. At this time point S increased serum UA by 25% and F + G by 40% relative to the control group, however only the latter reached statistical significance. Similarly, insulin was increased by S and F + G diets by 64 and 44% respectively, but only the S group was statistically significant (Table 3).
Table 3.
Laboratory tests obtained at 8 and 16 weeks in rats fed control, sucrose, or fructose–glucose diet in postprandial state
| TG (mg/dl) | Chol (mg/dl) | UA (mg/dl) | Gluc (mg/dl) | Insulin (ng/ml) | |
|---|---|---|---|---|---|
| 8 weeks | |||||
| C (n = 6) | 160 ± 97 | 81 ± 13 | 3.0 ± 0.2 | 164 ± 25 | |
| S (n = 6) | 284 ± 32 | 139 ± 26** | 3.4 ± 0.4 | 174 ± 18 | |
| FG (n = 6) | 422 ± 277* | 140 ± 34** | 3.0 ± 0.2 | 179 ± 26 | |
| 16 weeks | |||||
| C (n = 6) | 190 ± 103 | 85 ± 13 | 2.7 ± 0.4 | 193 ± 39 | 2.5 ± 0.7 |
| S (n = 6) | 294 ± 63 | 121 ± 13* | 3.4 ± 0.3 | 194 ± 19 | 4.1 ± 1.2* |
| FG (n = 6) | 486 ± 220* | 132 ± 21** | 3.8 ± 1.0* | 189 ± 32 | 3.6 ± 0.9 |
Values are mean ± SD
Chol, cholesterol, gluc, glucose, TG, triglycerides, UA, uric acid, C control diet, S sucrose diet, FG fructose + glucose diet
P < 0.05,
P < 0.02 compared to control
When all groups were combined, serum uric acid levels correlated with total fat (r = 0.74, P = 0.0004), abdominal fat (r = 0.74, P = 0.0005), TGs (r = 0.81, P < 0.0001), total cholesterol (r = 0.81, P < 0.0001), and insulin (r = 0.65, P = 0.004) (Fig. 1).
Fig. 1.
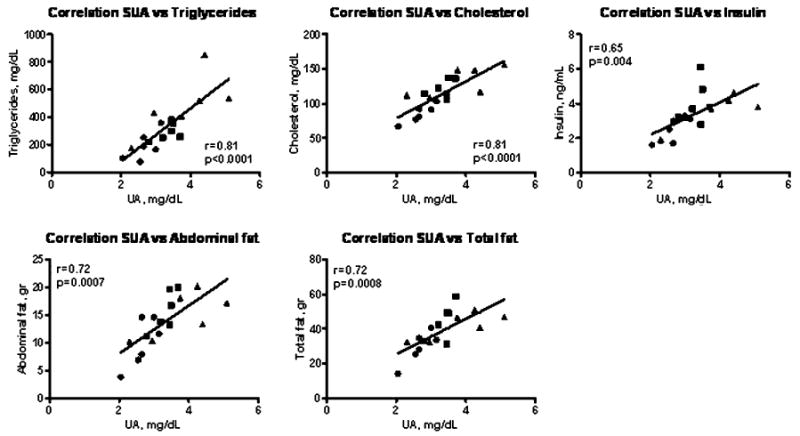
Correlations between serum uric acid at 16 weeks and various parameters. Control (dark filled circle), sucrose (dark filled square), and fructose + glucose (dark filled triangle) rats. UA uric acid
Effect of the different diets on the liver
The livers from both the FG and S groups showed evidence of fatty liver, with macro- and microvesicular steatosis (Fig. 2). Interestingly, the FG group had significantly greater fatty deposits than the S group (P < 0.02). Liver TG content as well as liver uric acid content were also higher in both FG and S groups, but only the FG group reached statistical significance compared with the C group (Fig. 2). In addition, a significant correlation between liver UA content and liver TGs was observed (r = 0.92, P < 0.0001). We also found a positive correlations between serum TGs levels measured at week 16 with liver TG content (r = 0.5, P = 0.04) and with fatty liver score (r = 0.7, P = 0.002).
Fig. 2.
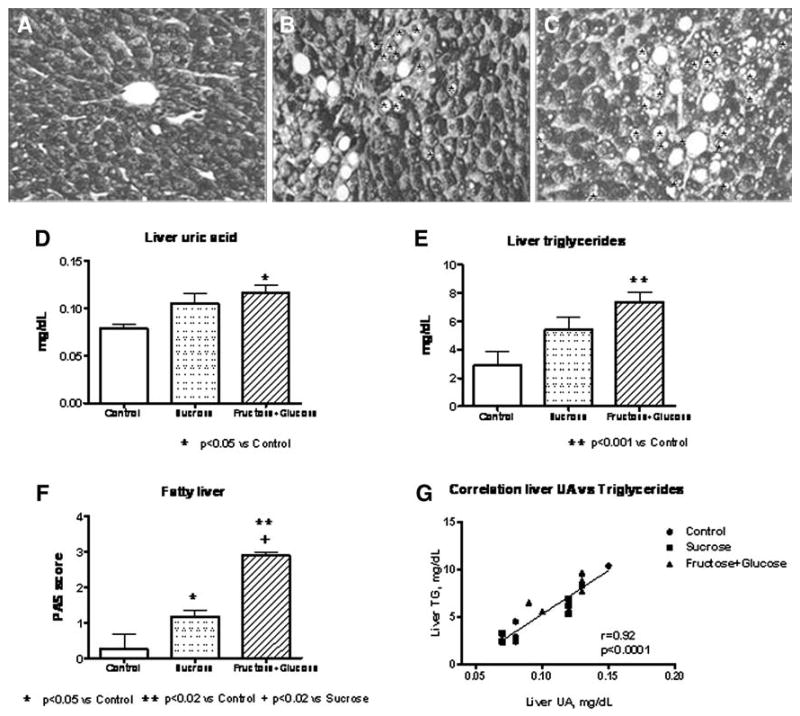
Liver histology (a, b, c, f) and liver homogenates uric acid and triglycerides (d, e) in rats fed control, sucrose, and fructose + glucose diets. a Control liver (magnification ×400). b Liver from sucrose fed rat (magnification ×400). c Liver from fructose + glucose rat (magnification ×400). Fat macrovesicles are indicated by Asterisks. g Correlation between intrahepatic uric acid and intrahepatic triglycerides
Induction of MCP-1 and TNF-α in liver tissue
Monocyte chemoattractant protein-1 and TNF-α levels were also measured in serum and in liver homogenates. Although no differences in serum MCP-1 were observed between groups, liver MCP-1 was increased by S and F + G diets, but only in S-fed rats was the difference significant. Serum TNF-α was also elevated by both experimental diets, and liver TNF-α was high in both S and FG rats (Fig. 3). Only a minimal inflammation was present in the liver despite the elevated levels of these inflammatory cytokines.
Fig. 3.
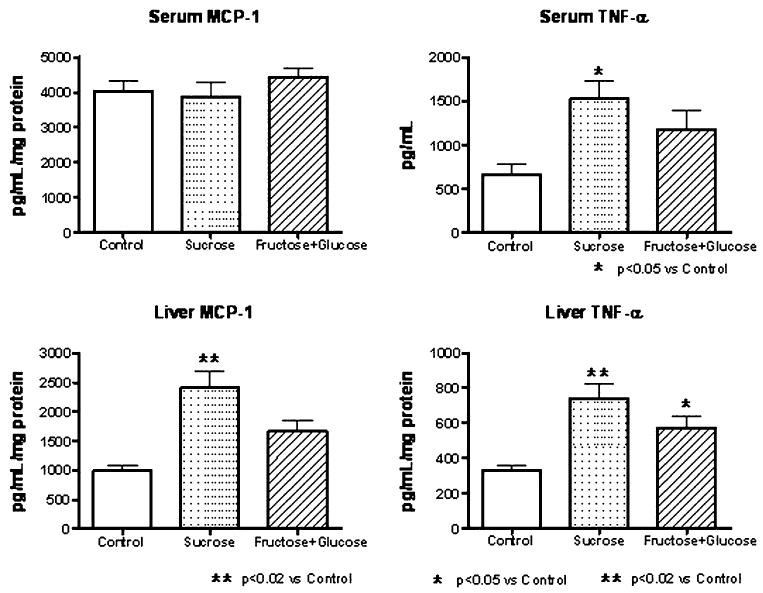
Serum and liver levels of MCP-1 and TNF-α in rats fed control, sucrose, and fructose + glucose diets
Most of the fat-laden hepatic parenchyma showed minimal inflammation and fibrosis. Although no difference in hepatic macrophage infiltration was observed among all groups (data not shown), there was focal inflammation consisting of CD8-positive T cells and ED-1-positive macrophages in the periportal regions in the FG and S groups. Periportal areas also stained for alpha actin pericytes (Ito cells) and type-III collagen, but no differences were noted in any of the three groups (data not shown).
Discussion
In the present study we tested the hypothesis whether sucrose is different from their individual components, fructose and glucose, in its metabolic effects, particularly as it relates to the development of fatty liver in rats. Rats were pair fed to assure equivalent energy intake, and the study was conducted over 4 months so that the chronic effects could be determined. Our hypothesis was that if there were to be observable differences between the two diets it would most likely be noted in the liver because this is the primary site of metabolism of fructose.
As expected, both groups showed systemic features consistent with metabolic syndrome, with elevations in serum TGs and with the development of insulin resistance. Serum uric acid levels were also increased, particularly in the FG group, consistent with the known effect of fructose to stimulate uric acid production [13]. Furthermore, the serum uric acid correlated with various markers of metabolic syndrome such as serum TGs (r = 0.81, P < 0.0001), cholesterol (r = 0.81, P < 0.0001), and insulin (r = 0.65, P = 0.004) as well as with abdominal (r = 0.72, P = 0.0007) and total fat (r = 0.72, P = 0.0008). These observations are consistent with our previous studies [28, 36]. In the present studies, we did not include a control group receiving a 60% glucose diet. In this regard, we have previously reported that rats consuming a 60% glucose diet had clinical chemistry values similar to rats receiving control regular diet [11, 28]. The use of diets containing 60% of energy derived from simple sugars has been questioned. However, it is a common maneuver in experimentation to use high doses as a way to induce disease conditions in a short period of time. For example, if rats are administered 60% of fructose, metabolic syndrome is induced within 4 weeks [28, 36], whereas if doses of 14% of total energy as fructose are administered, a period of 9 months is required for rats to develop insulin resistance [4].
The principal finding in this study was related to the effect of the diets on the development of fatty liver. Both diets induced fatty liver and increased hepatic TG content, although this tended to be worse in the FG group. We also documented that postprandial levels of TGs positively correlated with fatty liver score and intrahepatic TGs; similar findings have been observed in human studies [10, 30, 35]. In this regard, it is known that the maximal effects of fructose on TGs are best observed in the postprandial state, and recent studies by Stanhope et al. [41] have shown that this is particularly true in overweight humans fed fructose. In addition, postprandial hypertriglyceridemia is strongly associated with fat liver content [25, 30, 35], which was one of the main aims of the study.
Interestingly, sucrose- and glucose + fructose-fed animals also were found to have increased intrahepatic levels of uric acid. It has been established that the effects of uric acid on various cell types are mediated by intracellular uric acid [2, 4, 12, 16]. Importantly, we observed a parallel increase in uric acid and intrahepatic TGs as well as fatty liver. In addition, a strong positive correlation between liver uric acid and TG was observed (r = 0.92, P < 0.0001), despite the fact that we had a limited number of animals per group.
The role of uric acid as a biomarker of fatty liver has been recently underscored and there are epidemiological and clinical studies that support that concept [20, 21, 24, 37]. For instance, a recent large population study from China [20], reported that the prevalence rate of NAFLD was significantly higher in subjects with hyperuricemia than in those without hyperuricemia (24.75 vs. 9.54%; P < 0.001) and the prevalence increased with progressively higher serum uric acid levels (P value for trend <0.001) [20]. Moreover, the multiple regression analysis showed that hyperuricemia was associated with an increased risk of NAFLD (P < 0.001) [20]. There is also experimental evidence that chronic hyperuricemia may be able to cause hypertriglyceridemia and fatty liver in rats. In those studies, hyperuricemia was induced by the inhibition of uricase with allantoxanamide [45] or oxonic acid [46] and after 4 weeks the animals had developed elevated serum TGs and fatty liver. The mechanism by which uric acid might be able to cause fatty liver is unknown.
In the present studies, we also documented that both diets induced mild inflammation in the liver tissues which occurred in the periportal areas, and there was no fibrosis. However, subtle inflammatory processes could be discerned as noted by the higher hepatic and serum TNF-α levels in the FG- and S-fed rats and the higher MCP-1 levels in the S-fed rats. In this regard, data from animal and clinical studies indicate that TNF-α mediates not only the early stages of fatty liver disease but also the transition to more advanced stages of liver damage [8, 9, 23]; moreover serum levels of TNF-α strongly correlates with hepatic injury score in pediatric NAFLD [23]. Others have also reported that fructose feeding of rats can induce signaling pathways associated with inflammation in the liver [19]. Whether we would have observed more inflammatory and fibrotic changes if the diets had been continued for a longer period can only be speculated. These findings may be relevant to NAFLD, a condition commonly observed in subjects with metabolic syndrome. Whereas NAFLD was once rare, it has dramatically increased over the last several decades. Cirrhosis secondary to NAFLD has also increased and is now one of the more common reasons for liver transplantation in the United States. There is increasing evidence that this condition is due to increased fructose intake. In this regard, Ouyang et al. recently reported that subjects with NAFLD are ingesting more than twofold greater fructose from soft drinks than controls, amounting for 90 g of fructose per day from this source alone [32]. Recently Abdelmalek et al. have also reported a correlation between fructose intake and liver injury in subjects with NAFLD, as noted by the presence of ballooning degeneration, inflammation, and fibrosis in subjects with NAFLD [1].
In conclusion, we demonstrated that diets containing 30% fructose either as free fructose and glucose, or as sucrose, induce metabolic syndrome, intrahepatic accumulation of uric acid and TGs, increased MCP-1 and TNF-α as well as fatty liver in rats. Since uric acid can induce proinflammatory changes [17, 18], including in response to fructose [7], it will be important in future studies to determine if lowering uric acid can prevent the development of fatty liver or the proinflammatory changes that can accompany chronic fatty liver disease.
Acknowledgments
Supported by NIH grants HL-68607 and generous funds from Gatorade. Laura G. Sánchez-Lozada is supported by grant 081054 from CONACyT, Mexico.
Footnotes
Dr R. J. Johnson and Dr T. Nakagawa have patent applications related to lowering uric acid in the treatment of metabolic syndrome.
Dr Johnson also has a book, the Sugar Fix (Rodale, 2008; and Simon and Schuster, 2009) that discusses the potential role of fructose in the obesity epidemic.
Contributor Information
Laura G. Sánchez-Lozada, Division of Nephrology, Hypertension and Transplantation, University of Florida, Gainesville, FL, USA, Department of Nephrology, INC Ignacio Chavez, Mexico, Mexico.
Wei Mu, Division of Nephrology, Hypertension and Transplantation, University of Florida, Gainesville, FL, USA.
Carlos Roncal, Division of Nephrology, Hypertension and Transplantation, University of Florida, Gainesville, FL, USA.
Yuri Y. Sautin, Division of Nephrology, Hypertension and Transplantation, University of Florida, Gainesville, FL, USA
Manal Abdelmalek, Division of Gastroenterology and Hepatology, Duke University, Durham, NC, USA.
Sirirat Reungjui, Division of Nephrology, Hypertension and Transplantation, University of Florida, Gainesville, FL, USA.
MyPhuong Le, Division of Nephrology, Hypertension and Transplantation, University of Florida, Gainesville, FL, USA.
Takahiko Nakagawa, Division of Nephrology, Hypertension and Transplantation, University of Florida, Gainesville, FL, USA.
Hui Y. Lan, Department of Medicine and Therapeutics, Chinese University of Hong Kong, Hong Kong, China
Xuequing Yu, Department of Nephrology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China.
Richard J. Johnson, Division of Nephrology, Hypertension and Transplantation, University of Florida, Gainesville, FL, USA
References
- 1.Abdelmalek MF, Suzuki A, Guy C, Johnson RJ. Fructose induced hyperuricemia as a causal mechanism of nonalcoholic liver disease. Hepatology. 2007;46(Suppl 1):293A. [Google Scholar]
- 2.Ackerman Z, Oron-Herman M, Grozovski M, Rosenthal T, Pappo O, Link G, Sela BA. Fructose-induced fatty liver disease: hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension. 2005;45:1012–1018. doi: 10.1161/01.HYP.0000164570.20420.67. [DOI] [PubMed] [Google Scholar]
- 3.Beck-Nielsen H, Pedersen O, Lindskov HO. Impaired cellular insulin binding and insulin sensitivity induced by high-fructose feeding in normal subjects. Am J Clin Nutr. 1980;33:273–278. doi: 10.1093/ajcn/33.2.273. [DOI] [PubMed] [Google Scholar]
- 4.Blakely SR, Hallfrisch J, Reiser S, Prather ES. Long-term effects of moderate fructose feeding on glucose tolerance parameters in rats. J Nutr. 1981;111:307–314. doi: 10.1093/jn/111.2.307. [DOI] [PubMed] [Google Scholar]
- 5.Brown CM, Dulloo AG, Yepuri G, Montani JP. Fructose ingestion acutely elevates blood pressure in healthy young humans. Am J Physiol Regul Integr Comp Physiol. 2008;294:R730–R737. doi: 10.1152/ajpregu.00680.2007. [DOI] [PubMed] [Google Scholar]
- 6.Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis. 2001;21:3–16. doi: 10.1055/s-2001-12925. [DOI] [PubMed] [Google Scholar]
- 7.Cirillo P, Gersch MS, Mu W, Scherer PM, Kim KM, Gesualdo L, Henderson GN, Johnson RJ, Sautin YY. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J Am Soc Nephrol. 2009;20:545–553. doi: 10.1681/ASN.2008060576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crespo J, Cayon A, Fernandez-Gil P, Hernandez-Guerra M, Mayorga M, Dominguez-Diez A, Fernandez-Escalante JC, Pons-Romero F. Gene expression of tumor necrosis factor-alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158–1163. doi: 10.1053/jhep.2001.29628. [DOI] [PubMed] [Google Scholar]
- 9.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gersch MS, Mu W, Cirillo P, Reungjui S, Zhang L, Roncal C, Sautin YY, Johnson RJ, Nakagawa T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am J Physiol Renal Physiol. 2007;293:F1256–F1261. doi: 10.1152/ajprenal.00181.2007. [DOI] [PubMed] [Google Scholar]
- 12.Glushakova O, Kosugi T, Roncal C, Mu W, Heinig M, Cirillo P, Sanchez-Lozada LG, Johnson RJ, Nakagawa T. Fructose induces the inflammatory molecule ICAM-1 in endothelial cells. J Am Soc Nephrol. 2008;19:1712–1720. doi: 10.1681/ASN.2007121304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hallfrisch J. Metabolic effects of dietary fructose. FASEB J. 1990;4:2652–2660. doi: 10.1096/fasebj.4.9.2189777. [DOI] [PubMed] [Google Scholar]
- 14.Hallfrisch J, Ellwood KC, Michaelis OE, Reiser S, O'Dorisio TM, Prather ES. Effects of dietary fructose on plasma glucose and hormone responses in normal and hyperinsulinemic men. J Nutr. 1983;113:1819–1826. doi: 10.1093/jn/113.9.1819. [DOI] [PubMed] [Google Scholar]
- 15.Havel PJ. Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr Rev. 2005;63:133–157. doi: 10.1301/nr.2005.may.133-157. [DOI] [PubMed] [Google Scholar]
- 16.Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang DH, Gersch MS, Benner S, Sanchez-Lozada LG. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr. 2007;86:899–906. doi: 10.1093/ajcn/86.4.899. [DOI] [PubMed] [Google Scholar]
- 17.Kanellis J, Watanabe S, Li JH, Kang DH, Li P, Nakagawa T, Wamsley A, Sheikh-Hamad D, Lan HY, Feng L, Johnson RJ. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension. 2003;41:1287–1293. doi: 10.1161/01.HYP.0000072820.07472.3B. [DOI] [PubMed] [Google Scholar]
- 18.Kang DH, Park SK, Lee IK, Johnson RJ. Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol. 2005;16:3553–3562. doi: 10.1681/ASN.2005050572. [DOI] [PubMed] [Google Scholar]
- 19.Kelley GL, Allan G, Azhar S. High dietary fructose induces a hepatic stress response resulting in cholesterol and lipid dysregulation. Endocrinology. 2004;145:548–555. doi: 10.1210/en.2003-1167. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Xu C, Yu C, Xu L, Miao M. Association of serum uric acid level with non-alcoholic fatty liver disease: a cross-sectional study. J Hepatol. 2009;50:1029–1034. doi: 10.1016/j.jhep.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 21.Lonardo A, Loria P, Leonardi F, Borsatti A, Neri P, Pulvirenti M, Verrone AM, Bagni A, Bertolotti M, Ganazzi D, Carulli N. Fasting insulin and uric acid levels but not indices of iron metabolism are independent predictors of non-alcoholic fatty liver disease. A case–control study. Dig Liver Dis. 2002;34:204–211. doi: 10.1016/s1590-8658(02)80194-3. [DOI] [PubMed] [Google Scholar]
- 22.Macdonald I, Turner LJ. Serum-fructose levels after sucrose or its constituent monosaccharides. Lancet. 1968;291:841–843. doi: 10.1016/s0140-6736(68)90300-0. [DOI] [PubMed] [Google Scholar]
- 23.Manco M, Marcellini M, Giannone G, Nobili V. Correlation of serum TNF-alpha levels and histologic liver injury scores in pediatric nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;127:954–960. doi: 10.1309/6VJ4DWGYDU0XYJ8Q. [DOI] [PubMed] [Google Scholar]
- 24.Marchesini G, Babini M. Nonalcoholic fatty liver disease and the metabolic syndrome. Minerva Cardioangiol. 2006;54:229–239. [PubMed] [Google Scholar]
- 25.Matikainen N, Manttari S, Westerbacka J, Vehkavaara S, Lundbom N, Yki-Jarvinen H, Taskinen MR. Postprandial lipemia associates with liver fat content. J Clin Endocrinol Metab. 2007;92:3052–3059. doi: 10.1210/jc.2007-0187. [DOI] [PubMed] [Google Scholar]
- 26.Melanson KJ, Zukley L, Lowndes J, Nguyen V, Angelopoulos TJ, Rippe JM. Effects of high-fructose corn syrup and sucrose consumption on circulating glucose, insulin, leptin, and ghrelin and on appetite in normal-weight women. Nutrition. 2007;23:103–112. doi: 10.1016/j.nut.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Monsivais P, Perrigue MM, Drewnowski A. Sugars and satiety: does the type of sweetener make a difference? Am J Clin Nutr. 2007;86:116–123. doi: 10.1093/ajcn/86.1.116. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera-Acosta J, Patel JM, Johnson RJ. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290:F625–F631. doi: 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa T, Tuttle KR, Short RA, Johnson RJ. Hypothesis: fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol. 2005;1:80–86. doi: 10.1038/ncpneph0019. [DOI] [PubMed] [Google Scholar]
- 30.Nimitphong H, Phongkitkarun S, Rattarasarn C, Kongsooksai A, Chanprasertyothin S, Bunnag PA, Puavilai G. Hepatic fat content is a determinant of postprandial triglyceride levels in type 2 diabetes mellitus patients with normal fasting triglyceride. Metabolism. 2008;57:644–649. doi: 10.1016/j.metabol.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Oleszczuk A, Spannbauer M, Tannapfel A, Bluher M, Hengstler J, Pietsch UC, Schuhmacher A, Wittekind C, Hauss JP, Schon MR. Regenerative capacity differs between micro- and macrovesicular hepatic steatosis. Exp Toxicol Pathol. 2007;59:205–213. doi: 10.1016/j.etp.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, Johnson RJ, Abdelmalek MF. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. 2008;48:993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer JR, Boggs DA, Krishnan S, Hu FB, Singer M, Rosenberg L. Sugar-sweetened beverages and incidence of type 2 diabetes mellitus in African American women. Arch Intern Med. 2008;168:1487–1492. doi: 10.1001/archinte.168.14.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porikos KP, Van Itallie TB. Diet-induced changes in serum transaminase and triglyceride levels in healthy adult men. Role of sucrose and excess calories. Am J Med. 1983;75:624–630. doi: 10.1016/0002-9343(83)90444-8. [DOI] [PubMed] [Google Scholar]
- 35.Rijkelijkhuizen JM, Doesburg T, Girman CJ, Mari A, Rhodes T, Gastaldelli A, Nijpels G, Dekker JM. Hepatic fat is not associated with beta-cell function or postprandial free fatty acid response. Metabolism. 2009;58:196–203. doi: 10.1016/j.metabol.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Lozada LG, Tapia E, Bautista-Garcia P, Soto V, Ávila-Casado C, Vega-Campos IP, Nakagawa T, Zhao L, Franco M, Johnson RJ. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2008;294:F710–F718. doi: 10.1152/ajprenal.00454.2007. [DOI] [PubMed] [Google Scholar]
- 37.Sartorio A, Del CA, Agosti F, Mazzilli G, Bellentani S, Tiribelli C, Bedogni G. Predictors of non-alcoholic fatty liver disease in obese children. Eur J Clin Nutr. 2007;61:877–883. doi: 10.1038/sj.ejcn.1602588. [DOI] [PubMed] [Google Scholar]
- 38.Segal MS, Gollub E, Johnson RJ. Is the fructose index more relevant with regards to cardiovascular disease than the glycemic index? Eur J Nutr. 2007;46:406–417. doi: 10.1007/s00394-007-0680-9. [DOI] [PubMed] [Google Scholar]
- 39.Stanhope KL, Griffen SC, Bair BR, Swarbrick MM, Keim NL, Havel PJ. Twenty-four-hour endocrine and metabolic profiles following consumption of high-fructose corn syrup-, sucrose-, fructose-, and glucose-sweetened beverages with meals. Am J Clin Nutr. 2008;87:1194–1203. doi: 10.1093/ajcn/87.5.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanhope KL, Havel PJ. Fructose consumption: potential mechanisms for its effects to increase visceral adiposity and induce dyslipidemia and insulin resistance. Curr Opin Lipidol. 2008;19:16–24. doi: 10.1097/MOL.0b013e3282f2b24a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, Hatcher B, Cox CL, Dyachenko A, Zhang W, McGahan JP, Seibert A, Krauss RM, Chiu S, Schaefer EJ, Ai M, Otokozawa S, Nakajima K, Nakano T, Beysen C, Hellerstein MK, Berglund L, Havel PJ. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–1334. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swarbrick MM, Stanhope KL, Elliott SS, Graham JL, Krauss RM, Christiansen MP, Griffen SC, Keim NL, Havel PJ. Consumption of fructose-sweetened beverages for 10 weeks increases postprandial triacylglycerol and apolipoprotein-B concentrations in overweight and obese women. Br J Nutr. 2008;100:947–952. doi: 10.1017/S0007114508968252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teff KL, Elliott SS, Tschop M, Kieffer TJ, Rader D, Heiman M, Townsend RR, Keim NL, D'Alessio D, Havel PJ. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab. 2004;89:2963–2972. doi: 10.1210/jc.2003-031855. [DOI] [PubMed] [Google Scholar]
- 44.Thompson RG, Hayford JT, Hendrix JA. Triglyceride concentrations: the disaccharide effect. Science. 1979;206:838–839. doi: 10.1126/science.493983. [DOI] [PubMed] [Google Scholar]
- 45.Wexler BC. Allantoxanamide-induced myocardial necrosis in Sprague-Dawley vs spontaneously hypertensive rats. Proc Soc Exp Biol Med. 1982;170:476–485. doi: 10.3181/00379727-170-41462. [DOI] [PubMed] [Google Scholar]
- 46.Wexler BC, Greenberg BP. Effect of increased serum urate levels on virgin rats with no arteriosclerosis versus breeder rats with preexistent arteriosclerosis. Metabolism. 1977;26:1309–1320. doi: 10.1016/0026-0495(77)90027-0. [DOI] [PubMed] [Google Scholar]