

Abstract
Human skin is a large, heterogeneous organ that protects the body from pathogens while sustaining microorganisms that influence human health and disease. Our analysis of 16S ribosomal RNA gene sequences obtained from 20 distinct skin sites of healthy humans revealed that physiologically comparable sites harbor similar bacterial communities. The complexity and stability of the microbial community are dependent on the specific characteristics of the skin site. This topographical and temporal survey provides a baseline for studies that examine the role of bacterial communities in disease states and the microbial interdependencies required to maintain healthy skin.
The skin is a critical interface between the human body and its external environment, preventing loss of moisture and barring entry of pathogenic organisms (1). The skin is also an ecosystem, harboring microbial communities that live in a range of physiologically and topographically distinct niches (2). For example, hairy, moist underarms lie a short distance from smooth dry forearms, but these two niches are likely as ecologically dissimilar as rainforests are to deserts. Traditional culture-based characterizations of the skin microbiota are biased toward species that readily grow under standard laboratory conditions, such as Staphylococci spp. However, molecular approaches have revealed a greater diversity of skin microbiota within and between distinct topographical regions (3–5), underscoring the need to systematically survey multiple skin sites with the use of more contemporary genomic techniques.
Characterizing the microbiota that inhabit specific sites may provide insight into the delicate balance between skin health and disease. Certain dermatological disorders manifest at stereotypical skin sites [e.g., psoriasis on the outer elbow and atopic dermatitis (eczema) on the inner bend of the elbow]. Moreover, antibiotic exposure, modified hygienic practices, and lifestyle changes have the potential to alter the skin microbiome selectively and may underlie the increased incidence of human disorders such as atopic dermatitis. Understanding naturally occurring symbiotic microbial communities will provide insight into the conditions that favor the emergence of antibiotic-resistant organisms [e.g., the highly pathogenic strain of methicillin-resistant S. aureus, which acquired genes that promote growth on skin from the symbiont S. epidermidis (6)].
We characterized the topographical and temporal diversity of the human skin microbiome with the use of 16S rRNA gene phylotyping, and generated 112,283 near-full-length bacterial 16S gene sequences from samples of 20 diverse skin sites on each of 10 healthy humans (7) (fig. S1 and table S1). Nineteen bacterial phyla were detected, but most sequences were assigned to four phyla: Actinobacteria (51.8%), Firmicutes (24.4%), Proteobacteria (16.5%), and Bacteroidetes (6.3%). Of the 205 identified genera represented by at least five sequences, three were associated with more than 62% of the sequences: Corynebacteria (22.8%; Actinobacteria), Propionibacteria (23.0%; Actinobacteria), and Staphylococci (16.8%; Firmicutes). At the species level, we observed greater diversity than revealed by culture-based methods (2).
We selected skin sites that are not only representative of distinct niches but also characteristically affected by dermatologic disorders where microbes have been implicated in disease pathogenesis. We compared the relative abundance of major bacterial groups, as defined by the Ribosomal Database Project taxonomy (8), relative to three microenvironment types: (i) sebaceous [glabella (between the eyebrows), alar crease (beside the nostril), external auditory canal (inside the ear), occiput (back of the scalp), manubrium (upper chest), and back]; (ii) moist [nare (inside the nostril), axillary vault (armpit), antecubital fossa (inner elbow), interdigital web space (between the middle and ring fingers), inguinal crease (side of the groin), gluteal crease (topmost part of the fold between the buttocks), popliteal fossa (behind the knee), plantar heel (bottom of the heel), and umbilicus (navel)]; and (iii) dry [volar forearm (inside of the mid-forearm), hypothenar palm (palm of the hand proximal to the little finger), and buttock] (Fig. 1 and table S2). Propionibacteria species and Staphylococci species predominated in sebaceous sites (Fig. 1A). Corynebacteria species predominated in moist sites, although Staphylococci species were also represented (Fig. 1B). A mixed population of bacteria resided in dry sites, with a greater prevalence of β-Proteobacteria and Flavobacteriales (Fig. 1C). These observations were all significant (P < 0.05, one-tailed t test).
Fig. 1.
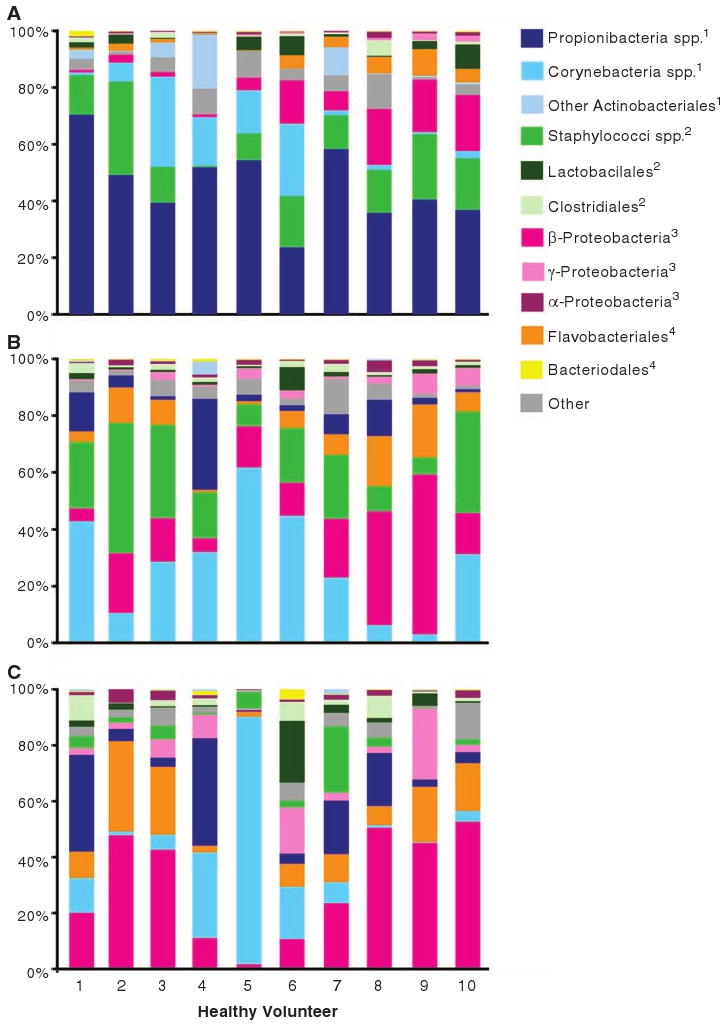
The 20 skin sites and associated microbiota are representative of three microenvironments: (A) sebaceous, (B) moist, and (C) dry. The relative abundance of the most abundant bacterial groups associated with each microenvironment is depicted for each healthy volunteer. Superscripts indicate phylum: 1, Actinobacteria; 2, Firmicutes; 3, Proteobacteria; 4, Bacteroidetes.
The taxonomic diversity, evenness, and richness of each site's microbiome were assessed using ecological diversity statistics (7). To perform these analyses, we clustered sequences into species-level operational taxonomic units (OTUs) of 99% sequence similarity by the furthest-neighbor method, using the DOTUR (Distance-based OTU and Richness) program (9). The richest site, or the site with the greatest number of OTUs, was the volar forearm with 44 median OTUs; the least rich site was the retroauricular crease with 15 median OTUs (fig. S3A). Taxonomic evenness, or the relative distribution of sequences among the OTUs, was assessed by the Shannon Equitability Index. The most even site was the popliteal fossa, followed by plantar heel and antecubital fossa; the least even sites were back, retroauricular crease, and toe web space (fig. S3B). The Shannon Diversity Index accounts for both richness and evenness of OTUs and largely mirrors the evenness findings of our data set (Fig. 2). In general, sebaceous sites were less diverse, less even, and less rich than moist and dry sites (P < 0.05, one-tailed t test).
Fig. 2.
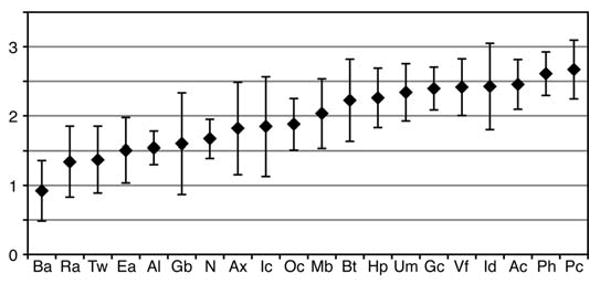
Median diversity of sites as measured by the Shannon Diversity Index. Error bars represent median absolute deviation. See fig. S1 for key to site codes displayed on the x axis.
To assess interpersonal variation, we used two OTU-based measurements: (i) community membership, a measure of shared OTUs, and (ii) community structure, which accounts for relative OTU abundance in addition to community membership. By these measures, the degree of interpersonal variation depended on skin site (table S5). The least similar sites were interdigital web spaces, toe webs, axillae, and umbilici. The most similar sites were alar creases, nares, and backs. We analyzed three paired symmetric sites (left and right antecubital fossae, axillae, and volar forearms) to assess intrapersonal variation. Subjects were more similar to themselves than to others (fig. S4 and table S3). A similar analysis of sampling techniques showed that swabbing was a suitable surrogate for scraping as a method for sample collection to assess microbial diversity (fig. S5 and table S4).
Microbes are predicted to play a role in the pathophysiology of many common dermatoses with predilection for specific skin sites (e.g., atopic dermatitis, psoriasis, acne, seborrheic dermatitis). Atopic dermatitis preferentially involves the antecubital and popliteal fossae, sites that were highly diverse, even, and rich relative to other sites (Fig. 2 and fig. S3). These sites also harbored similar ranges of organisms, but community membership was better preserved than community structure (table S6A). Psoriasis also occurs at stereotypical sites: umbilici, gluteal creases, occiputs, elbows, and knees (10). We did not identify common characteristics between bacterial communities at psoriasis-associated sites (table S6B).
Because of increasing public health concerns regarding methicillin-resistant S. aureus infections, we included the anterior nares in our survey. A measurable proportion of the population harbors S. aureus here, and this constitutes a risk factor for the development of localized skin, soft tissue, and systemic infections (10). The unique microenvironment of the anterior nares consists of moist, hair-bearing, keratinized epithelia contiguous with both noncornified nasal mucosa and drier keratinized skin surfaces. When comparing community structure and membership, we found that microbes in the nares most closely resemble those collected from the contiguous alar crease (table S6C).
To characterize the temporal variation of the 20 skin sites, we collected follow-up samples 4 to 6 months after the initial visit from 5 of the 10 healthy volunteers. The most consistent sites with respect to community membership and structure were the external auditory canal, inguinal crease, alar crease, and nare, whereas we found appreciable variation on the second sampling of the popliteal fossa, volar forearm, and buttock (Fig. 3 and table S7), which suggests that longitudinal stability of the skin microbiome was site-dependent. Overall, four of the five resampled volunteers were significantly more like themselves over time than they were like other volunteers (P < 0.005, two-tailed t test) (Fig. 3 and table S7).
Fig. 3.
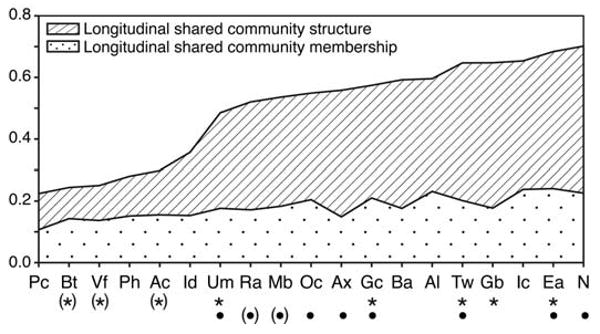
Longitudinal stability of the skin microbiome. A higher number corresponds to greater shared community membership or structure over time. Sites with an asterisk below the site code retained significant community membership over time; those with a bullet retained significant community structure over time, as compared to interpersonal variation at the same site (P ≤ 0.05). Parentheses around asterisks or bullets indicate that significance was achieved for that site when the outlier (volunteer 2) was removed from the analysis. See fig. S1 for key to site codes.
The recently launched Human Microbiome Project aims to characterize the human microbiota and its role in health and disease (11). The study reported here contributes to the broad goals of this project and may have implications for the future treatment of skin disorders. We have gained insights into the normal skin bacterial variation (intrapersonal, interpersonal, temporal, topographical), which can be used as a reference to calculate the statistical power required to carry out disease-related studies. Although multiple factors, including local skin anatomy, lipid content, pH, sweat, and sebum secretion, have long been recognized as contributing to certain skin disorders, we have shown that these factors correlate with the predominant microbiota. For example, the sebaceous glands of the face, scalp, chest, and back produce large amounts of oily sebum and are the sites where the lipophilic anaerobe Propionibacterium acnes predominates.
The effectiveness of antimicrobial agents in the management of some common skin disorders supports a role for microbes in pathophysiology. Elucidation of the baseline skin microbiome is a step toward testing the therapeutic potential of manipulating the microbiome in skin disorders. Indeed, an initial study of psoriasis (12) and an animal model of ichthyosis (13) describe selective microbial shifts associated with skin diseases. Targeted therapies to maintain healthy skin might require not only inhibiting the growth of pathogenic bacteria, but also promoting the growth of symbiotic bacteria.
Supplementary Material
Acknowledgments
We thank the volunteers who participated in this study; E. Bassett for assistance with sample collection; members of the Segre laboratory for their underlying contributions; M. Udey, N. Salafsky, and E. Lander for critical reading of the manuscript; and J. Fekecs and D. Leja for graphical assistance. Supported by a NIGMS Pharmacology Research Associate Training Fellowship (E.A.G.) and by the NHGRI and NCI Center for Cancer Research Intramural Research Programs. All sequences are deposited under NCBI Genome Project 30125, GenBank accession numbers GQ000001 to GQ116391.
Footnotes
References and Notes
- 1.Segre JA. J Clin Invest. 2006;116:1150. doi: 10.1172/JCI28521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marples M. The Ecology of the Human Skin. Bannerstone; Springfield, IL: 1965. [Google Scholar]
- 3.Fierer N, Hamady M, Lauber CL, Knight R. Proc Natl Acad Sci USA. 2008;105:17994. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao Z, Tseng CH, Pei Z, Blaser MJ. Proc Natl Acad Sci USA. 2007;104:2927. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grice EA, et al. Genome Res. 2008;18:1043. doi: 10.1101/gr.075549.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diep BA, et al. Lancet. 2006;367:731. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 7.See supporting material on Science Online.
- 8.Cole JR, et al. Nucleic Acids Res. 2007;35:D169. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schloss PD, Handelsman J. Appl Environ Microbiol. 2005;71:1501. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schon MP, Boehncke WH. N Engl J Med. 2005;352:1899. doi: 10.1056/NEJMra041320. [DOI] [PubMed] [Google Scholar]
- 11.NIH Human Microbiome Project. ( http://nihroadmap.nih.gov/hmp)
- 12.Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. PLoS One. 2008;3:e2719. doi: 10.1371/journal.pone.0002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scharschmidt TC, et al. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.