

KIT is a receptor tyrosine kinase (RTK), the interaction of which with its ligand, stem cell factor (SCF), is essential for growth and differentiation of various cells.[1] SCF binding promotes KIT dimerization,[2] transphosphorylation, and activation of downstream cell signaling pathways essential for cell proliferation, differentiation, and survival. Gain-of-function mutations in KIT have been identified in human cancers such as gastrointestinal stromal tumors (GIST).[3,4] It was also demonstrated that autocrine or paracrine mechanisms mediated by aberrant expression of SCF and/or KIT might also lead to oncogenesis.[5–7] Because most cases of GIST are driven by oncogenic KIT mutations resulting in enhanced tyrosine kinase activity, inhibitors of the tyrosine kinase[8] activity of KIT, such as Gleevec® (imatinib mesylate) and Sutent® (sunitinib), have been successfully applied for the treatment of GIST patients.
Another way to inhibit the activity of RTKs such as KIT is to block the binding of the ligand to the receptor.[9] Although this can effectively be achieved by using monoclonal antibodies,[10] the development of nonpeptidic protein binders that might be less susceptible to enzymatic degradation, could be taken orally, and would be more amenable to modification is highly desired.[11,12] Realizing such inhibitors, however, is a challenging goal in molecular recognition and drug discovery because the interactions between two proteins normally involve complex, large, and shallow interfaces that lack grooves and pockets to accommodate traditional small-molecule inhibitors. Targeting key interaction sites, so called “hot-spots”, which contribute significantly to the stability of protein–protein complexes, is one approach[13] that could be taken to disrupt ligand–receptor contacts. A systematic method for finding and targeting these sites with synthetic agents remains to be developed.
An alternative strategy has recently emerged from the field of supramolecular chemistry.[14–19] Rather than trying to minimize the complexity of the target into a “lock and key” problem, a supramolecular design takes its inspiration from the natural protein partners or their antibodies. Specifically, it offers the possibility of systematically functionalizing molecular scaffolds with a variety of hydrogen-bonding motifs, hydrophobic, and charged groups to match the complementary surface of a target protein. In this way, a wealth of allosteric inhibitions also becomes available.
Despite difficulties in creating synthetic receptors that can recognize protein surfaces in aqueous environments with high affinity and selectivity, a number of inhibitors based on synthetic agents,[12,14–17] unnatural peptides,[17–19] and nanoparticles[20,21] have been developed, which demonstrate the viability of this approach. We have previously taken two strategies in the design of synthetic receptors that can be modified to recognize protein surfaces. One is based on scaffolds that mimic structural features of important α-helix motifs,[15,22–26] and the other focuses on surface binding agents designed to complement larger portions of the interface.[14,27–35]
Here, we report the identification of potent scaffolds capable of interfering with the binding of SCF to the extracellular ligand-binding domain of KIT and in doing so disrupting cell signaling stimulation by SCF.
Inspection of the crystal structure of the extracellular ligand-binding domain of KIT in complex with SCF[36] shows that the SCF-binding domain of KIT can be divided into three sites (I, II, and III) having an overall buried surface area of 2060 Å2. Figure 1A shows the electrostatic surface potentials in sites II (770 Å2) and III (1010 Å2) on SCF and KIT, which together constitute the most part of the SCF–KIT interface. While in site II the binding is dominated by complementary electrostatic interactions between basic amino acids on KIT and acidic amino acids on SCF, in site III the interactions are principally mediated by hydrogen bonds and van der Waals contacts between polar and hydrophobic amino acids.
Figure 1.

A) Electrostatic potential map of KIT (left) and SCF monomer (right) at the interface. The negatively charged surface is shown in red and the positive in blue. B) Electrostatic surface potential (left) of a porphyrin-based synthetic receptor (P2; right) possessing two distinct hydrophobic (highlighted by the circle) and charged domains. P2 is presented on the same scale as SCF and KIT in Figure 1A.
The distinct forces that govern the interaction in the two sites, as well as the large and generally flat interfaces, suggest that potential inhibitors might be based on relatively planar molecules that cover a large surface area, and are composed of two distinct hydrophobic and charged domains (Figure 1B). A few examples by Hamilton[27–31] and others[37,38] of porphyrin-based receptors that bind to cytochrome c,[27–31] VEGF,[37] as well as Kv1.3 potassium channels,[38] have provided proofs-of-concept for the applicability of such systems in recognizing shallow and hydrophobic protein surfaces. In targeting the SCF–KIT interaction, meso-tetrakis (4-carboxyphenyl)porphine (TCPP) modified with four aspartic acids (P9) or aminobenzyl groups (P1–8) appended with alcohols, amines, and carboxylic acids afforded various binding agents composed of a flat hydrophobic core and combinations of a cationic, anionic and zwitterionic periphery (Figure 2, Table 1, P1–9). Figure 1B shows the electrostatic potential of one of these receptors (P2) on the same scale as SCF and KIT in Figure 1A. The distance between the two diagonal carboxylic acids is ~35 Å, which enables the molecule to span about 1200 Å2 of protein surface area; this is potentially more than a single site at the interface.
Figure 2.

Three synthetic scaffolds were used to generate 18 potential inhibitors; their functional groups are shown in Table 1. Arrow heads and tails correspond to charged functional groups and hydrophobic domains, respectively. An additional compound, P9, is a simple meso-tetrakis (4-carboxyphenyl)porphine (TCPP) to which four aspartic acids are appended.
Table 1.
IC50 range estimated by an ELISA assay. Inhibition was tested at five different concentrations (100, 10, 1, 0.1 and 0.01 µm) for each synthetic receptor.
| In.[a] | R[b] | IC50[c] [µm] |
In.[a] | R[b] | IC50[c] [µm] |
|---|---|---|---|---|---|
| P1 | 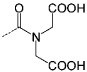 |
<0.1 | P9 | TCPP(Asp)4 | <10 |
| P2 | 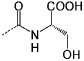 |
<0.1 | C10 | <10 | |
| P3 | 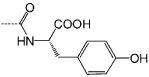 |
<0.1 | C11 | 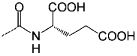 |
<100 |
| P4 | 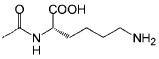 |
<1 | C12 | 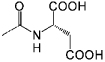 |
<100 |
| P5 | 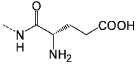 |
<10 | C13 | 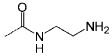 |
– |
| P6 | 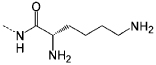 |
– | C14 | 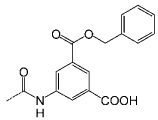 |
– |
| P7 | 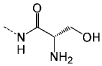 |
– | O15–17 | 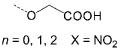 |
– |
| P8 | 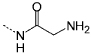 |
– | O18 | – |
Inhibitor;
modifying groups;
concentration [µm] that led to more than 50% inhibition of the SCF*–KIT interaction.
To assess the efficiency of the different molecules (P1–9) to inhibit the SCF–KIT interaction, an enzyme-linked immunosorbent assay (ELISA), which uses a whole extracellular KIT receptor and horseradish peroxidase (HRP)-labeled SCF (SCF*), was developed (see the Supporting Information). The synthetic molecules were then incubated along with SCF* at five different concentrations (100 to 0.01 µm, every tenfold) with surface-bound KIT to evaluate their efficiency in achieving 50% effect in disrupting the interactions (IC50). Table 1 (P1–9) summarizes the results of this initial screen, from which three lead compounds P1–3 (0.01 µm < IC50 < 0.1 µm) were identified and structure–activity relationships (SAR) could be deduced. The importance of an overall negative charge is clearly observed because the zwitterionic receptors (P4, P5) were less effective than the anionic binders (P1–3), and all of the positively charged amino-modified compounds (P6–8) showed no inhibitory effect. Nevertheless, these data also show that increasing the number of carboxy groups is not the only requirement for realizing enhanced affinity. For example P9, which carries eight carboxylic acids, was 100-fold less potent than P1, and also P2 and P3, which possess only four negatively charged groups. This suggests that the nature of the hydrophobic domain is a significant factor in the potency of surface binding inhibitors.
To further evaluate the structural requirements in the design of surface binding inhibitors of the SCF–KIT interactions, we prepared additional classes of synthetic agents (C10–14 and O15–18) that, similar to porphyrins (P1–9), possess distinct hydrophobic and charged regions (Figure 2, Table 1). In this way, the three receptor types carry similar charged aromatic functionalities, and yet differ in their surface area and topology, as well as the orientation of the modifying groups. In the porphyrin-based receptors (P1–9) the four functional groups are projected around a flat hydrophobic core. Introducing similar substituents onto calixarene-based scaffolds (C10–13) generates a nonplanar structure possessing hydrophobic and charged domains below and above the rim, respectively. Compound C14, which is a potent inhibitor of the PDGF–PDGFR interaction,[35] provides an example of a receptor with both a charged and hydrophobic upper rim. In the oligopyridyl-amides (O15–18)[39] internal hydrogen bonds hold the charged carboxy groups in a linear topology on the same face of the oligomeric and hydrophobic scaffold.
The structure–activity relationship for these receptors (Table 1, C10–O18) was consistent with that of the porphyrins, and showed a correlation between the orientation of charged groups, topology, surface area, and inhibition. For example, the best carboxy-modified calixarene inhibitor (C10) was 100-fold less potent than the larger porphyrin receptors (P1–3). Similarly, none of the oligopyridyl amides (O15–18), possessing the smallest surface area, affected the SCF–KIT interaction even at 100 µm concentration. The lack of activity of amino-modified calixarene C13, as well as hydrophobic C14, further underlined the importance of a well-defined, negatively charged region. Finally, in agreement with previous observations, increasing the number of carboxy groups from four (C10) to eight (C11 and C12) did not improve the potency of the inhibitors.
These data point to important structural features that should influence the identification of surface binding inhibitors for the SCF–KIT interaction. Specifically, a large and planar hydrophobic patch affords high affinity, whereas a separate region with an overall negative charge facilitates recognition and selectivity. Figures 3A and B show ligand-saturation and competition-binding curves, respectively, for the lead compounds that fulfill those requirements. The apparent dissociation constant of the SCF*–KIT interaction (KD(app) = 16 nm) is in the range of values previously reported (2–55 nm),[2] depending on experimental conditions (Figure 3A). Following the change in ELISA signal with increasing concentrations of synthetic receptors, P2 and P3 allowed us to evaluate the potency of the lead agents (Figure 3B); the results showed apparent inhibition constants of KI(app) = 31 and 33 nm, respectively.
Figure 3.

A) Saturation-binding curve of HRP-labeled SCF (SCF*) binding to ELISA plates coated with the whole extracellular ligand-binding domain of KIT (KD(app) = 16 nm). B) Competition-binding curves for the lead inhibitors (In.) P2 ( ) and P3 (●); KI(app) = 31 and 33 nm, respectively. C) Cell-based assays testing the efficiency of the lead compounds (IC50 < 10 µm) to inhibit SCF-induced KIT phosphorylation. Four compounds (P1–3, C10) strongly inhibited cell stimulation; P2 completely abolished KIT phosphorylation (p-KIT).
) and P3 (●); KI(app) = 31 and 33 nm, respectively. C) Cell-based assays testing the efficiency of the lead compounds (IC50 < 10 µm) to inhibit SCF-induced KIT phosphorylation. Four compounds (P1–3, C10) strongly inhibited cell stimulation; P2 completely abolished KIT phosphorylation (p-KIT).
Next, we tested the ability of these lead inhibitors to block the stimulation of cell signaling by SCF (Figure 3C). Starved 3T3 cells that stably express KIT were incubated with compounds that possess IC50 < 10 µm (P1–5, P9 and C10) in a final concentration of 10 µm. SCF was then added for five minutes and the cells were lysed, followed by KIT immunoprecipitation and analysis by SDS-PAGE and immunobloting. Figure 3C shows the results of the cell-based assays, in which KIT expression levels were visualized by using anti-KIT antibodies with comparable KIT expression levels in all eight experiments. In contrast, SCF-induced KIT phosphorylation (p-KIT), as visualized by anti-phosphotyrosine antibodies, was strongly inhibited by four compounds (P1–3 and C10). It is noteworthy that P2 completely abolished KIT phosphorylation. Further studies regarding the mechanism of inhibition, as well as the potential of the lead compounds to inhibit tumor growth, are underway.
In summary, we have shown the regulation of KIT tyrosine phosphorylation, previously inhibited at the tyrosine kinase domain, by protein surface-binding molecules that interrupt the SCF–KIT interaction at the extracellular region. Synthetic inhibitors of this class might open new possibilities in overcoming drug resistance by KIT mutations. In addition, this work demonstrates how new structural insights on protein–protein interfaces,[36] when coupled with a supramolecular design,[14] can provide a systematic approach for disrupting the interaction with synthetic agents.
Supplementary Material
Acknowledgements
This work was supported by NIH grant GM35208 to A.D.H. and NIH grants R01-AR051448, R01-AR051886 and P50-AR054086 to J.S. D.M. thanks the Human Frontier Science Program Organization for a cross-disciplinary postdoctoral fellowship. Y.O. was a Cancer research Institute postdoctoral fellow.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.200900079.
References
- 1.Ashman LK. Int. J. Biochem. Cell Biol. 1999;31:1037–1051. doi: 10.1016/s1357-2725(99)00076-x. [DOI] [PubMed] [Google Scholar]
- 2.Lemmon MA, Pinchasi D, Zhou M, Lax I, Schlessinger J. J. Biol. Chem. 1997;272:6311–6317. doi: 10.1074/jbc.272.10.6311. [DOI] [PubMed] [Google Scholar]
- 3.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Tunio GM, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 4.Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y. Nat. Genet. 1998;19:323–324. doi: 10.1038/1209. [DOI] [PubMed] [Google Scholar]
- 5.Zheng R, Klang K, Gorin N, Small D. Leuk. Res. 2004;28:121–126. doi: 10.1016/s0145-2126(03)00184-x. [DOI] [PubMed] [Google Scholar]
- 6.Hines SJ, Organ C, Kornstein MJ, Krystal GW. Cell Growth Differ. 1995;6:769–779. [PubMed] [Google Scholar]
- 7.Reis RM, Martins A, Ribeiro SA, Basto D, Longatto-Filho A, Schmitt FC, Lopes JM. Cell. Oncol. 2005;27:319–326. doi: 10.1155/2005/347863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kung C, Shokat KM. ChemBioChem. 2005;6:523–526. doi: 10.1002/cbic.200400393. [DOI] [PubMed] [Google Scholar]
- 9.Fischer OM, Streit S, Hart S, Ullrich A. Curr. Opin. Chem. Biol. 2003;7:490–495. doi: 10.1016/s1367-5931(03)00082-6. [DOI] [PubMed] [Google Scholar]
- 10.Reichert JM, Valge-Archer VE. Nat. Rev. Drug Discovery. 2007;6:349–356. doi: 10.1038/nrd2241. [DOI] [PubMed] [Google Scholar]
- 11.Arkin MR, Wells JA. Nat. Rev. Drug Discovery. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 12.Yin H, Hamilton AD. Angew. Chem. 2005;117:4200–4235. [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:4130–4163. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 13.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 14.Fletcher S, Hamilton AD. J. R. Soc. Interf. 2006;3:215–233. doi: 10.1098/rsif.2006.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis JM, Tsou LK, Hamilton AD. Chem. Soc. Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 16.Dings RPM, Mayo KH. Acc. Chem. Res. 2007;40:1057–1065. doi: 10.1021/ar700086k. [DOI] [PubMed] [Google Scholar]
- 17.Goodman CM, Choi S, Shandler S, DeGrado WF. Nat. Chem. Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kritzer JA, Stephens OM, Guarracino DA, Reznik SK, Schepartz A. Bioorg. Med. Chem. 2005;13:11–16. doi: 10.1016/j.bmc.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horne WS, Gellman SH. Acc. Chem. Res. 2008;41:1399–1408. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayraktar H, Ghosh PS, Rotello VM, Knapp MJ. Chem. Commun. (Cambridge) 2006:1390–1392. doi: 10.1039/b516096k. [DOI] [PubMed] [Google Scholar]
- 21.Bayraktar H, You C-C, Rotello VM, Knapp MJ. J. Am. Chem. Soc. 2007;129:2732–2733. doi: 10.1021/ja067497i. [DOI] [PubMed] [Google Scholar]
- 22.Orner BP, Ernst JT, Hamilton AD. J. Am. Chem. Soc. 2001;123:5382–5383. doi: 10.1021/ja0025548. [DOI] [PubMed] [Google Scholar]
- 23.Yin H, Lee G-I, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD. Angew. Chem. 2005;117:2764. doi: 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:2704–2707. doi: 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]
- 24.Yin H, Lee G-I, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst JT, Wang H-G, Sebti SM, Hamilton AD. J. Am. Chem. Soc. 2005;127:10191–10196. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]
- 25.Becerril J, Hamilton AD. Angew. Chem. 2007;119:4555. doi: 10.1002/anie.200700657. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:4471–4473. doi: 10.1002/anie.200700657. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez JM, Hamilton AD. Angew. Chem. 2007;119:8768. doi: 10.1002/anie.200701869. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:8614–8617. doi: 10.1002/anie.200701869. [DOI] [PubMed] [Google Scholar]
- 27.Jain RK, Hamilton AD. Org. Lett. 2000;2:1721–1723. doi: 10.1021/ol005871s. [DOI] [PubMed] [Google Scholar]
- 28.Jain RK, Hamilton AD. Angew. Chem. 2002;114:663–665. [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:641–643. [Google Scholar]
- 29.Wilson AJ, Groves K, Jain RK, Park HS, Hamilton AD. J. Am. Chem. Soc. 2003;125:4420–4421. doi: 10.1021/ja028574m. [DOI] [PubMed] [Google Scholar]
- 30.Groves K, Wilson AJ, Hamilton AD. J. Am. Chem. Soc. 2004;126:12833–12842. doi: 10.1021/ja0317731. [DOI] [PubMed] [Google Scholar]
- 31.Fletcher S, Hamilton AD. New J. Chem. 2007;31:623–627. [Google Scholar]
- 32.Hamuro Y, Calama MC, Park HS, Hamilton AD. Angew. Chem. 1997;109:2797–2800. [Google Scholar]
- 33.Blaskovich MA, Lin Q, Delarue FL, Sun J, Park HS, Coppola D, Hamilton AD, Sebti SM. Nat. Biotechnol. 2000;18:1065–1070. doi: 10.1038/80257. [DOI] [PubMed] [Google Scholar]
- 34.Park HS, Lin Q, Hamilton AD. Proc. Natl. Acad. Sci. USA. 2002;99:5105–5109. doi: 10.1073/pnas.082675899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou H, Wang D-a, Baldini L, Ennis E, Jain R, Carie A, Sebti SM, Hamilton AD. Org. Biomol. Chem. 2006;4:2376–2386. doi: 10.1039/b515483a. [DOI] [PubMed] [Google Scholar]
- 36.Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Cell. 2007;130:323–334. doi: 10.1016/j.cell.2007.05.055. [DOI] [PubMed] [Google Scholar]
- 37.Aviezer D, Cotton S, David M, Segev A, Khaselev N, Galili N, Gross Z, Yayon A. Cancer Res. 2000;60:2973–2980. [PubMed] [Google Scholar]
- 38.Gradl SN, Felix JP, Isacoff EY, Garcia ML, Trauner D. J. Am. Chem. Soc. 2003;125:12668–12669. doi: 10.1021/ja036155z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saraogi I, Incarvito CD, Hamilton AD. Angew. Chem. 2008;120:9837–9840. doi: 10.1002/anie.200803778. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:9691–9694. doi: 10.1002/anie.200803778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.