

Abstract
The ubiquitin-proteasome system plays key roles in the control of cell growth. The cell cycle in particular is highly regulated by the functions of the SCF and APC/C ubiquitin ligases, and perturbation of their function can result in tumorigenesis. Although the SCF and APC/C complexes are well-established in growth control pathways, many aspects of their function remain unknown. Recent studies have shed light on the mechanism of SCF-mediated ubiquitination and new functions for the SCF complex and APC/C. Our expanding understanding of the roles of the SCF and APC/C complexes highlight the potential for targeted molecular therapies.
Introduction
The ubiquitin-proteasome system allows for precise spatial and temporal control of many diverse cellular processes ranging from the control of cell proliferation to cell death to circadian clock cycles. The assembly of polyubiquitin chains on the lysine of target proteins can produce many different outcomes based on the topology of the chains [1]. Chains extended using lysines 11 and 48 of ubiquitin lead to recognition and degradation by the proteasome, while mono-ubiquitination or chains extended via lysines 63 and 29 of ubiquitin can play a role in signal transduction. Although all these modification types are reversible through the action of deubiquitinating enzymes (DUBs), ubiquitin-mediated proteolysis by the proteasome is irreversible, providing directionality to cyclical events, such as the cell cycle.
The ubiquitination of target proteins occurs through an enzymatic cascade in which ubiquitin is covalently activated by an E1 ubiquitin activating enzyme and transferred to an E2 ubiquitin conjugating enzyme before a subsequent transfer mediated by an E3 ubiquitin ligase [2,3]. While the human genome encodes only two E1s and a handful of E2s, there are many hundreds of E3s, consistent with their role in the ultimate selection of substrates. E3s can broadly be grouped into two discreet classes based on both homology domains and biochemical function [4]. The Homologous to E6-AP C-Terminus (HECT) domain proteins are structurally similar to E6-AP, which mediates the destruction of p53 by HPV E6. During the final transfer of ubiquitin to substrates, the HECT family ligases form a transient, covalent linkage with ubiquitin using a conserved cysteine. In contrast, the RING family E3s, which use the RING domain to recruit E2s, do not play a direct role in the ubiquitination reaction; instead, they facilitate the transfer of ubiquitin from the E2 to the substrate. RING E3s can be further subdivided into individual proteins that contain both the RING and substrate adapter domains in a single polypeptide or multi-subunit protein complexes that use distinct RING and substrate adapter proteins to recruit the E2 and substrate.
The canonical multisubunit RING complex is the Skp1/Cul1/F-box (SCF) complex, in which the Cul1 scaffold binds the RING protein Rbx1 (to recruit the E2) and Skp1, to bridge to a variable F-box protein, which determines substrate specificity [5–7]. The human genome encodes eight Cullin proteins (CUL1-4A, 4B, 5, 7, and 9 [formerly PARC]) that form similar Cullin-RING Ligase (CRL) complexes [7,8]. Additionally, the more distantly related APC2 forms the scaffold of the SCF-related Anaphase Promoting Complex/Cyclosome (APC/C) [9,10]. Each complex (excluding the less well-defined CUL7 and CUL9 complexes) can utilize multiple substrate specificity subunits, allowing a single core to target multiple substrates. For example, the human genome contains 69 F-box proteins [11]. Additionally, a single substrate adapter can recognize multiple substrates, which allows even the APC/C, with only two substrate adapters, to target many substrates.
Research into the functions of ubiquitin ligases has focused largely on their control of cell growth and division, where they play key roles in the targeted proteolysis of growth control and cell cycle regulatory proteins. The SCF complex and the APC/C have been demonstrated to be the major E3s controlling the degradation of cell cycle regulators, although they play additional, largely unexplored roles in other aspects of cell physiology [6,12]. This review will focus on the current literature concerning: the mechanism of CRL ligase function; biological functions for the SCF and APC/C ligases; and the future challenges remaining for the SCF and APC/C field.
Controlling CRL function: the role of NEDD8
The ubiquitin-like protein NEDD8 is an essential activator of CRLs, but the biochemical basis for this activation has remained mysterious. Neddylation, the covalent linkage of NEDD8 to another protein, has been demonstrated for all but one cullin and, like ubiquitination, results from an enzymatic cascade involving an E1, E2, and E3 (in this case, the CRL itself) [13,14]. Additionally, NEDD8 can be removed from cullins by the COP9 signalosome[15]. Due to the involvement of multiple CRLs in cell growth control and tumorigenesis, inhibitors of the neddylation pathway have been developed and appear to be effective anti-tumor compounds [16].
Neddylation has been proposed to have many functional consequences for CRL complex activity, beginning with the regulation of CRL assembly. Unmodified CUL1 binds to CAND1, a large sequestration factor, blocking the binding of the Skp1-F-box protein pair to the N-terminus [17–20]. Nedd8 conjugation dissociates CAND1, allowing formation of an active complex. CAND1 appears to function in this manner for all cullins. Despite the appeal of this model for explaining NEDD8’s essential function, CAND1 displacement cannot explain the stimulatory effects of NEDD8. NEDD8 conjugation remains essential in plants deficient in CAND1, and in vitro ubiquitination reactions (devoid of CAND1) can be stimulated by the addition of the neddylation machinery, suggesting a catalytic role in the ubiquitination reaction [21–26].
In isolation, the crystal structure of SCF offers little insight into the role of NEDD8 modification, as the structure was solved using non-neddylated CUL1[27]. The SCF complex structure and the resulting models addressed two key components of the ubiquitination reaction (i.e. substrate binding and E2 recruitment), but many mechanistic questions remained unanswered. With a large gap between the F-box protein and the presumptive E2 binding site, how is ubiquitin transferred from the E2 to substrates? Furthermore, with CUL1 forming an apparently rigid backbone, how could the complex adjust to facilitate extension of the ubiquitin chain? These questions were compounded by a lack of the knowledge of the chemistry of CRL-mediated ubiquitination. To date, no CRL structure has been solved with an intact E2 and substrate, making detailed evaluations of the ubiquitination mechanism difficult.
In principle, ubiquitination of substrates by CRLs can be broken into several steps: substrate binding, E2 recruitment, and ubiquitin transfer, with the transfer reaction further broken down into monoubiquitination and polyubiquitination. Notably, substrates for monoubiquitination and polyubiquitination are very different in terms of geometry, making them chemically distinct steps despite processive polyubiquitination occurring during one substrate binding event. The role of NEDD8 in substrate binding (via CAND1 displacment) has previously been addressed, but the impact of neddylation on other steps in CRL-mediated ubiquitination remained unknown until recently.
Using multiple in vitro assays to probe SCF complex function, Deshaies and colleagues found that neddylation impacts virtually every step of polyubiquitination by the SCF complex [28]. As previously observed, neddylation increases the binding of E2 enzymes to the SCF complex, but it also has larger and more pleiotropic effects [29]. In general, neddylation enhanced not only the addition of the first ubiquitin to substrates but also the extension of the polyubiquitin chain [28]. The effect of neddylation appears to be conformational in nature and to affect three distinct processes. First, neddylation modestly increases the intrinsic activity of the CRL-E2 complex, as evidenced by enhanced transfer of ubiquitin from the E2 in a target-independent manner to a small molecule substrate. This generic ubiquitin transfer reaction suggests a stabilization of the transition state in the transfer reaction, which likely applies equally for the mono- and polyubiquitination steps. Second, modification of CUL1 with NEDD8 appears to close the 50 angstrom gap between the F-box protein and the activated E2, greatly facilitating the initial ubiquitin transfer. Finally, neddylation also affects ubiquitin chain extension, suggesting that the conformation change also allows the activated E2 greater access to the end of the nascent polyubiquitin chain.
These predicted conformation changes predicted were fully illuminated by crystal structures of the NEDD8-conjugated C-terminus of CUL5 produced by the Schulman group [30]. Neddylation provokes a large conformation change in the CUL5-RBX1 complex, during which the CAND1 binding site is destroyed and RBX1 is released from its tight binding of the cullin on a flexible linker into an “open” form. The flexible RBX1 linker allows the bound E2 to bridge the 50 angstrom gap present in the original SCF complex structure, explaining the ability of neddylation to stimulate the initial ubiquitin transfer to substrate. Furthermore, after the addition of ubiquitin to the substrate, the flexibility allows the E2 to adjust to the end of the chain to allow efficient polyubiquitination. Mutational analysis suggests that the RBX1 linker evolved for the optimal efficiency of three distinct reactions: cullin neddylation, substrate monoubiquitination, and substrate polyubiquitination. The conformational change mechanism captured in the CUL5-NEDD8 structure appears to be a general mechanism that explains the efficient initiation and extension of ubiquitin chains on substrates by CRLs (Fig. 1).
Figure 1. Neddylation Stimulates SCF-mediated ubiquitination through a conformational change.

Non-neddylated CUL1 holds RBX1 in a closed form and is bound by CAND1, preventing SKP1-F-box protein binding. Neddylation of CUL1 releases RBX1 on a flexible linker and dissociates CAND1, allowing binding of the SKP1-F-box protein pair. The flexible RBX1 linker bridges the gap between the E2 and substrate (that would be present in non-neddylated SCF), facilitating both the initial ubiquitination and subsequent polyubiquitination.
However, many questions about neddylation and CRL-mediated ubiquitination remain. Although the neddylated C-terminus of CUL5 was crystallized in complex with RBX1, CUL5 actually utilizes the highly related protein RBX2 in vivo [31]. Notably, while the other cullins utilize RBX1 and UBE2M in the neddylation reaction, CUL5 uses RBX2 and UBE2F, a recently discovered second E2 for NEDD8 [32]. The existence of a two systems for cullin neddylation and a large complex (the COP9 signalosome) devoted to removal of NEDD8 suggests that neddylation is more tightly regulated than previously thought and that other factors affecting neddylation remain unidentified. The structural insights into NEDD8 function also highlight another gap in our current knowledge of SCF. Despite the generation of models of SCF bound to E2s, a definitive structure has not been determined, hiding aspects of the ubiquitination reaction.
New functions for the APC/C and the SCF ligases
The APC/C controls the onset of anaphase and the resetting of the cell cycle through the Cdc20 and Cdh1 substrate adapters, respectively (Fig. 2). APC/CCdc20 triggers anaphase by mediating the degradation of Securin and Shugoshin and the exit from mitosis by mediating the degradation of cyclin B [9,10,33]. APC/CCdh1 cooperates with APC/CCdc20 in controlling the exit from mitosis, but it plays a larger role in the maintenance of the G0 and G1 phase by mediating the degradation of multiple substrates [9,10,34]. As the controller of anaphase entry, APC/CCdc20 is the primary target of the spindle assembly checkpoint (SAC), which ensures the proper attachment of the chromosomes to the mitotic spindle before chromosome separation begins [35]. Entrance into anaphase with unattached chromosomes or in the presence of mitotic spindle poisons could lead to chromosome loss and/or breakage, creating aneuploidy and genomic instability. When the SAC is activated, Cdc20 is inhibited in a Mad2-dependent process.
Figure 2. Regulation of the APC/C during the cell cycle.
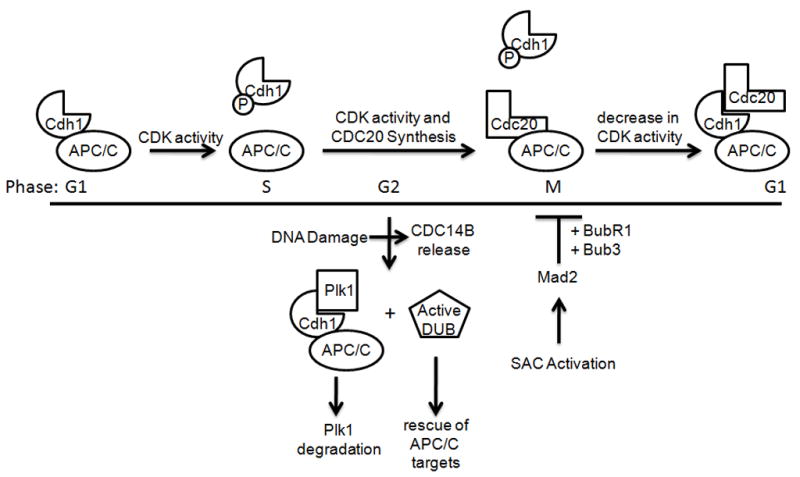
APC/C is activity is highly regulated during the cell cycle through the binding of the Cdc20 and Cdh1 activating subunits. APC/CCdh1 is active in G1, but Cdh1 is inactivated by increasing amounts of Cdk activity as the cell cycle progresses, preventing binding to the APC/C core. (Other means of Cdh1 inactivation, including Emi1 binding and degradation are not depicted here.) Cdc20 levels are low in G1 due to APC/CCdh1-mediated degradation, but they increase through S and G2. In M, APC/CCdc20 forms, but it remains inactive until bipolar attachment of all chromosomes to the spindle and the Spindle Assembly Checkpoint (SAC) is satisfied. Then, APC/CCdc20 targets Securin, Shugoshin, Cyclin B, and other proteins for degradation. With decreasing Cdk activity and Cdc14 phosphatase activity, unphosphorylated Cdh1 binds the APC/C core, resulting in the targeting of Cdc20 and other substrates (many of which are also APC/CCdc20 substrates) whose degradation is required to exit mitosis, enter G1, and maintain the G1 state. Although Cdh1 is inactive due to phosphorylation in G2, DNA damage releases the Cdc14B phosphatase from the nucleolus, causing Cdh1 dephosphorylation, resulting in active APC/CCdh1, which targets Plk1 for degradation. Notably, certain APC/CCdh1 substrates, such as Claspin, are spared, likely through the action of deubiquitinating enzymes (DUBs).
Intuitively, the SAC appears to inhibit APC/C activity, but recent reports suggest that APC/C activity is actually crucial for maintenance of the SAC [36,37]. Upon SAC activation, Cdc20 is subject to an immediate inhibition that remains mechanistically unclear, requiring Mad2 binding. Despite this initial inhibition, Cdc20 may threaten the ability of the initial response to keep APC/CCdc20 activity below the threshold level required for anaphase entry. However, a second stage of the SAC, utilizing Mad2, BubR1 and Bub3, directs the APC/C-dependent degradation of Cdc20, preventing premature override of the SAC. The exact mechanisms that allow Mad2, BubR1 and Bub3 to redirect APC/C activity to Cdc20 remain unclear, but these studies, in conjunction with previous studies conducted in S.cerevisiae [38], clearly demonstrate a new role for the APC/C core complex as both a target and an effector of the SAC, implicating the APC/C in the control of genome integrity.
While the core of the APC/C helps maintain genome integrity during SAC activation, a new role for APC/CCdh1 in the DNA damage response was recently revealed. APC/CCdh1 contributes to mitotic exit and is essential for the maintenance of the G0/G1 phase of the cell cycle [34]. This well-established function is dependent upon the degradation of cell cycle activators and DNA replication machinery. In addition, a role for Cdh1 in maintaining genome stability was uncovered by the development of Cdh1 null mice [39,40]. Cdh1 null cells exhibit defects in mitotic exit and cytokinesis and display many chromosome abnormalities, ranging from misaligned metaphase chromosomes to multi-polar spindles to an increased rate of chromosome bridge formation, all of which can generate aneuploid cells with up to 150 chromosomes. The precise underlying mechanisms of these effects are unknown, and it has been suggested to result from dysregulation of the DNA replication machinery. However, the role of APC/CCdh1 in the response to DNA damage may also contribute to the genomic instability of Cdh1 null cells.
During the cell cycle, APC/CCdh1 is kept inactive from late G1 through late mitosis by Cdk-mediated, inhibitory phosphorylation of Cdh1, which prevents binding to the APC/C core (Fig. 2) [9,10]. After Cdk activity is sufficiently reduced in mitosis, Cdh1 binds APC/C to form the active complex. Despite this activation profile, APC/CCdh1 activity has been observed in G2 following DNA damage, and the function and mechanism of this reactivation was recently elucidated, revealing an ancient DNA damage response pathway [41,42].
In the G2 phase of the cell cycle, Plk1 kinase activity controls both the activation of Cdk1 (through phosphorylation-directed Wee1 degradation) and the recovery from DNA replication stresses (through phosphorylation-directed Claspin degradation) before inactivation via APC/C-mediated degradation in late mitosis [43–46]. Both functions of Plk1 are problematic for cells exposed to genotoxic stress in G2, which need to arrest the cell cycle and repair damaged DNA. New evidence demonstrates that activation of APC/CCdh1 in DNA-damaged cells targets Plk1 [42]. Following DNA damage stimuli, the Cdc14B phosphatase is released from the nucleolus to dephosphorylate Cdh1, permitting the formation of active APC/CCdh1, which directs the degradation of Plk1. Although the activation of APC/CCdh1 appears general in nature, other substrates are protected by either deubiquitinating enzymes (e.g., Claspin, which is required for the DNA damage response) and, likely, Emi1 (e.g., cyclin A and cyclin B, which may be necessary for restarting the cell cycle after DNA damage repair). The participation of APC/CCdh1 in the G2 DNA damage response suggests another mechanism for the genome instability observed in Cdh1 null cells.
Other unique aspects of the biology of APC/C function are also becoming apparent. Polyubiquitin chains assembled through Lysine 48 (K48) of ubiquitin target substrates to the proteasome; however, several recent studies have now demonstrated that the APC/C can assemble polyubiquitin chains in vitro using alternative Lysines, particularly Lysine 11 (K11), which also target substrates for degradation [47,48]. Furthermore, at least in purified systems, APC/C substrates can be targeted to the proteasome through very short ubiquitin chains originating from multiple target Lysines [48]. This mechanism is in contrast to polyubiquitination by SCF (using Cdc34), which forms long, predominantly K48-linked ubiquitin chains. The use of alternative chains, perhaps of even of mixed morphology, suggests key mechanistic differences between the APC/C and other ubiquitin ligases that require further investigation.
Similar to the APC/C, the biological functions of the SCF scaffold continue to expand. While the large number of F-box proteins predicts a role for the SCF complex in many processes, the identification of multiple substrates for a given F-box protein often allows the assignment of a generalized function for that F-box protein. For example, Skp2, which degrades inhibitors of cell cycle progression, is an established oncogene [49]. By analogy, Fbxw7, which degrades activators of cell cycle progression, including c-Myc, c-Jun, and Cyclin E, should be a tumor suppressor, and, indeed, Fbxw7 mutations are common in T-ALL [50,51]. However, new mouse models of Fbxw7 inactivation reveal a more nuanced role for Fbxw7 in regulating stem cell quiescence and tumor progression.
The deletion of Fbxw7 is embryonic lethal, but conditional deletion models have been constructed to explore the role of Fbxw7 in the hematopoietic system [52–55]. Hematopoietic Stem Cells are thought to remain in the G0 phase of the cell cycle until departing from the stem cell niche to proliferate and differentiate. In the absence of Fbxw7, elevated c-Myc levels drive HSCs through the cell cycle, eventually leading to exhaustion, as evidenced by the inability of Fbxw7 null HSCs to reconstitute bone marrow [53,54]. The enforced cycling of Fbxw7 null HSCs also leads to elevated levels of certain progenitor cells, such as double positive (CD4+/CD8+)T cells, but the generation and survival of differentiated single positive cells is compromised by activation of p53, likely through oncogene-induced stress [56]. After losing p53 function, these cells drive T-ALL, demonstrating the tumor suppressor function of Fbxw7.
Unlike Skp2 and Fbxw7, β-TRCP appears to target substrates with a broader range of functions [49]. However, with the identification of many β-TRCP substrates, it now appears that β-TRCP function as an integrator of signaling pathways (Fig. 3). β-TRCP is well-known as an exquisitely sensitive rheostat for Cdk1 function through canonical substrates such as Cdc25A and Wee1, and newly identified substrates, such as Bora and Claspin, also contribute to feedback loops controlling Cdk1 [44,57]. Claspin mediates the ATR-dependent activation of Chk1, leading to Cdc25A degradation and Cdk1 inhibition. Diametrically opposed to this pathway, Bora, with the Aurora A kinase, activates Plk1 in late G2 to drive cells into mitosis through Wee1 degradation and consequent Cdk1 activation [58,59]. As cells progress into mitosis, both pathways are reset by Plk1 activity, which causes the β-TRCP-dependent degradation of Claspin (in G2) and Bora (in M).
Figure 3. β-TRCP integrates multiple stimuli for global control of the cell cycle.

β-TRCP controls early events in the cell cycle, such as the response to mitogens, but it also controls later events in the cell cycle through Cdk1, the APC/C, and Plk1. Notably, feedback loops are common in the control of the Cdk1 and Plk1 kinases. During DNA damage responses, β-TRCP targets for Cdc25A for degradation, but during the recovery from DNA damage, β-TRCP targets Clapsin for degradation, blocking Chk1 activation and Cdc25A degradation. Additionally, Plk1 is involved in the degradation of multiple β-TRCP substrates, and Bora, which activates Plk1, is targeted for β-TRCP-dependent degradation by Plk1, attenuating Plk1 activity at the end of mitosis. Notably, there is also much crosstalk between β-TRCP targets, with APC/C activity controlled by Cdk1 (via phosphorylation) and Plk1 (via phosphorylation and induction of Emi1 degradation). At the same time, APC/C is required for the inactivation of Cdk1 and Plk1 activators (cyclins and Bora, respectively). Finally, Plk1 is a positive regulator of Cdk1 through the induction of Claspin and Wee1 degradation.
Although β-TRCP tightly controls Cdk1 function, β-TRCP function is not restricted to mitosis. In response to mitogens, β-TRCP targets PDCD4, an inhibitor of protein translation, for degradation at the G0/G1 transition, and more recent data demonstrate that β-TRCP also degrades BimEL, a pro-apoptotic protein, in response to mitogens [60,61]. Therefore, in response to cell growth stimuli, β-TRCP upregulates protein translation (PDCD4) while promoting cell survival (BimEL). As mitogen-stimulated cells enter the cell cycle, β-TRCP further promotes cell cycle progression through the activation of Cdk1 (Wee1), activation of the APC/C (Emi1), and de-repression of mitotic checkpoint proteins (REST) [43,62–64]. At the same time, β-TRCP can also block cell cycle progression in response to genotoxic stress (Cdc25A) [65,66]. In this manner, β-TRCP integrates multiple stimuli to tightly control cell proliferation: all the proper signals must be present for progression through the cell cycle. Notably, multiple β-TRCP substrates, such as Period, are not part of the core cell cycle machinery, suggesting that β-TRCP may coordinate other processes with the cell cycle [49].
New challenges for the SCF: finding substrates
One of the main challenges for the SCF field – in fact for the ubiquitin ligase field in general - remains the identification of substrates. The search for SCF complex substrates is highlighted by the large number of “orphan” F-box proteins that have not been paired with substrates [11]. Each year, multiple new F-box substrate pairs involved in growth control are proposed, but the rate of discovery remains low for the effort invested. The challenge of identifying and validating substrates for orphan F-box proteins will hopefully be aided by new techniques designed explicitly for the investigation of protein stability. The Elledge laboratory recently developed a system for proteome-scale stability profiling (Global Protein Stability profiling; GPS), and this system has already been applied to screen for SCF substrates [67,68]. The initial screen cannot directly pair a substrate with its F-box protein, but similar screens, in conjunction with current discovery techniques will hopefully aid the identification of ubiquitin ligase-substrate pairs.
An additional problem inherent in establishing F-box protein substrates is the requirement of understanding the biology behind substrate degradation. Because substrates require posttranslational modifications, the identities and activities of substrate kinases are extremely important for placing substrates in biological networks. An additional layer of complexity is added when substrates are targeted by multiple ubiquitin ligases. These issues are exemplified in multiple recent studies of SCF-mediated Cyclin D1 degradation. To date, six F-box proteins (Fbxo4, Fbxo31, β-TRCP1/2, Fbxw8, and Skp2) have been reported to target Cyclin D1, and the reports suggest that each F-box protein binds a similar degron [69–73]. However, the stimuli and associated-kinase involved appear different in each case. For example, Fbxo31 has been reported to participate in the DNA damage-stimulated degradation of Cyclin D1 in a manner dependent on MAP kinase (MAPK) (which phosphorylates Cyclin D1) and ATM (which directly phosphorylates Fbxo31). In contrast, Fbxo4 and Fbxw8 have been reported to control the cell cycle dependent degradation of Cyclin D1 via GSK3 or MAPK, respectively. How different signals coordinate the action of six different F-box proteins on a single substrate is not yet completely understood. Therefore, it is vital to not only pair an F-box protein with a substrate but also pair the degradation event with a biological function.
Conclusions
The SCF and APC/C ligases play wide-ranging – and continually expanding – roles in cell growth control. However, despite the clinical success of general inhibitors of ubiquitin-mediated proteolysis and the potential for targeting the NEDD8 pathway for general inhibition of CRLs, the inhibition of specific ligase complexes has remained a largely unexplored therapeutic option. Continued research into the molecular mechanisms and biological function of SCF and APC/C ligases presents the opportunity to develop novel therapeutics with greater efficacy than general proteasome inhibitors.
Acknowledgments
JRS was supported by the American Cancer Society – Mr. and Mrs. William G. Campbell Postdoctoral Fellowship in Memory of Carolyn Cabott. MP is grateful to T.M. Thor for continuous support. Work in the Pagano laboratory is supported by the National Institutes of Health (R01-GM057587, R37-CA076584, and R21-CA125173) and the Multiple Myeloma Research Foundation. MP is an Investigator with the Howard Hughes Medical Institute. The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- 4.Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–178. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 5.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 6.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 7.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 8.Skaar JR, Florens L, Tsutsumi T, Arai T, Tron A, Swanson SK, Washburn MP, DeCaprio JA. PARC and CUL7 form atypical cullin RING ligase complexes. Cancer Res. 2007;67:2006–2014. doi: 10.1158/0008-5472.CAN-06-3241. [DOI] [PubMed] [Google Scholar]
- 9.Acquaviva C, Pines J. The anaphase-promoting complex/cyclosome: APC/C. J Cell Sci. 2006;119:2401–2404. doi: 10.1242/jcs.02937. [DOI] [PubMed] [Google Scholar]
- 10.Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- 11.Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ang XL, Harper JW. Interwoven ubiquitination oscillators and control of cell cycle transitions. Sci STKE 2004. 2004:pe31. doi: 10.1126/stke.2422004pe31. [DOI] [PubMed] [Google Scholar]
- 13.Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23:1985–1997. doi: 10.1038/sj.onc.1207414. [DOI] [PubMed] [Google Scholar]
- 14.Rabut G, Peter M. Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:969–976. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *15.Wei N, Serino G, Deng XW. The COP9 signalosome: more than a protease. Trends Biochem Sci. 2008;33:592–600. doi: 10.1016/j.tibs.2008.09.004. This review provides an excellent introduction to the COP9 signalosome, an important regulator of cullin neddylation that was not addressed by this review due to space constraints. [DOI] [PubMed] [Google Scholar]
- *16.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. This report not only illustrates the role of CRLs in growth control, but it also suggests that NEDD8 inhibitors are clinically viable chemotherapeutics. [DOI] [PubMed] [Google Scholar]
- 17.Goldenberg SJ, Cascio TC, Shumway SD, Garbutt KC, Liu J, Xiong Y, Zheng N. Structure of the Cand1-Cul1-Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell. 2004;119:517–528. doi: 10.1016/j.cell.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Furukawa M, Matsumoto T, Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell. 2002;10:1511–1518. doi: 10.1016/s1097-2765(02)00783-9. [DOI] [PubMed] [Google Scholar]
- 19.Zheng J, Yang X, Harrell JM, Ryzhikov S, Shim EH, Lykke-Andersen K, Wei N, Sun H, Kobayashi R, Zhang H. CAND1 binds to unneddylated CUL1 and regulates the formation of SCF ubiquitin E3 ligase complex. Mol Cell. 2002;10:1519–1526. doi: 10.1016/s1097-2765(02)00784-0. [DOI] [PubMed] [Google Scholar]
- 20.Oshikawa K, Matsumoto M, Yada M, Kamura T, Hatakeyama S, Nakayama KI. Preferential interaction of TIP120A with Cul1 that is not modified by NEDD8 and not associated with Skp1. Biochem Biophys Res Commun. 2003;303:1209–1216. doi: 10.1016/s0006-291x(03)00501-1. [DOI] [PubMed] [Google Scholar]
- 21.Chuang HW, Zhang W, Gray WM. Arabidopsis ETA2, an apparent ortholog of the human cullin-interacting protein CAND1, is required for auxin responses mediated by the SCF(TIR1) ubiquitin ligase. Plant Cell. 2004;16:1883–1897. doi: 10.1105/tpc.021923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furukawa M, Zhang Y, McCarville J, Ohta T, Xiong Y. The CUL1 C-terminal sequence and ROC1 are required for efficient nuclear accumulation, NEDD8 modification, and ubiquitin ligase activity of CUL1. Mol Cell Biol. 2000;20:8185–8197. doi: 10.1128/mcb.20.21.8185-8197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Podust VN, Brownell JE, Gladysheva TB, Luo RS, Wang C, Coggins MB, Pierce JW, Lightcap ES, Chau V. A Nedd8 conjugation pathway is essential for proteolytic targeting of p27Kip1 by ubiquitination. Proc Natl Acad Sci U S A. 2000;97:4579–4584. doi: 10.1073/pnas.090465597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, Pierce JW, Podust VN, Luo RS, Chau V, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20:2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu K, Chen A, Pan ZQ. Conjugation of Nedd8 to CUL1 enhances the ability of the ROC1-CUL1 complex to promote ubiquitin polymerization. J Biol Chem. 2000;275:32317–32324. doi: 10.1074/jbc.M004847200. [DOI] [PubMed] [Google Scholar]
- 26.Morimoto M, Nishida T, Honda R, Yasuda H. Modification of cullin-1 by ubiquitin-like protein Nedd8 enhances the activity of SCF(skp2) toward p27(kip1) Biochem Biophys Res Commun. 2000;270:1093–1096. doi: 10.1006/bbrc.2000.2576. [DOI] [PubMed] [Google Scholar]
- **27.Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. The crystal structure presented in this study is a landmark in the study of the SCF complex. Although the structure is now understood to represent an inactive, closed form, it is still the most complete SCF structure, giving valuable architectural information. Additionally, it introduces key questions about SCF complex-mediated ubiquitination that lead to the more recent structural studies of NEDD8-conjugated cullins. [DOI] [PubMed] [Google Scholar]
- **28.Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. This study offers an extremely detailed biochemical dissection of SCF complex-mediated ubiquitination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawakami T, Chiba T, Suzuki T, Iwai K, Yamanaka K, Minato N, Suzuki H, Shimbara N, Hidaka Y, Osaka F, et al. NEDD8 recruits E2-ubiquitin to SCF E3 ligase. EMBO J. 2001;20:4003–4012. doi: 10.1093/emboj/20.15.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **30.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. This important study reveals the structural nature of NEDD8 conjugation’s stimulation of CRL function, which also solves several mechanistic questions posed by the original SCF complex structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, Conaway JW, Nakayama KI. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004;18:3055–3065. doi: 10.1101/gad.1252404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, Borg LA, Neale G, Murray PJ, Roussel MF, et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. 2009;33:483–495. doi: 10.1016/j.molcel.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H. Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell. 2007;27:3–16. doi: 10.1016/j.molcel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Li M, Zhang P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div. 2009;4:2. doi: 10.1186/1747-1028-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- **36.Nilsson J, Yekezare M, Minshull J, Pines J. The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat Cell Biol. 2008;10:1411–1420. doi: 10.1038/ncb1799. This report demonstrates that APC/C is both a target and effector of the spindle assembly checkpoint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ge S, Skaar JR, Pagano M. APC/C- and Mad2-mediated degradation of Cdc20 during spindle checkpoint activation. Cell Cycle. 2009;8:167–171. doi: 10.4161/cc.8.1.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan J, Chen RH. Spindle checkpoint regulates Cdc20p stability in Saccharomyces cerevisiae. Genes Dev. 2004;18:1439–1451. doi: 10.1101/gad.1184204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **39.Garcia-Higuera I, Manchado E, Dubus P, Canamero M, Mendez J, Moreno S, Malumbres M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802–811. doi: 10.1038/ncb1742. In addition to demonstrating a role for Cdh1 in maintaining genome stability, this study also provides evidence that Cdh1 likely functions as a tumor suppressor. Somewhat surprisingly, Cdh1 function is not essential for mouse development. [DOI] [PubMed] [Google Scholar]
- 40.Li M, Shin YH, Hou L, Huang X, Wei Z, Klann E, Zhang P. The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat Cell Biol. 2008 doi: 10.1038/ncb1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sudo T, Ota Y, Kotani S, Nakao M, Takami Y, Takeda S, Saya H. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J. 2001;20:6499–6508. doi: 10.1093/emboj/20.22.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134:256–267. doi: 10.1016/j.cell.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe N, Arai H, Nishihara Y, Taniguchi M, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFβ-TRCP. Proc Natl Acad Sci U S A. 2004;101:4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006;23:319–329. doi: 10.1016/j.molcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 45.Mailand N, Bekker-Jensen S, Bartek J, Lukas J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell. 2006;23:307–318. doi: 10.1016/j.molcel.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 46.Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A, Medema RH, Freire R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr Biol. 2006;16:1950–1955. doi: 10.1016/j.cub.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 47.Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653–665. doi: 10.1016/j.cell.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, Gygi SP. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8:700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 49.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and β-TRCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 51.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 52.Tsunematsu R, Nakayama K, Oike Y, Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T, Nakayama KI. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J Biol Chem. 2004;279:9417–9423. doi: 10.1074/jbc.M312337200. [DOI] [PubMed] [Google Scholar]
- *53.Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, Zavadil J, Nimer SD, Aifantis I. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. 2008;205:1395–1408. doi: 10.1084/jem.20080277. This report (and the subsequent reference) detail the control of hematopoietic stems by Fbxw7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *54.Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008;22:986–991. doi: 10.1101/gad.1621808. This report (and the previous reference) detail the control of hematopoietic stems by Fbxw7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tetzlaff MT, Yu W, Li M, Zhang P, Finegold M, Mahon K, Harper JW, Schwartz RJ, Elledge SJ. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc Natl Acad Sci U S A. 2004;101:3338–3345. doi: 10.1073/pnas.0307875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alboran IM, Nakayama K, Nakayama KI. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med. 2007;204:2875–2888. doi: 10.1084/jem.20062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seki A, Coppinger JA, Du H, Jang CY, Yates JR, 3rd, Fang G. Plk1- and β-TRCP-dependent degradation of Bora controls mitotic progression. J Cell Biol. 2008;181:65–78. doi: 10.1083/jcb.200712027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- 59.Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655–1658. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 61.Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, Dustin M, Huang DC, Taunton J, Pagano M. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell. 2009;33:109–116. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, Margottin-Goguet F, Jackson PK, Yamasaki L, Pagano M. Control of meiotic and mitotic progression by the F box protein β-TRCP1 in vivo. Dev Cell. 2003;4:799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 63.Margottin-Goguet F, Hsu JY, Loktev A, Hsieh HM, Reimann JD, Jackson PK. Prophase destruction of Emi1 by the SCF(betaTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev Cell. 2003;4:813–826. doi: 10.1016/s1534-5807(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 64.Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, Lasorella A, Iavarone A, Chang S, Hernando E, et al. Control of chromosome stability by the β-TRCP-REST-Mad2 axis. Nature. 2008;452:365–369. doi: 10.1038/nature06641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF. Degradation of Cdc25A by β-TRCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- 66.Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. SCFβ-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yen HC, Elledge SJ. Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science. 2008;322:923–929. doi: 10.1126/science.1160462. [DOI] [PubMed] [Google Scholar]
- **68.Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science. 2008;322:918–923. doi: 10.1126/science.1160489. This study introduces a method for measuring the stability of proteins on a global scale. The use of such methods in new screens for substrates of SCF complexes (see reference 58) or other ubiquitin ligases may faciliate the future identification of substrates. Notably, GPS will likely not help in the identification of substrates for non-degradation types of ubiquitin modification. [DOI] [PubMed] [Google Scholar]
- 69.Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, Diehl JA. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355–366. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009 doi: 10.1038/nature08011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei S, Yang HC, Chuang HC, Yang J, Kulp SK, Lu PJ, Lai MD, Chen CS. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J Biol Chem. 2008;283:26759–26770. doi: 10.1074/jbc.M802160200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE. 2006;1:e128. doi: 10.1371/journal.pone.0000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci U S A. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]