

Abstract
Radiotracers for imaging protein targets in living human brain with positron emission tomography (PET) are increasingly useful in clinical research and in drug development. Such radiotracers must fulfill many criteria, among which an ability to enter brain adequately and reversibly without contamination by troublesome radiometabolites is desirable for accurate measurement of the density of a target protein (e.g., neuroreceptor, transporter, enzyme or plaque). Candidate radiotracers may fail as a result of poor passive brain entry, rejection from brain by efflux transporters or undesirable metabolism. These issues are reviewed. Emerging PET radiotracers for measuring efflux transporter function, and new strategies for ameliorating radiotracer metabolism are discussed. A growing understanding of the molecular features affecting the brain penetration, metabolism and efflux transporter sensitivity of prospective radiotracers should ultimately lead to their more rational and efficient design, and also to their greater efficacy.
Background
Positron emission tomography (PET) is a powerful molecular imaging modality, which when applied with specific radiopharmaceuticals, can deliver quantitative measures of biochemical parameters, such as the concentrations or functions of neurotransmitter receptors, transporters or enzymes in living subjects, including human beings. Consequently, PET is increasingly useful for both clinical research [1] and also for drug discovery and development [2].
PET is commonly applied to studies of brain, and the required radiopharmaceuticals are usually labeled with one of the short-lived positron-emitters, carbon-11 (t1/2 = 20 min) or fluorine-18 (t1/2 = 110 min). Brain PET radiopharmaceuticals may have different principles of action. For example, some are designed to enter brain, often by active transport, and to accumulate in brain after a metabolic transformation. These are often analogues of endogenous compounds. The classic example is 2-[18F]fluoro-2-deoxy-D-glucose, which enters brain by the action of a glucose transporter and is then phosphorylated by hexokinase to give the 18F-labeled 6-phosphate [3]. The latter accumulates in brain to provide a measure of hexokinase action (energy metabolism). Members of another major class of radiopharmaceuticals are characterized by having no recognized transporter for brain entry and are intended to interact structurally-unchanged and by reversible non-covalent binding with a protein target (e.g., receptor, transporter, enzyme or plaque). This much larger class of radiopharmaceutical is the main focus of this review, and for want of a more specific term, will be referred to throughout as ‘radiotracers’ to distinguish them from other types of brain radiopharmaceutical. Only a low fraction of potentially interesting protein targets can presently be imaged effectively. Hence, there is a great need to expand the range of such PET radiotracers.
For PET radiotracers to be useful for quantitative imaging of protein targets in brain, they must fulfill a wide range of demanding criteria [4, 5] (Box 1). Therefore, much like CNS drug discovery, PET radiotracer development for brain is scientifically challenging with success being quite sporadic. Primarily, the radiotracer must show high affinity and selectivity for the target protein. Moreover, the radiotracer must be able to reach its target in vivo. This includes running the gauntlet of the blood-brain barrier (BBB) which is well-equipped with efflux pumps, such as P-glycoprotein (P-gp) [6] and multi-drug resistance-associated proteins [7], that function to repel many unwanted or xenobiotic compounds.
Box 1. Criteria to be met ideally by candidate PET radiotracers. The highlighted criteria are discussed in this review.
High affinity (usually in nM range for KD)
Selectivity for target
Ability to penetrate blood-brain barrier
Non-substrate for efflux transporters
Lack of troublesome radiometabolites
Low non-specific binding
Suitable brain pharmacokinetics in relation to radiolabel half-life (observable brain uptake and washout kinetics)
Amenability to labeling with 11C or 18F at high specific radioactivity
Safe for administration at low tracer dose
Intravenously administered PET radiotracers are immediately exposed to an array of metabolizing enzymes in blood and other tissues. Almost without exception, PET radiotracers are metabolized significantly over minutes such that the fraction of radioactivity present in blood as unchanged radiotracer very quickly diminishes. The radiometabolites are usually less lipophilic than the parent radiotracer with reduced propensity to enter brain. Nevertheless, not all radiometabolites are adequately excluded from brain, and in some cases radiometabolites may also be formed within the brain. All of these brain radiometabolites may be termed ‘troublesome’; they sully the nature of the signal being recorded, because PET has no means to discriminate between the chemical sources of detected radioactivity. Ideally, a PET radiotracer should readily enter brain and interact selectively with its protein target in the absence of troublesome radiometabolites (Figure 1).
Figure 1.

PET Radiotracers: crossing the blood-brain barrier and surviving metabolism. The cartoon represents a voxel in brain, with edge dimensions of a millimeter or so, as seen with a PET camera after intravenous administration of a radiotracer. Blood represents about 5% of the total volume. The red circles indicate some of the radioactive entities that might be generated from the parent radiotracer (Rf) and their locations. The green areas represent efflux transporters at the blood-brain barrier (e.g., P-gp). Key: R = radiotracer; M = radiometabolite. Subscripts: f = free; b = plasma protein-bound; np = non BBB-penetrant; ns = non-specifically bound to brain; sp = specifically bound to brain. Radiotracer development for a protein target within brain aims to achieve ready passive diffusion of the radiotracer across the BBB, without impediment by efflux transporters, to give a high ratio of specifically bound radiotracer (Rsp) to non-specifically bound (Rnp) plus free (Rf) radiotracer, while suppressing the presence of radiometabolite entities in brain.
This review discusses factors affecting the brain entry of PET radiotracers, including the action of efflux pumps. Emerging radiotracers for imaging efflux pump function are also discussed. Finally, radiotracer metabolism is discussed indicating tactics to avoid troublesome radiometabolites. Increasing appreciation of radiotracer brain entry and metabolism will enable the more efficient development of much-needed PET radiotracers for delivering meaningful quantitative output measures (e.g., regional values of volumes of distribution (VT) or binding potentials (BP) for target proteins [8]; see Glossary).
Radiotracer brain entry
A PET radiotracer must exhibit adequate brain entry to be useful for quantitative measurements. A convenient measure of brain tissue radiotracer concentration is the standardized uptake value (SUV), which normalizes radioactivity concentration (e.g., Bq/mL) to injected radioactive dose and subject body weight. An SUV value of 1 corresponds to the concentration of radiotracer that would result from uniform distribution of the injected dose throughout the entire body. SUV values may be compared between tissues and species. For brain tissue, peak SUV values greater than 2 are generally desirable, with the best radiotracers showing much higher values (Table 1). The ability of a radiotracer molecule to enter brain from plasma essentially depends on its capacity for passive entry versus its susceptibility to rejection by efflux pumps [9].
Table 1.
A selection of effective radiotracers for studying human brain receptors and other protein targets with PET: lipophilicity (cLogD and clogP) and peak brain concentration (SUV). Data are listed in ascending order of cLogD value.
| Radiotracer | Target proteina | cLogDb | cLogPb | Peakc conc’n. (SUV). | Reference |
|---|---|---|---|---|---|
| [18F]6-A-85380 | α4β2 nAChR | − 1.93 | 0.57 | 3.5 | [65] |
| [11C]CFT | DAT | 0.01 | 2.42 | 7.0 | [66] |
| [11C]Flumazenil | Bz | 0.87 | 0.87 | 5.0d | [52] |
| [18F]SPA-RQ | NK1 | 1.00 | 3.23 | 3.0 | [67] |
| [18F]Fallypride | D2/D3 | 1.08 | 2.12 | 9.3 | [68] |
| [11C]Diprenorphine | OR | 1.30 | 2.21 | 4.1 | [69] |
| [11C](R)-Rolipram | PDE4 | 1.43 | 1.43 | 3.5 | [70] |
| [11C]Raclopride | D2 | 1.50 | 3.96 | 2.9 | [71] |
| [11C]DWAY | 5-HT1A | 1.60 | 1.70 | 6.0 | [72] |
| [11C]MDL 100907 | 5-HT2A | 1.87 | 3.56 | 4.9 | [73] |
| [18F]MPPF | 5-HT1A | 2.27 | 2.42 | 2.7 | [29] |
| [11C]WAY-100635 | 5-HT1A | 2.51 | 2.66 | 3.4 | [72] |
| [18F]SP203 | mGluR5 | 2.97 | 2.97 | 5.8 | [49] |
| [11C]PBR28 | TSPO | 3.29 | 3.30 | 2.0 | [74] |
| [11C]PIB | Aβ-plaque | 3.31 | 3.33 | 4.0 | [75] |
| [11C]DASB | SERT | 3.35 | 3.35 | 7.8 | [76] |
| [18F]PBR06 | TSPO | 4.09 | 4.09 | 1.7 | [47] |
| [11C](R)-PK 11195 | TSPO | 4.58 | 4.58 | 1.3 | [77] |
| [11C]NNC 112 | D1 | 4.90 | 5.53 | 4.0 | [78] |
| [11C]MePPEP | CB1 | 5.42 | 5.46 | 3.7 | [16] |
Target abbreviations: α4β2 nAChR, α4β2 sub-type of nicotinic acetylcholine receptor; DAT, dopamine transporter; Bz, benzodiazepine binding site of GABAA receptors; NK1, neurokinin-1 receptor; D2/D3, dopamine sub-type 2 and 3 receptors; OR, opiate receptor; PDE4, phosphodiesterase-4; 5-HT1A, serotonin receptor sub-type-1; mGluR5, metabotropic glutamate sub-type 5 receptor; TSPO, translocator protein (18 kDa), (formerly known as the ‘peripheral benzodiazepine receptor’ or PBR); Aβ-plaque, brain amyloid plaque; SERT, serotonin transporter; D1 dopamine sub-type 1 receptor; CB1, cannabinoid sub-type 1 receptor.
Calculated with ACD/logD software (Advanced Chemistry Development Inc.).
Values are for brain regions reported with highest concentration, unless indicated for whole brain. Where values were estimated from radiotracer dose and radioactivity concentration, a body weight of 70 kg was assumed.
Estimated for whole brain.
Passive entry
Most PET brain radiotracers are necessarily drug-like molecules. Hence, factors influencing the ability of these radiotracers to enter brain are very similar to those that influence CNS drugs. Several molecular features of a radiotracer may govern its passive transfer across the blood-brain barrier in either direction. Generally, passive transfer is promoted by low molecular weight (< 500 Da), small cross-sectional area (< 80 Å2), low hydrogen bonding capacity and lack of formal charge [10–12]. Lipophilicity is a major factor influencing passive brain entry. The most commonly used index of compound lipophilicity is LogP where P is the n-octanol/water partition coefficient of the unionized species. The corresponding distribution coefficient for all derived species (e.g., unionized and ionized species) at physiological pH is termed D7.4. In general, radiotracers with moderate lipophilicity, indicated by LogD7.4 values in the approximate range 2.0–3.5, show optimal passive brain entry in vivo [13]. Exceptionally, some useful radiotracers with lower or higher LogD7.4 values also readily enter brain, but for unclear reasons [13]. Table 1 includes many examples. Passive entry into brain might be expected to increase with radiotracer lipophilicity, since entry first requires partitioning of the radiotracer from plasma into the lipid bilayer of the blood-brain barrier. However, a radiotracer with high lipophilicity may also become extensively bound to blood proteins, thereby reducing the fraction of radiotracer that is freely available in plasma (fP) to traverse the barrier. Thus, in general, highly lipophilic radiotracers with low fp values are expected to have low brain uptake. This is not always observed. For example, a recently developed radiotracer for brain cannabinoid sub-type-1 (CB1) receptors, [11C]MePPEP [14], readily enters monkey (peak SUV ~ 6 [15]) and human brain (peak SUV ~ 3.7 [16]), despite being highly lipophilic (cLogP = 5.7) with a low fP value (0.05%) (Table 2). In this case, it may be deduced that the extensive binding of [11C]MePPEP to blood proteins is readily reversible.
Table 2.
Structures and targets for PET radiotracers discussed in this review.
| Radiotracer | Target | Structure |
|---|---|---|
| [18F]FCWAY | 5-HT1A | 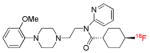 |
| [18F]MPPF | 5-HT1A | 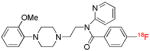 |
| [carbonyl-11C]WAY- 100635 | 5-HT1A | 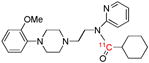 |
| [O-methyl-11C](R)-RWAY | 5-HT1A | 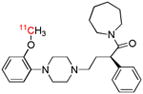 |
| [O-methyl-11C]WAY- 100635 | 5-HT1A | 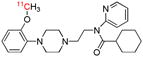 |
| [11C]Flumazenil | Bz | 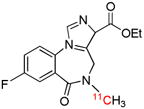 |
| [18F]Flumazenil | Bz | 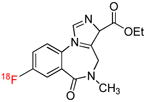 |
| [18F]CPFX | A1 | 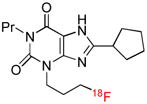 |
| [11C]MePPEP | CB1 | 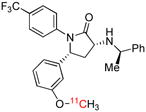 |
| [11C]PE2I | DAT | 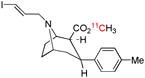 |
| [18F]FECNT | DAT | 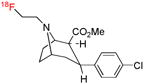 |
| [N-methyl-11C]cocaine | DAT | 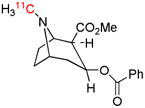 |
| [O-methyl-11C]cocaine | DAT | 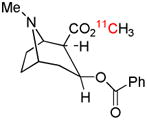 |
| [18F]SP203 | mGluR5 | 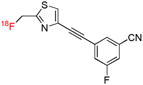 |
| [18F]FMeNER-d2 | NET | 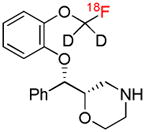 |
| [11C]dLop | P-gp function | 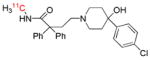 |
| [11C]Loperamide | P-gp function | 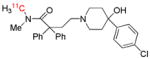 |
| [11C]Verapamil | P-gp function | 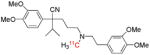 |
| [11C]PBR28 | TSPO | 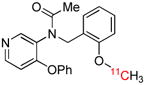 |
| [18F]PBR06 | TSPO | 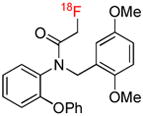 |
Abbreviations for target site: A1, adenosine sub-type 1 receptor; MRP-1, multi-drug resistance –associated protein 1; NET, noradrenalin transporter. See legend to Table 1 for explanation of other abbreviations.
The development of candidate brain PET radiotracers with high lipophilicity has usually been regarded as undesirable not only for avoiding slow brain entry but also for avoiding high non-specific binding to brain fats and proteins and consequent attenuation of specific signal [4, 5]. However, one of the reasons that [11C]MePPEP works as a radiotracer, besides its high affinity and despite its high lipophilicity, is that CB1 receptors are one of the most abundant G-protein-coupled receptors in brain, so giving a high receptor-specific to non-specific signal.
Influence of efflux transporters
Many PET radiotracers are now known to be substrates for brain efflux transporters, and especially P-gp [17, 18], which is highly prevalent at the blood-brain barrier. In general, substrate behavior for P-gp is promoted by high lipophilicity, positive charge at pH 7.4 and multiple aromatic groups [9]. P-gp substrates therefore show high structural diversity. The degree to which P-gp restricts brain entry varies with the radiotracer and may also vary across species [19]. Quite small structural differences may have a large and unpredictable impact on radiotracer susceptibility to brain efflux in a single animal species. Closely related radiotracers, developed for imaging 5-HT1A receptors, exemplify these points [Table 2]. In rats, [11C]WAY-100635 is a weak substrate for P-gp [20], while [11C](R)-RWAY [21] and [18F]MPPF [22, 23] are strong substrates and well excluded from brain. However, all three radiotracers have acceptable brain accumulation (> 2 SUV) in both monkey [24–26] and human [27–29]. In fact, [11C](R)-RWAY was shown not to be a P-gp substrate in monkey [25]. Such species differences clearly exacerbate the severe challenge of circumventing P-gp recognition through rational radiotracer design [9].
The possible consequences of radiotracer susceptibility to brain efflux transporters on PET measurements of protein targets are not yet fully clarified; it has generally been assumed that any effects of efflux transporters are similar across brain regions. However, a recent study reports heterogeneity of P-gp distribution in rat brain and consequently regional effects on radiotracer ([18F]MPPF) uptake in rat brain [23]. Such heterogeneity in P-gp distribution and action may become amplified in some diseases and would need to be factored into the biomathematical analysis of P-gp-sensitive PET radiotracers.
Successful drug inhibition of P-gp, resulting in the increased brain uptake of several radiotracers, has clearly been shown in rodents [18–23, 30] and non-human primates [19, 31]. Cyclosporin A has been the most widely used inhibitor, but is not regarded as safe for human use. Drugs that are safe and capable of completely inhibiting brain P-gp would now be useful for clarifying radiotracer susceptibility to efflux in human subjects, including patients with possibly compromised P-gp function.
Finally, with regard to brain entry, it may be noted that a PET scan aims to measure the net brain uptake and subsequently the net washout of radiotracer, with each depending on the interplay of several possible factors, including blood flow, passive blood-brain barrier permeability, efflux transporter action, target protein concentration, free fractions in plasma and brain [32, 33], and the concentration of radiotracer in plasma. Full kinetic compartmental analysis, using a measured arterial input function of unchanged radiotracer is required to establish the rate at which radiotracer traverses the blood-brain barrier in each direction, and to derive parameters related to measurement of target protein density [34]. In view of the multiple factors affecting peak radiotracer uptake in brain, the wide range of SUV values seen in Table 1 is unsurprising. The full influence of efflux transporters on the peak SUV data in Table 1 is unknown; a few of the radiotracers are known to be P-gp substrates in at least one species (e.g., [18F]MPPF), but as far as has been reported, not all the listed radiotracers have been tested for P-gp sensitivity.
Imaging of P-gp and other efflux transporter function
The efflux transporter behavior of some radiotracers raises the possibility to study efflux transporter function in vivo with PET. The study of P-gp function has immediate interest for understanding the suspected roles of P-gp in the unfolding of some neuropsychiatric disorders (e.g., Alzheimer’s disease and Parkinson’s disease) and in the efficacy of treatment of other conditions (e.g., epilepsy) [6]. P-gp also plays a major role in multi-drug resistance [6], the cause of a high proportion of failed cancer chemotherapy; here, molecular imaging in vivo would be valuable for deepening our understanding of the occurrence of drug resistance and for developing improved cancer treatment strategies.
Several radiotracers have been proposed for imaging P-gp function with PET [17]. These radiotracers are all transport substrates rather than compounds that bind reversibly and avidly to P-gp. The most studied radiotracer is [11C]verapamil either as racemate or single R-enantiomer [20, 35, 36] (Table 2). However, this radiotracer is extensively metabolized in human subjects [35]. Some metabolites of verapamil are known also to be P-gp substrates [37]. The major radiometabolite emerging in human plasma enters normal brain at a similar but non-identical rate to parent radiotracer [35]. Consequently, a two-input (radiotracer and metabolite) compartmental model best describes the brain time-activity curve. Such a model requires the measurement of two arterial inputs, one for radiotracer and the other for radiometabolite. This raises a question over the practical utility of [11C]verapamil for quantitative measurement of P-gp function. An especially promising radiotracer of P-gp function, which seems devoid of major problems from radiometabolites, is [11C]dLop [31] (Table 2), an N-methyl-11C-labeled metabolite of the over-the-counter anti-diarrheal agent, loperamide [38]. [11C]dLop (LogD7.4 = 3.49) mainly gives rise to radiometabolites that are relatively less lipophilic with low access to brain. Accumulation of [11C]dLop in brain increases several-fold in P-gp knockout mice, or in mice or monkeys in which P-gp has been blocked with an inhibitor, such as cyclosporin A, DCPQ or tariquidar [31, 39]. [11C]dLop shows promise for studying P-gp function in human subjects [40].
An interesting approach to measure the function of an efflux transporter with PET is to use a labeled compound that enters brain readily by passive diffusion to be converted into a radiometabolite that is then expelled by the efflux transporter of interest [41]. Thus, [11C]6-bromo-7-methylpurine readily enters mouse brain, becomes converted into its S-glutathione conjugate and is then expelled by the efflux transporter, MRP1 (multi-drug resistance-associated protein 1). Absence of MRP1 function in knockout mice resulted in 90% reduction in the rate of radioactivity efflux.
Radiotracers that bind avidly to an efflux transporter, as opposed to acting as transport substrates, would now be useful for measuring the regional brain concentration of transporter as a complement to measuring function.
The preceding discussion has considered how to ensure radiotracer entry into brain; the following discussion considers the impact of troublesome radiometabolites and how to ensure their adequate exclusion from brain.
Radiotracer metabolism
Radiotracers that resist significant metabolism over the time-span of a PET scanning session are rare. Moreover, the nature and degree of metabolism may vary appreciably across species with less extensive metabolism expected in higher species. Hence, potential problems from troublesome radiometabolites must be limited through effective radiotracer design. Ideally, for brain radiotracers, metabolism should not occur within brain but outside to produce less lipophilic radiometabolites with poor brain entry and little or no interaction with the target protein (Figure 1). Radiotracer metabolism that occurs solely outside brain to produce non brain-penetrant radiometabolites helps to eliminate parent radiotracer from plasma and thus from brain. This may actually be beneficial for observing brain radiotracer washout during a PET scanning session. Therefore, not all peripheral radiotracer metabolism is necessarily detrimental to accurate biomathematical analysis of radiotracer binding to target.
Some common patterns of PET radiotracer metabolism and their consequences
11C-Labeled PET radiotracers are often produced by relatively easy 11C-methylation at hetero atoms (e.g., O, N or S) [4]. Table 2 lists many examples. Often a fortuitous advantage of this type of radiolabel is that the radiotracer is metabolized by demethylation, leading to small polar radiometabolites (e.g., [11C]formaldehyde, 11CO2). These radiometabolites are well excluded by the blood-brain barrier and therefore have little interference with brain measurements. However, labeling of a radiotracer at an N,N-dimethylamino or N,N-dimethylamido group should usually be avoided since mono-demethylation may give rise to a radiometabolite with structure and properties similar to the parent radiotracer. Such a radiometabolite may confound quantitative measurements. Thus, the quantification of P-gp function with the tertiary amide, [11C]loperamide, is confounded by its major N-desmethyl radiometabolite, [11C]dLop [38] (Table 2). Use of the secondary amide [11C]dLop as the radiotracer avoids this problem [31].
Compounds labeled with an N-[18F]2-fluoroethyl group, instead of perhaps an N-[11C]methyl group, may be prone to dealkylation to give 18F-labeled two-carbon species capable of entering human brain. A prominent example is the DAT radiotracer, [18F]FECNT (Table 2), for which the brain ingress of radiometabolites mandates the use of a biomathematical model with a measured arterial input function [42].
Metabolic pathways that derivatize the radiotracer, rather than sequester the radiolabel into small, more polar fragments, may produce troublesome radiometabolites. For example, the DAT radiotracer, [11C]PE2I (Table 2), is metabolized by hydroxylation of its tolyl methyl group in rats [43]. The resultant radiometabolite is brain-penetrant and binds to DAT. This radiometabolite is also further metabolized to a radioactive carboxylic acid. Similar metabolism occurs in human subjects with an undetermined impact on the accuracy of DAT quantification [44]. Another example is provided by [11C](R)-RWAY (Table 2). The reversed direction of the amide bond in this radiotracer resists the amide hydrolysis seen in related radiotracers, such as [11C]WAY-100635. Nevertheless, [11C](R)-RWAY is primarily metabolized by derivatization in monkey [45] and fails as a quantifiable radiotracer in human, most likely because of the significant formation and brain ingress of such radiometabolites [28].
Defluorination is a major issue for some 18F-labeled radiotracers. Defluorination produces [18F]fluoride ion which binds avidly to bone, including skull. The accumulation of radioactivity in skull is problematic. Since the spatial resolution of PET is limited to a millimeter or so, there will be a ‘spill-over’ of radioactivity from the skull to nearby brain tissue (and vice versa) through a partial volume effect, rendering quantitation of radiotracer binding to be inaccurate. In general, defluorination can be avoided by 18F-labeling at an aromatic carbon atom within a phenyl or pyridinyl group. However, 18F bound to an aliphatic carbon atom is often prone to defluorination. Exceptions have been noted. For example, the translocator protein (18 kDa) (TSPO) radiotracer, [18F]PBR06 [46] (Table 2), in which the 18F is contained in an N-fluoroacetyl group, is not noticeably defluorinated in human subjects [47]. Species differences in susceptibility to radiodefluorination exist for many radiotracers. Thus, the mGluR5 receptor radiotracer [18F]SP203 [48] (Table 2), which has the radiolabel in its 2-fluoromethyl group is defluorinated in rat brain and blood, and also in monkey blood, but fortuitously is not at all defluorinated in human subjects [49]. Unexpectedly, defluorination of [18F]SP203 in rats was found to be due to glutathione S-transferase in brain and periphery [50].
Countering troublesome radiometabolites
The metabolic fate of PET radiotracers in lower species, such as rodents, can be determined by ex vivo analyses (e.g., [50]). In higher species, including human, labeling of a molecule at different sites and comparison of the brain uptake of the resultant radiotracers can provide a check for the absence of troublesome brain radiometabolites. If the site of radiolabel has minimal effect on brain radioactivity kinetics, then the presence of troublesome radiometabolites in brain is deemed unlikely. In this way, [O-methyl-11C]cocaine and [N-methyl-11C]cocaine (Table 2), as radiotracers for the dopamine transporter (DAT), were deduced to be free of troublesome radiometabolites in baboon [51]. Likewise, a recent comparison of [18F]flumazenil and [11C]flumazenil (Table 2) in human subjects showed virtually identical brain kinetics, so providing convincing evidence of the absence of troublesome brain radiometabolites for these radiotracers [52].
Different brain kinetics resulting from a change in position of the label in a radiotracer reveals the presence of troublesome brain radiometabolites for at least one version of the radiotracer. This is the case for two versions of [11C]WAY-100635 [53] (Figure 2). The major metabolic pathway for WAY-100635 is not the expected demethylation, but amide hydrolysis outside brain [54]. In primates, including humans, [O-methyl-11C]WAY-100635 consequently gives rise to a brain-penetrant and pharmacologically active amine as a troublesome radiometabolite. The entry of this radiometabolite into brain attenuates specific signal through non-specific binding and thwarts attempts at quantitative analysis. This problem is circumvented by labeling WAY-100635 in its amido carbonyl group [27]. [11C]Cyclohexanecarboxylic acid is then formed as the major radiometabolite, which has low brain entry because of its almost complete ionization at physiological pH [55]. Consequently, the non-specific binding of radiometabolites is reduced, and a several-fold greater proportion of radioactivity in brain represents the specific binding of radiotracer to 5-HT1A receptors [27]. In general, judicious choice of the site of radiolabel in a candidate radiotracer molecule is the most effective strategy for countering troublesome radiometabolites, although some other strategies are available, as follows.
Figure 2.

[11C]WAY-110635 for imaging brain 5-HT1A receptors: avoiding troublesome radiometabolites by judicious choice of position of radiolabel. WAY-100635 is metabolized in the periphery by hydrolysis of its amide bond. When WAY-100635 is labeled with carbon-11 in its O-methyl group, metabolism produces [11C]A as a major radiometabolite in plasma. [11C]A is able to enter brain to bind non-specifically, and also potentially specifically to some proteins for which it has moderately high affinity (5-HT1A receptors and α1 adrenoceptors) [54]. This radiometabolite therefore attenuates the signal from the unchanged radiotracer, [O-methyl-11C]WAY-100635, and exacerbates biomathematical analysis. By placing the radiolabel in the carbonyl position, metabolism of the radiotracer, [carbonyl-11C]WAY-100635, to [11C]A is avoided. The major radiometabolite becomes [11C]cyclohexanecarboxylate anion, which has only low transient entry into brain [55]. Consequently, with this radiotracer a much greater proportion of the radioactivity in brain is specifically bound unchanged radioligand [27], and biomathematical modeling of the acquired PET data is possible to deliver measures of receptor density [79, 80].
Radiotracers have been deuterated with the aim of exploiting a primary isotope effect to retard metabolism through specific pathways [56, 57]. A recent example is di-deuteration of the O-[18F]fluoromethyl group in the noradrenalin transporter (NET) radiotracer, [18F]FMeNER-d2, (Table 2), which retards undesirable defluorination [58]. A deuteration strategy can suppress but not abolish unwanted metabolism. Of course, introduction of deuterium remote from the site of primary metabolism will be ineffective. Thus, trideuteration of the O-methyl-11C group in the TPSO radiotracer, [11C]PBR28 (Table 2), had no effect [59] because this radiotracer is not primarily metabolized, as expected by demethylation, but by debenzylation [60].
Where the enzyme on the major pathway of radiotracer metabolism is known or can be predicted with reasonable confidence, it may be possible to block metabolism by specifically inhibiting the responsible enzyme. Thus, the 5-HT1A receptor radioligand [18F]FCWAY (Table 2) is extensively defluorinated in rodents [61] and in human subjects [62]. The enzyme CYP2EI was considered likely to perform the defluorination. Defluorination of [18F]FCWAY in rats was virtually abolished by pre-administration of the CYP2EI inhibitor, miconazole [61]. Similarly, a different inhibitor, disulfiram, was fully effective in blocking [18F]FCWAY defluorination in human subjects. As a result brain 5-HT1A receptor imaging was greatly improved [62]. A wider successful application of this type of strategy will depend on correct identification of the enzyme(s) involved in the major step of radiotracer metabolism and availability of safe and effective inhibitors of the identified enzymes. Recent studies of the adenosine A1 receptor radiotracer, [18F]CPFPX [63] (Table 2) demonstrate the feasibility of this approach. Oxidation of the cyclopentyl ring was established as the route of metabolism [63], and CYP1A2 was identified as the metabolizing enzyme. Inhibition of this enzyme with fluovoxamine greatly reduced radiotracer metabolism in human subjects [64].
In the absence of a wide array of safe inhibitors for the main pathways of radiotracer metabolism, the molecular design of the radiotracer, including its position of label, remains the main measure for avoiding troublesome radiometabolites.
Conclusions
The design of PET radiotracers for imaging protein targets in animal or human brain is challenging with respect to ensuring adequate brain entry in the absence of troublesome radiometabolites. Steadily accumulating knowledge on the behavior of existing and candidate radiotracers in animal and human subjects, and on the influence and possible modulation of efflux transporters and metabolizing enzymes, will be invaluable for guiding improved radiotracer design. Future extensive studies are required to build sufficient information that might eventually allow a priori prediction of radiotracer brain penetration, efflux transporter action and metabolism. The extent to which efflux transporter action affects the regional brain kinetics of established and candidate radiotracers is at present mostly unknown, and needs to be addressed to ensure that appropriate biomathematical analyses are applied.
Acknowledgments
This work was supported by the Intramural Research Program of NIH (NIMH), project number Z01 MH 002793. The author thanks members of the Molecular Imaging Branch (NIMH) for their collaboration on topics discussed in this review.
Glossary
- BP
the binding potential (BP) of a radiotracer at a target binding site may be defined as the ratio of the target concentration (Bmax) to the radiotracer equilibrium dissociation constant (KD) [8]. When Bmax and KD are expressed in the same units (e.g., nM), BP is unitless. Since KD is the inverse of ligand affinity, BP may also be considered to be the product of target density and radiotracer affinity. KD is normally considered to have a single, usually unknown, value in vivo. Hence, BP measures obtained with PET and a specific radiotracer are considered to be proportional to receptor density in vivo
- cLogD7.4
computed LogD7.4 value. See below for a definition of LogD7.4. cLogD7.4 is the LogD7.4 computed theoretically from molecular structure. A number of software programs are available for this purpose, which do not necessarily give the same answers, or answers agreeing with experiment
- cLogP
computed LogP value. See below for a definition of LogP. cLogP is the LogP computed theoretically from molecular structure. A number of software programs are available for this purpose, which do not necessarily give the same answers, or answers agreeing with experiment
- LogD7.4
Log of the distribution coefficient (D) of a compound, including all protonated and deprotonated species, between n-octanol and a buffer (usually 0.1 M sodium phosphate buffer) at pH 7.4 (to represent physiological pH), normally measured at ambient temperature
- LogP
Log of the partition coefficient (P) of a compound (as a single microspecies) between n-octanol and water, normally measured at ambient temperature. LogP is the most common index of compound lipophilicity
- fp
this is the fraction of radiotracer in plasma that is bound to plasma proteins under equilibrium conditions [8]
- Radiotracer
For the purpose of this review, the term radiotracer is restricted to radioactive probes which are designed to bind structurally-unchanged and reversibly with a target protein through non-covalent interactions e.g. a ligand-receptor interaction. In the literature, this type of radiotracer is also often called a radioligand, and the term radiotracer is also applied to radiopharmaceuticals acting by other mechanisms
- SUV
Standardized uptake value, which may be defined as, [Tissue radioactivity concentration (Bq/mL)/Administered dose (Bq)]/Body weight (g). SUV values normalize radioactivity concentration for administered dose and subject body weight and therefore allow comparisons of radioactivity concentrations between tissues, subjects and species
- VT
the ratio of the concentration of radiotracer in a region of tissue to that in plasma at equilibrium [8]
References
- 1.Jacobs AH, et al. PET-based molecular imaging in neuroscience. Eur J Nucl Med & Mol Imaging. 2003;30:1051–1065. doi: 10.1007/s00259-003-1202-5. [DOI] [PubMed] [Google Scholar]
- 2.Lee CM, Farde L. Using positron emission tomography to facilitate drug development. TiPs. 2006;27:310–316. doi: 10.1016/j.tips.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Fowler JS, Ido T. In: Handbook of Radiopharmaceuticals, Radiochemistry and Applications. Welch MJ, Redvanly CS, editors. Chapter 9. Wiley; Chichester: 2003. pp. 307–321. [Google Scholar]
- 4.Pike VW. Positron-emitting radioligands for studies in vivo Probes for human psychopharmacology. J Psychopharmacology. 1993;7:139–158. doi: 10.1177/026988119300700202. [DOI] [PubMed] [Google Scholar]
- 5.Laruelle M, et al. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol Imaging Biol. 2003;5:363–375. doi: 10.1016/j.mibio.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Lee G, Bendayan R. Functional expression and localization of P-glycoprotein in the central nervous system: relevance to the pathogenesis and treatment of neurological disorders. Pharm Res. 2004;21:1313–1330. doi: 10.1023/b:pham.0000036905.82914.8e. [DOI] [PubMed] [Google Scholar]
- 7.Dallas S, et al. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58:140–161. doi: 10.1124/pr.58.2.3. [DOI] [PubMed] [Google Scholar]
- 8.Innis RB, et al. Concensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cerebr Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 9.Raub TJ. P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol Pharmaceutics. 2006;3:3–25. doi: 10.1021/mp0500871. [DOI] [PubMed] [Google Scholar]
- 10.Fischer H, et al. Blood-brain barrier permeation: molecular parameters governing passive diffusion. J Membrane Biol. 1998;165:201–211. doi: 10.1007/s002329900434. [DOI] [PubMed] [Google Scholar]
- 11.Gerebtzoff G, Seelig A. In silico prediction of blood-brain barrier permeation using the calculated molecular cross-sectional area as main parameter. J Chem Inf Model. 2006;46:2638–2650. doi: 10.1021/ci0600814. [DOI] [PubMed] [Google Scholar]
- 12.Seelig A. The role of size and charge for blood-brain barrier permeation of drugs and fatty acids. J Mol Neurosci. 2007;33:323–341. doi: 10.1007/s12031-007-0055-y. [DOI] [PubMed] [Google Scholar]
- 13.Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 14.Donohue SR, et al. Synthesis, ex vivo evaluation and radiolabeling of potent 1,5-diphenyl-pyrrolidin-2-one cannabinoid subtype-1 (CB1) receptor ligands as candidates for in vivo imaging. J Med Chem. 2008;51:5833–5842. doi: 10.1021/jm800416m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yasuno F, et al. The PET radioligand [11C]MePPEP binds reversibly and with high specific signal to cannabinoid CB1 receptors in nonhuman primate brain. Neuropsychopharmacology. 2008;33:259–269. doi: 10.1038/sj.npp.1301402. [DOI] [PubMed] [Google Scholar]
- 16.Terry GE, et al. Evaluation of an inverse agonist with high affinity for the CB1 receptor, [11C]MePPEP, in rodents and humans. NeuroImage. 2008;41 (Suppl 2):T197. [Google Scholar]
- 17.Elsinga PH, et al. PET studies on P-glycoprotein function in the blood-brain barrier: how it affects uptake and binding of drugs within the CNS. Curr Pharm Design. 2004;10:1493–1503. doi: 10.2174/1381612043384736. [DOI] [PubMed] [Google Scholar]
- 18.Ishiwata K, et al. In vivo evaluation of P-glycoprotein modulation of 8 PET radioligands used clinically. J Nucl Med. 2007;48:81–87. [PubMed] [Google Scholar]
- 19.Syvänen S, et al. Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab Disposition. 2009;37:635–643. doi: 10.1124/dmd.108.024745. [DOI] [PubMed] [Google Scholar]
- 20.Elsinga PH, et al. Positron emission tomography studies on binding of central nervous system drugs and P-glycoprotein function in the rodent brain. Mol Imaging Biol. 2005;7:37–44. doi: 10.1007/s11307-005-0951-x. [DOI] [PubMed] [Google Scholar]
- 21.Liow JS, et al. Effect of a P-glycoprotein inhibitor, cyclosporin A, on the disposition in rodent brain and blood of the 5-HT1A receptor radioligand, [11C](R)-(−)-RWAY. Synapse. 2007;61:96–105. doi: 10.1002/syn.20348. [DOI] [PubMed] [Google Scholar]
- 22.Passchier J, et al. Influence of P-glycoprotein on brain uptake of [18F]MPPF in rats. Eur J Pharmacol. 2000;407:273–280. doi: 10.1016/s0014-2999(00)00752-4. [DOI] [PubMed] [Google Scholar]
- 23.Lacan G, et al. Cyclosporine, a P-glycoprotein modulator, increases [18F]MPPF uptake in rat brain and peripheral tissues: microPET and ex vivo studies. Eur J Nucl Med & Mol Imaging. 2008;35:2256–2266. doi: 10.1007/s00259-008-0832-z. [DOI] [PubMed] [Google Scholar]
- 24.Farde L, et al. PET characterisation of [carbonyl-11C]WAY-100635 binding to 5-HT1A receptors in the primate brain. Psychopharmacology. 1997;133:196–202. doi: 10.1007/s002130050391. [DOI] [PubMed] [Google Scholar]
- 25.Yasuno F, et al. Quantification of serotonin 5-HT1A receptors in monkey brain with [11C](−)-RWAY. Synapse. 2006;60:510–520. doi: 10.1002/syn.20327. [DOI] [PubMed] [Google Scholar]
- 26.Shiue CY, et al. p-[18F]-MPPF: a potential radioligand for PET studies of 5-HT1A receptors in humans. Synapse. 1997;25:147–154. doi: 10.1002/(SICI)1098-2396(199702)25:2<147::AID-SYN5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 27.Pike VW, et al. Exquisite delineation of 5-HT1A receptors in human brain with PET and [carbonyl-11C]WAY-100635. Eur J Pharmacol. 1996;301:R5–R7. doi: 10.1016/0014-2999(96)00079-9. [DOI] [PubMed] [Google Scholar]
- 28.Zhang XY, et al. Quantification of serotonin 5-HT1A receptors in humans with [11C](R)-(−)-RWAY: radiometabolite(s) likely confound brain measurements. Synapse. 2007;61:469–477. doi: 10.1002/syn.20392. [DOI] [PubMed] [Google Scholar]
- 29.Passchier J, et al. In vivo delineation of 5-HT1A receptors in human brain with p-[18F]MPPF. J Nucl Med. 2000;41:1830–1835. [PubMed] [Google Scholar]
- 30.Doze P, et al. Enhanced cerebral uptake of receptor ligands by modulation of P-glycoprotein function in the blood brain barrier. Synapse. 2000;36:66–74. doi: 10.1002/(SICI)1098-2396(200004)36:1<66::AID-SYN7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 31.Lazarova N, et al. Synthesis and evaluation of [N-methyl-11C]N-desmethyl-loperamide as a new and improved PET radiotracer for imaging P-gp function. J Med Chem. 2008;51:6034–6043. doi: 10.1021/jm800510m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Summerfield SG, et al. Improving the in vitro prediction of in vivo central nervous system penetration: integrating permeability, P-glycoprotein efflux, and free fractions in blood and brain. J Pharmacol Exp Ther. 2006;316:1282–1290. doi: 10.1124/jpet.105.092916. [DOI] [PubMed] [Google Scholar]
- 33.Summerfield SG, et al. Toward an improved prediction of human in vivo brain penetration. Xenobiotica. 2008;38:1518–1535. doi: 10.1080/00498250802499459. [DOI] [PubMed] [Google Scholar]
- 34.Willemsen ATM, van den Hoff J. Fundamentals of quantitative PET data analysis. Curr Pharm Design. 2002;8:1513–1526. doi: 10.2174/1381612023394359. [DOI] [PubMed] [Google Scholar]
- 35.Ikoma Y, et al. Quantitative analysis of [11C]verapamil transfer at the human blood-brain barrier for evaluation of P-glycoprotein function. J Nucl Med. 2006;47:1531–1537. [PubMed] [Google Scholar]
- 36.Luurtsma G, et al. Evaluation of (R)-[11C]verapamil as PET tracer of P-glycoprotein function at the blood-brain barrier: kinetics and metabolism in the rat. Nucl Med Biol. 2005;32:87–93. doi: 10.1016/j.nucmedbio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Pauli-Magnus C, et al. Characterization of the major metabolites of verapamil as substrates and inhibitors of P-glycoprotein. J Pharmacol Exp Ther. 2000;293:376–382. [PubMed] [Google Scholar]
- 38.Zoghbi SS, et al. 11C-Loperamide and its N-desmethyl radiometabolite are avid substrates for brain P-glycoprotein efflux. J Nucl Med. 2008;49:649–656. doi: 10.2967/jnumed.107.047308. [DOI] [PubMed] [Google Scholar]
- 39.Liow JS, et al. P-glycoprotein function at the blood-brain barrier imaged in monkey with 11C-N-desmethyl-loperamide. J Nucl Med. 2009;50:108–115. doi: 10.2967/jnumed.108.056226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seneca N, et al. Human brain imaging and radiation dosimetry of 11C-N-desmethyl-loperamide, a positron emission tomography radioligand to measure the function of P-glycoprotein. J Nucl Med. 2009;50:807–813. doi: 10.2967/jnumed.108.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okamura T, et al. Noninvasive and quantitative assessment of the function of multidrug resistance-associated protein 1 in the living brain. J Cerebr Blood Flow Metab. 2009;29:504–511. doi: 10.1038/jcbfm.2008.135. [DOI] [PubMed] [Google Scholar]
- 42.Zoghbi SS, et al. PET imaging of the dopamine transporter with [18F]FECNT: a polar radioactive metabolite confounds brain activity measurements. J Nucl Med. 2006;47:520–527. [PubMed] [Google Scholar]
- 43.Shetty HU, et al. Identification and regional distribution in rat brain of radiometabolites of the dopamine transporter PET radioligand, [11C]PE2I. Eur J Nucl Med & Mol Imaging. 2007;34:667–678. doi: 10.1007/s00259-006-0277-1. [DOI] [PubMed] [Google Scholar]
- 44.Hirvonen J, et al. Measurement of striatal and extrastriatal dopamine transporter binding with high-resolution PET and [11C]PE2I: quantitative modeling and test-retest reproducibility. J Cerebr Blood Flow Metab. 2008;28:1059–1069. doi: 10.1038/sj.jcbfm.9600607. [DOI] [PubMed] [Google Scholar]
- 45.McCarron JA, et al. Synthesis and preliminary evaluation of [11C](R)-RWAY in monkey a new simply labeled PET radioligand for imaging brain 5-HT1A receptors. Eur J Nucl Med & Mol Imaging. 2007;34:1670–1682. doi: 10.1007/s00259-007-0460-z. [DOI] [PubMed] [Google Scholar]
- 46.Briard E, et al. Single-step high-yield radiosynthesis and evaluation of a sensitive 18F-labeled ligand for imaging brain peripheral benzodiazepine receptors with PET. J Med Chem. 2009;52:688–699. doi: 10.1021/jm8011855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujimura Y, et al. Quantification of peripheral benzodiazepine receptors in human brain with positron emission tomography and 18F-PBR06. J Nucl Med (In press) [Google Scholar]
- 48.Siméon F, et al. Synthesis and simple 18F-labeling of a high affinity 2-(fluoromethyl)thiazole derivative ([18F]SP203) as a radioligand for imaging brain metabotropic glutamate subtype-5 receptors with PET. J Med Chem. 2007;50:3256–3266. doi: 10.1021/jm0701268. [DOI] [PubMed] [Google Scholar]
- 49.Brown AK, et al. Metabotropic glutamate subtype-5 (mGluR5) receptors quantified in human brain with a novel radioligand for positron emission tomography. J Nucl Med. 2008;49:2042–2048. doi: 10.2967/jnumed.108.056291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shetty HU, et al. 3-Fluoro-5-(2-(2-([18F]fluoromethyl)thiazol-4-yl)ethynyl)benzonitrile ([18F]SP203), a radioligand for imaging brain mGluR5 in vivo, is defluorinated by glutathionylation in rat brain. J Pharmacol Exp Therapeutics. 2008;327:727–735. doi: 10.1124/jpet.108.143347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gatley SJ, et al. Studies with differentially labeled [11C]cocaine, [11C]norcocaine, [11C]benzoylecgonine, and [11C] and 4′-[18F]fluorococaine to probe the extent to which [11C]cocaine metabolites contribute to PET images of the baboon brain. J Neurochem. 1994;62:1154–1162. doi: 10.1046/j.1471-4159.1994.62031154.x. [DOI] [PubMed] [Google Scholar]
- 52.Odano I, et al. [18F]Flumazenil binding to central benzodiazepine receptor studies by PET Quantitative analysis and comparisons with [11C]flumazenil. NeuroImage. 2009;45:891–902. doi: 10.1016/j.neuroimage.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 53.Pike VW, et al. Pre-clinical development of a radioligand for studies of central 5-HT1A receptors in vivo [11C]WAY-100635. Med Chem Res. 1995;5:208–227. [Google Scholar]
- 54.Osman S, et al. Characterization of the radioactive metabolites of the 5-HT1A receptor radioligand, [O-methyl-11C]WAY-100635, in monkey and human plasma by HPLC Comparison of the behaviour of an identified radioactive metabolite with parent radioligand in monkey using PET. Nucl Med Biol. 1996;23:627–634. doi: 10.1016/0969-8051(96)00061-3. [DOI] [PubMed] [Google Scholar]
- 55.Osman S, et al. Characterisation of the appearance of radioactive metabolites in monkey and human plasma from the 5-HT1A receptor radioligand, [carbonyl-11C]WAY-100635 Explanation of high signal in PET and an aid to biomathematical modelling. Nucl Med Biol. 1998;25:215–223. doi: 10.1016/s0969-8051(97)00206-0. [DOI] [PubMed] [Google Scholar]
- 56.Hashimoto K, et al. Deuterium-isotope effect of [11C]N,N-dimethylphenethylamine-α,α-d2 reduction in metabolic trapping rate in brain. Nucl Med Biol. 1986;13:79–80. doi: 10.1016/0883-2897(86)90256-4. [DOI] [PubMed] [Google Scholar]
- 57.Fowler JS, et al. Mechanistic positron emission tomography studies: demonstration of a deuterium isotope effect in the monoamine oxidase-catalyzed binding of [11C]L-deprenyl in living baboon brain. J Neurochem. 1988;51:1524–1534. doi: 10.1111/j.1471-4159.1988.tb01121.x. [DOI] [PubMed] [Google Scholar]
- 58.Schou M, et al. PET evaluation of novel radiofluorinated reboxetine analogs as norepeienphrine transporter probes in the monkey brain. Synapse. 2004;53:57–67. doi: 10.1002/syn.20031. [DOI] [PubMed] [Google Scholar]
- 59.Wilson AA, et al. Radiosynthesis and initial evaluation of [18F]FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl Med Biol. 2008;35:305–314. doi: 10.1016/j.nucmedbio.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 60.Briard E, et al. Synthesis and evaluation in monkey of two sensitive 11C-labeled aryloxyanilide ligands for imaging brain peripheral benzodiazepine receptors in vivo. J Med Chem. 2008;51:17–30. doi: 10.1021/jm0707370. [DOI] [PubMed] [Google Scholar]
- 61.Tipre DN, et al. PET imaging of brain 5-HT1A receptors in rat in vivo with [18F]FCWAY and improvement by successful inhibition of defluorination with miconazole. J Nucl Med. 2006;47:1–9. [PubMed] [Google Scholar]
- 62.Ryu YH, et al. Disulfiram inhibits defluorination of [18F]FCWAY, reduces bone radioactivity, and enhances visualization of radioligand binding to serotonin 5-HT1A receptors in human brain. J Nucl Med. 2007;48:1154–1161. doi: 10.2967/jnumed.107.039933. [DOI] [PubMed] [Google Scholar]
- 63.Matusch A, et al. Metabolism of the A1 adenosine receptor PET ligand [18F]CPFPX by CYP1A2: implications of bolus/infusion studies. Nucl Med Biol. 2006;33:891–898. doi: 10.1016/j.nucmedbio.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 64.Bier D, et al. Metabolism of the A1 adenosine receptor positron emission tomography ligand [18F]8-cyclopentyl-3(-3-fluoropropyl)-1-propylxanthine ([18F]CPFPX) in rodents and humans. Drug Metab Disposition. 2006;34:570–576. doi: 10.1124/dmd.105.006411. [DOI] [PubMed] [Google Scholar]
- 65.Ding YS, et al. 6-[18F]Fluoro-A-85380, a new PET tracer for the nicotonic acetylcholine receptor: studies in the human brain and in vivo demonstration of specific binding in white matter. Synapse. 2004;53:184–189. doi: 10.1002/syn.20051. [DOI] [PubMed] [Google Scholar]
- 66.Rinne JO, et al. PET examination of the monoamine transporter with [11C]β-CIT and [11C]β-CFT in early Parkinson’s disease. Synapse. 1995;21:97–103. doi: 10.1002/syn.890210202. [DOI] [PubMed] [Google Scholar]
- 67.Yasuno F, et al. PET Imaging of neurokinin-1 receptors with [18F]SPA-RQ in human subjects: assessment of reference tissue models and their test-test reproducibility. Synapse. 2007;61:242–251. doi: 10.1002/syn.20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mukherjee J, et al. Brain imaging of 18F-fallypride in normal volunteers: blood analysis, distribution, test-retest studies, and preliminary assessment of sensitivity to aging effects on dopamine D-2/D-3 receptors. Synapse. 2002;46:170–188. doi: 10.1002/syn.10128. [DOI] [PubMed] [Google Scholar]
- 69.Jones AKP, et al. Quantification of [11C]diprenorphine cerebral kinetics in man acquired by PET using presaturation, pulse-chase and tracer only protocols. J Neurosci Methods. 1994;51:123–134. doi: 10.1016/0165-0270(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 70.DaSilva JN, et al. Imaging cAMP-specific phosphodiesterases-4 in human brain with R-[11C]rolipram and positron emission tomography. Eur J Nucl Med. 2002;29:1680–1683. doi: 10.1007/s00259-002-0950-y. [DOI] [PubMed] [Google Scholar]
- 71.Farde L, et al. Subsituted benzamides as ligands for visualization of dopamine receptor binding in the human brain by positron emission tomography. Proc Natl Acad Sci USA. 1985;82:3863–3867. doi: 10.1073/pnas.82.11.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andrée B, et al. The PET radioligand [carbonyl-11C]desmethyl-WAY-100635 binds to 5-HT1A receptors and provides a higher radioactive signal than [carbonyl-11C]WAY-100635 in the human brain. J Nucl Med. 2002;43:292–303. [PubMed] [Google Scholar]
- 73.Hinz R, et al. Validation of a tracer kinetic model for the quantification of 5-HT2A receptors in human brain with [11C]MDL 100,907. J Cerebr Blood Flow Metab. 2007;27:161–172. doi: 10.1038/sj.jcbfm.9600323. [DOI] [PubMed] [Google Scholar]
- 74.Fujita M, et al. Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential marker for inflammation. NeuroImage. 2008;40:43–52. doi: 10.1016/j.neuroimage.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klunk WE. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 76.Ginovart N, et al. Positron emission tomography quantification of [11C]-DASB binding to the human serotonin transporter: modeling strategies. J Cerebr Blood Flow Metab. 2001;21:1342–1353. doi: 10.1097/00004647-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 77.Kropholler MA, et al. Development of a tracer kinetic plasma input model for (R)-[11C]PK11195 brain studies. J Cerebr Blood Flow Metab. 2005;25:842–851. doi: 10.1038/sj.jcbfm.9600092. [DOI] [PubMed] [Google Scholar]
- 78.Halldin C, et al. Carbon-11-NNC 112: a radioligand for PET examination of striatal and neocortical D1-dopamine receptors. J Nucl Med. 1998;39:2061–2068. [PubMed] [Google Scholar]
- 79.Gunn RN, et al. Tracer kinetic modeling of the 5-HT1A receptor ligand [carbonyl-11C]WAY-100635. NeuroImage. 1998;8:426–440. doi: 10.1006/nimg.1998.0379. [DOI] [PubMed] [Google Scholar]
- 80.Farde L, et al. Quantitative analyses of [carbonyl-11C]WAY-100635 binding to central 5-hydroxytryptamine 1A receptors in man. J Nucl Med. 1998;39:1965–1971. [PubMed] [Google Scholar]