

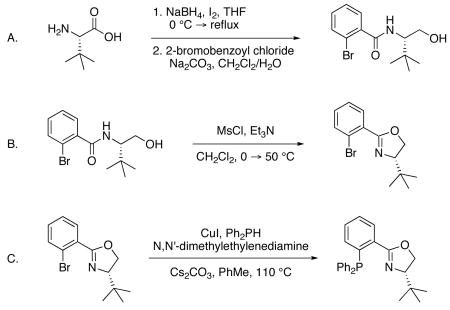
1. Procedure
Caution! This procedure should be carried out in an efficient fume hood due to the evolution of hydrogen gas during the reaction.
A. 2-Bromo-N-[(1S)-1-(hydroxymethyl)-2,2-dimethylpropyl]-benzamide
An oven-dried 1 L, 3-neck flask equipped with a magnetic stir bar, pressure-equalizing addition funnel, and reflux condenser were assembled hot and cooled under a stream of dry nitrogen. The flask was charged with 10.00 g of (L)-tert-leucine (76.24 mmol, 1.0 equiv, 99% ee) (Note 1) and 200 mL of tetrahydrofuran (0.38 M) (Note 2) under a positive pressure of nitrogen. The resulting suspension was cooled to ca. 4 °C in an ice-water bath and 6.922 g of sodium borohydride (183.0 mmol, 2.4 equiv) (Note 1) was added in one portion (Note 3). The addition funnel was charged with a solution of 19.35 g iodine (76.24 mmol, 1.0 equiv) (Note 1) in 50 mL of tetrahydrofuran (Note 2) via syringe and added dropwise to the suspension over 1 h. After complete addition, the bath was removed and the reaction was warmed to reflux in an oil bath at 80 °C. The reaction was allowed to cool to ambient temperature after 18 h and 100 mL of methanol (Note 2) was added slowly until the contents were mostly clear (Note 4). After stirring for 30 min the solution was quantitatively transferred to a 1 L, 1-neck flask with ca. 100–150 mL of methanol and concentrated under reduced pressure to a white semi-solid. The resulting material was dissolved in 150 mL of 20% aqueous potassium hydroxide and stirred for 5 h at ambient temperature. The aqueous phase was extracted with dichloromethane (6 × 125 mL) and the combined organics were dried over sodium sulfate, filtered, and concentrated under reduced pressure to yield 8.83 g (75.35 mmol, 98.8% yield) of crude (S)-tert-leucinol as a colorless semi-solid (Note 5). This material was used in the following step without purification.
A 1 L flask containing a magnetic stirbar and crude (S)-tert-leucinol was diluted with 250 mL of dichloromethane (0.30 M) (Note 2) and a solution of 23.96 g sodium carbonate (226.1 mmol, 3 equiv) (Note 1) in 190 mL of distilled water was added at ambient temperature. The biphasic mixture was stirred vigorously to emulsify and 11.33 mL of 2-bromobenzoyl chloride (86.65 mmol, 1.15 equiv) (Note 1) was added dropwise neat via syringe over ca. 10–15 minutes. The reaction flask was capped with a yellow plastic stopper and stirred for 10 h, after which time the layers were partitioned in a 1 L separatory funnel and the aqueous phase was extracted with dichloromethane (4 × 100 mL). The combined organics were stirred with 38 mL of 1 N potassium hydroxide solution in methanol in a 1 L Erlenmeyer flask with a magnetic stir bar for 30 min at ambient temperature and acidified to neutral pH with 1 N hydrochloric acid (ca. 35 mL). Water (50 mL) was added, the phases were partitioned in a 2 L separatory funnel, and the aqueous phase was extracted with dichloromethane (4 × 75 mL). The combined organics were washed with saturated brine (150 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure to an off-white solid. The crude white solid was dissolved in a minimal amount of hot acetone (ca. 25 mL) (Note 2) and hexanes (Note 2) was added until a cloudy solution was obtained (ca. 90 mL). The crystals formed upon cooling and aging for a few hours were collected, washed with hexanes, and dried under vacuum to afford 18.413 g of 2-bromo-N-[(1S)-1-(hydroxymethyl)-2,2-dimethylpropyl]-benzamide (61.34 mmol) as white blocks (Note 6). The filtrate was concentrated and recrystallized in a similar manner (with ca. 7 mL acetone and 20 mL hexanes) to provide an additional 2.251 g of product as white blocks (7.50 mmol) (Note 7), for a combined yield of 20.664 g (68.84 mmol, 90.3% yield over two steps).
B. 2-(2-Bromophenyl)-4-(1,1-dimethylethyl)-4,5-dihydro-(4S)-oxazole
An oven-dried 1 L, 3-neck flask equipped with a stir bar and reflux condenser were assembled hot and cooled under a stream of dry nitrogen. The flask was charged with 20.631 g of 2-bromo-N-[(1S)-1-(hydroxymethyl)-2,2-dimethylpropyl]-benzamide (68.73 mmol, 1.0 equiv), 345 mL of dichloromethane (0.2 M) (Note 2), and 23.0 mL triethylamine (164.9 mmol, 2.4 equiv) (Note 8) under a positive pressure of nitrogen. The resulting colorless solution was cooled to ca. 4 °C in an ice-water bath and 6.12 mL of methanesulfonyl chloride (79.04 mmol, 1.15 equiv) (Note 1) was added dropwise neat via syringe over 5 min, at which point the solution turns slightly yellow. Following consumption by TLC (Note 9), the reaction was warmed to 50 °C in a water bath. Upon completed cyclization by TLC (Note 9), the reaction was allowed to cool to ambient temperature and 120 mL of saturated sodium bicarbonate was added with vigorous stirring for 5 min. The layers were partitioned in a 2 L separatory funnel, the aqueous phase was extracted with dichloromethane (2 × 70 mL), the combined organics were washed with saturated brine (150 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure to afford a pale yellow semi-solid. The residue was dissolved in a minimal amount of dichloromethane (ca. 75 mL), dry-loaded onto 25 g of silica gel, and purified by silica gel chromatography (Note 10) to afford 18.642 g (66.06 mmol, 96.1% yield) of 2-(2-bromophenyl)-4-(1,1-dimethylethyl)-4,5-dihydro-(4S)-oxazole as a pale yellow oil. This material solidifies when placed in a −20 °C freezer and is preferred in this state for the subsequent reaction (Note 11).
Caution! This procedure should be carried out with the protection of a blast shield due to the heating of the reaction in a sealed flask at the boiling point of toluene.
C. 4-(1,1-dimethylethyl)-2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-(4S)-oxazole ((S)-tert-ButylPHOX)
A 250 mL Schlenk flask equipped with a Teflon valve, a 14/20 glass joint, and a large stir bar was flame-dried under vacuum and cooled under a dry nitrogen atmosphere. The Teflon valve was removed under a positive pressure of nitrogen and the flask was charged with 19.0 mg copper(I) iodide (0.10 mmol, 0.005 equiv) (Note 1), 4.35 mL diphenylphosphine (25.0 mmol, 1.25 equiv) (Note 12), 53.3 μL N,N′-dimethylethylenediamine (0.50 mmol, 0.025 equiv) (Note 1) and 20 mL toluene (Note 2). The valve was then replaced and the colorless contents were stirred at ambient temperature for 20 min. The valve was then removed under a positive pressure of nitrogen and the flask was charged with 5.642 g of 2-(2-bromophenyl)-4-(1,1-dimethylethyl)-4,5-dihydro-(4S)-oxazole (20.0 mmol, 1.0 equiv), 9.775 g of cesium carbonate (30.0 mmol, 1.5 equiv) (Note 1), and 20 mL toluene (0.50 M, total) to wash the neck and walls of the flask. The Teflon valve was sealed and the now yellow heterogeneous reaction was placed in a 110 °C oil bath and vigorously stirred (Note 13). Following consumption of starting material (Note 14), the reaction was allowed to cool to ambient temperature, filtered through a pad of celite, and the filter cake washed with dichloromethane (2 × 40 mL). The filtrate was concentrated under reduced pressure to a pale yellow semi-solid, dissolved in a minimal amount of dichloromethane (ca. 40 mL) and ethyl ether (ca. 50 mL), and dry-loaded onto 10 g of silica gel. This material was flushed through a silica gel plug eluting with 24:1 hexanes-ethyl ether until excess Ph2PH elutes, then with a 9:1 dichloromethane-ethyl ether mixture until the desire product elutes (Note 15). The combined fractions are concentrated to a viscous pale yellow oil and layered with ca. 5 mL acetonitrile to facilitate crystallization (Note 2)(Note 16). The flask was swirled while crystals form within seconds (Note 17). After ca. 15 minutes, the flask is placed under high vacuum to remove volatiles to afford 7.033 g (18.15 mmol, 90.8% yield) of (S)-tert-ButylPHOX as white blocks (Note 18).
2. Notes
(L)-tert-leucine (99%, 99% ee), sodium borohydride (98%), iodine (≥99%), sodium carbonate (99%), cesium carbonate (99%), and N,N′-dimethylethylenediamine (99%) were purchased from Aldrich and used as received. (2)-Bromobenzoyl chloride (98%) and methanesulfonyl chloride (99.5%) were purchased from Acros and used as received. Copper iodide (98%) was purchased from Strem and used as received.
Tetrahydrofuran was distilled from sodium 9-fluorenone ketyl2 prior to use. Methylene chloride, toluene, and acetonitrile were purified by passage through an activated alumina column under argon.3 Reagent grade acetone was purchased from EMD, and hexanes and methanol (both ACS grade) were purchased from Fisher and used as received.
The evolution of hydrogen gas during the addition of sodium borohydride is minor due to the adequate size of the reaction flask and the surface area of cooling. This is readily vented through the oil bubbler.
The initial reaction quench with methanol proceeds with vigorous gas evolution. Methanol should be added dropwise until the intensity of gas evolution abates.
The reduction product, (S)-tert-leucinol, can be purified by distillation,4 but was not necessary for this application. The material showed the following characterization data: 1H NMR (500 MHz, CDCl3) δ 3.70 (dd, J = 10.4, 4.0 Hz, 1H), 3.19 (app. t, J = 10.1 Hz, 1H), 2.49 (dd, J = 10.1, 3.7 Hz, 1H), 1.71 (br s, 3H), 0.89 (s, 9H). This material may also be purchased from commercial sources, but is less expensive in the amino acid form. Similar amino acid reductions have appeared in Organic Syntheses.5
2-bromo-N-[(1S)-1-(hydroxymethyl)-2,2-dimethylpropyl]-benzamide showed the following characterization data: mp 50–51 °C from acetone/hexanes; Rf = 0.28 (2:1 hexanes-acetone); 1H NMR (500 MHz, CDCl3) δ 7.60 (app. d, J = 8.1 Hz, 1H), 7.57 (dd, J = 7.6, 1.5 Hz, 1H), 7.36 (app. t, J = 7.3 Hz, 1H), 7.28 (app. dt, J = 8.1, 1.7 Hz, 1H), 6.17 (br d, J = 8.3 Hz, 1H), 4.07 (ddd, J = 11.0, 7.6, 3.7 Hz, 1H), 3.95 (dd, J = 11.2, 3.4 Hz, 1H), 3.69 (dd, J = 11.5, 7.6 Hz, 1H), 2.33 (br s, 1H), 1.04 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 168.8, 138.1, 133.5, 131.5, 129.9, 127.8, 119.2, 63.2, 60.5, 33.9, 27.2; IR (Neat Film NaCl) 3245, 3070, 2963, 1640, 1557 cm−1; HRMS (FAB+) m/z calc’d for C13H19NO2Br [M+H]+: 300.0599, found 300.0590; [α]D29 +20.2 ° (c 2.38, methanol, >99 % ee)); Anal. calc’d. for C13H19NO2Br: C, 52.01; H, 6.04; N, 4.67. Found: C, 51.81; H, 5.87; N, 4.56.
In some cases the resulting filtrate can be purified by silica gel flash chromatography, eluting with a 3:1 → 2:1 hexanes-acetone gradient to afford an additional 2–6 % of an off-white amorphous solid that is spectroscopically identical to the crystalline material.
Triethylamine (99.5%) was purchased from Aldrich and distilled from calcium hydride prior to use.
Reaction progress can be monitored by TLC analysis using 2:1 ethyl acetate-hexanes as the eluent with UV visualization (Rf amide = 0.39, Rf mesylate = 0.60, Rf bromooxazoline = 0.89). Mesylate formation is typically complete upon final addition of methanesulfonyl chloride, whereas cyclization to the bromooxazoline typically requires ca. 5 h at 50 °C to complete.
Flash chromatography column dimensions: 5 cm diameter × 20 cm height of silica gel, eluting with 400 mL of 9:1 hexanes-ethyl acetate, then 900 mL of 6:1 hexanes-ethyl acetate, collecting ca. 40–50 mL fractions. Fraction purity can be assayed by TLC analysis using 4:1 hexanes-ethyl acetate with UV visualization. This method of purification removes color from the crude material and a minor impurity at Rf = 0.46.
2-(2-bromophenyl)-4-(1,1-dimethylethyl)-4,5-dihydro-(4S)-oxazole showed the following characterization data: mp 45–48 °C; Rf = 0.38 (4:1 hexanes-ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 7.64 (app. dt, J = 8.0, 2.1 Hz, 2H), 7.34 (app. dt, J = 7.5, 1.3 Hz, 1H), 7.27 (app. dt, J = 7.4, 2.1 Hz, 1H), 4.39 (dd, J = 10.4, 8.8 Hz, 1H), 4.26 (dd, J = 8.0, 8.0 Hz, 1H), 4.11 (dd, J = 10.4, 8.0 Hz, 1H), 1.00 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 162.8, 133.6, 131.4, 131.2, 130.2, 127.0, 121.8, 76.6, 69.0, 34.0, 25.9; IR (Neat Film NaCl) 2956, 1661, 1478, 1354, 1099, 1022, 963 cm−1; HRMS (FAB+) m/z calc’d for C13H17NOBr [M+H]+: 282.0493, found 282.0488; [α]D29 −48.3 ° (c 3.77, hexane, >99 % ee)); Anal. calc’d. for C13H17NOBr: C, 55.33; H, 5.72; N, 4.96. Found: C, 55.38; H, 5.73; N, 4.87.
Diphenylphosphine (99%) was purchased from Strem and cannula transferred to a dry Schlenk tube under nitrogen to prolong reagent life.
Reactions performed with minimal stirring or that cease to stir result in incomplete conversion. The preferred stirring rate of the coupling reaction is ca. 700 setting (ca. 700 rpm) on an IKAmag RET basic stir/hot plate (a range between 500–800 rpm is sufficient). Additionally, the color of the reaction becomes an intense yellow within 5–10 minutes of heating. The color of the inorganic base then dominates as it turns to light gray, and finally to a dark maroon/purple color after several hours.
The reaction typically requires 21 h to reach complete conversion. Reaction progress can be monitored by TLC analysis using 4:1 hexanes-ethyl ether as the eluent (developed twice) with UV visualization (Rf bromooxazoline = 0.34, Rf reduced oxazoline = 0.51, Rf tert-butylPHOX = 0.64, Rf Ph2PH = 0.89).
Flash chromatography column dimensions: 5 cm diameter × 16 cm height of silica gel, collecting ca. 50 mL fractions. Fraction purity can be assayed by TLC analysis using 4:1 hexanes-ethyl ether. The mixture of products may contain reduced arene, starting bromooxazoline, and desired (S)-tert-ButylPHOX, depending on the extent of reaction.
As the percentage of desired (S)-tert-ButylPHOX in the crude mixture increases, the oil readily solidifies upon concentration under reduced pressure. To decrease the time required to induce crystallization, this oil can then be dissolved in diethyl ether and further concentrated. Additionally, acetonitrile efficiently promotes crystallization of (S)-tert-ButylPHOX in concentrated solutions.
If the reaction is pushed to completion, the material obtained from this simple purification is typically =99% pure (by 1H NMR analysis of crude oil). If the purity is unsatisfactory, this crystalline material can be recrystallized with hot acetonitrile. A typical recrystallization is performed as follows: 7.033 g of crude (S)-tert-ButylPHOX (i.e., the crude material after the silica gel plug) is dissolved in a minimal amount (ca. 8–10 mL) of boiling acetonitrile and allowed to cool slowly to ambient temperature. The crystals are then filtered and washed with ca. 15–25 mL hexanes, then dried under high vacuum to yield 6.613 g (17.07 mmol, 85.3% yield) of white blocks. This material is >99% pure by 1H NMR and all other spectroscopic data (see Note 18).
(S)-tert-ButylPHOX showed the following characterization data; mp 114–115 °C from acetonitrile; Rf = 0.64 (4:1 hexanes-ethyl ether, developed twice); 31P NMR (121 MHz, CDCl3) δ −5.33 (s); 1H NMR (300 MHz, CDCl3) δ 7.94 (ddd, J = 7.4, 3.5, 1.3 Hz, 1H), 7.36 (app. dt, J = 7.4, 1.3 Hz, 1H), 7.33-7.21 (comp. m, 11H), 6.86 (ddd, J = 7.4, 4.0, 1.3 Hz, 1H), 4.08 (dd, J = 10.1, 8.2 Hz, 1H), 4.01 (dd, J = 8.0, 8.0 Hz, 1H), 3.88 (dd, 10.1, 8.0 Hz, 1H), 0.73 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 162.8 (d, JCP = 2.8 Hz), 138.9 (d, JCP = 25.3 Hz), 138.7 (d, JCP = 12.4 Hz), 138.4 (d, JCP = 9.7 Hz), 134.5 (d, JCP = 21.2 Hz), 134.2, 133.7 (d, JCP = 20.3 Hz), 132.1 (d, JCP = 19.8 Hz), 130.5, 130.0 (d, JCP = 3.2 Hz), 128.6 (d, JCP = 20.2 Hz), 128.5, 128.4 (2 lines), 128.2, 76.8, 68.4, 33.7, 25.9; IR (Neat Film NaCl) 3053, 2954, 2902, 2867, 1652, 1477, 1434, 1353, 1336, 1091, 1025, 966, 743, 696, 503 cm−1; HRMS (FAB+) m/z calc’d for C25H27NOP [M+H]+: 388.1830, found 388.1831; [α]D23 −61.5 ° (c 0.925, CHCl3, >99 % ee); Anal. calc’d. for C25H27NOP: C, 77.50; H, 6.76; N, 3.62. Found: C, 77.10; H, 6.62; N, 3.71. This crystalline material is stable indefinitely at ambient temperatures in a closed container under an atmosphere of nitrogen or argon.
Waste Disposal Information
All toxic materials were disposed of in accordance with “Prudent Practice in the Laboratory”, National Academy Press; Washington, D. C., 1995.
3. Discussion
This synthesis of (S)-tert-ButylPHOX (4-(1,1-dimethylethyl)-2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-(4S)-oxazole) is a modification of our previously reported procedure.6 Several improvements have been implemented that facilitate large-scale preparation of this ligand. Recrystallization of 2-bromo-N-[(1S)-1-(hydroxymethyl)-2,2-dimethylpropyl]-benzamide obviates the previous need for flash column chromatography. Subsequent oxazoline formation is now accomplished via mesylate displacement with improved efficiency and yield. The use of methanesulfonyl chloride enables rapid mesylate formation at milder temperatures, and aqueous reaction workup is favored over the previous method, where incomplete hydrolysis of p-toluenesulfonyl chloride complicated purification. The copper(I) iodide-catalyzed phosphine coupling7 has been optimized to maximize the efficiency of the reaction by minimizing the use catalyst and diamine ligand, as well as reducing the quantities of phosphine, cesium carbonate, and solvent. Finally, a procedure to purify (S)-tert-ButylPHOX is described, using a simple silica gel plug, followed by crystallization with acetonitrile (or recrystallization when necessary), to afford the ligand as a white crystalline solid in four steps from (L)-tert-leucine in excellent overall yield (78.8% over four steps).
The phosphinooxazoline8 (S)-tert-ButylPHOX is a chiral N,P-ligand useful for an array of organometallic transformations, including alkylations,9 desymmetrizations of meso-anhydrides,10 Heck reactions,11 hetero-Diels–Alder,12 and hydrogenations.13 Our laboratory has recently described its use as a uniquely effective ligand for the palladium-catalyzed asymmetric decarboxylative allylation6,14 and protonation15 of prochiral ketone enolates. This synthesis of (S)-tert-ButylPHOX highlights improvements of a general and efficient strategy to access PHOX ligands of varied structure and electronics in substantial quantities.6b
Appendix Chemical Abstracts Nomenclature; (Registry Number)
(L)-tert-Leucine: L-Valine, 3-methyl-; (20859-02-3)
(S)-tert-Leucinol: 1-Butanol, 2-amino-3,3-dimethyl-, (2S)-; (112245-13-3)
Sodium borohydride: Borate(1-), tetrahydro-, sodium (1:1); (16940-66-2)
Iodine; (7553-56-2)
2-Bromobenzoyl chloride: Benzoyl chloride, 2-bromo-; (7154-66-7)
Sodium carbonate: Carbonic acid sodium salt (1:2); (497-19-8)
Triethylamine: Ethanamine, N,N-diethyl-; (121-44-8)
Methansulfonyl chloride; (124-63-0)
Copper iodide; (1335-23-5)
Diphenylphosphine: Phosphine, diphenyl-; (829-85-6)
N,N′-dimethylethylenediamine: 1,2-Ethanediamine, N1,N2-dimethyl-; (110-70-3)
Cesium carbonate: Carbonic acid, cesium salt (1:2); (534-17-8)
(S)-tert-ButylPHOX: Oxazole, 4-(1,1-dimethylethyl)-2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-, (4S)-; (148461-16-9)
References
- 1.Department of Chemistry and Chemical Engineering, M/C 164-30, California Institute of Technology, Pasadena, California, 91125; E-mail: stoltz@caltech.edu.
- 2.Kamaura M, Inanaga J. Tetrahedron Lett. 1999;40:7347–7350. [Google Scholar]
- 3.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 4.McKennon MJ, Meyers AI. J Org Chem. 1993;58:3568–3571. [Google Scholar]
- 5.(a) Dickman DA, Meyers AI, Smith GA, Gawley RE. Org Synth Coll. 1990;VII:530–532. [Google Scholar]; (b) Gage JR, Evans DA. Org Synth Coll. 1993;VIII:528–531. [Google Scholar]
- 6.(a) Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; (b) Tani K, Behenna DC, McFadden RM, Stoltz BM. Org Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]
- 7.Gelman D, Jiang L, Buchwald SL. Org Lett. 2003;5:2315–2318. doi: 10.1021/ol0346640. [DOI] [PubMed] [Google Scholar]
- 8.(a) von Matt P, Pfaltz A. Angew Chem, Int Ed Engl. 1993;32:566–568. [Google Scholar]; (b) Sprinz J, Helmchen G. Tetrahedron Lett. 1993;34:1769–1772. [Google Scholar]; (c) Dawson GJ, Frost CG, Coote SJ, Williams JMJ. Tetrahedron Lett. 1993;34:3149–3150. [Google Scholar]
- 9.(a) Dawson GJ, Frost CG, Williams JMJ. Tetrahedron Lett. 1993;34:3149–3150. [Google Scholar]; (b) Helmchen G, Pfaltz A. Acc Chem Res. 2000;33:336–345. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]; (c) Weiβ TD, Helmchen G, Kazmaier U. Chem Commun. 2002;12:1270–1271. doi: 10.1039/b203791m. [DOI] [PubMed] [Google Scholar]
- 10.Cook MJ, Rovis T. J Am Chem Soc. 2007;129:9302–9303. doi: 10.1021/ja073269s. [DOI] [PubMed] [Google Scholar]
- 11.Loiseleur O, Hayashi M, Schmees N, Pfaltz A. Synthesis. 1997;11:1338–1345. [Google Scholar]
- 12.Yao S, Saaby S, Hazell RG, Jørgensen KA. Chem–Eur J. 2000;6:2435–2448. doi: 10.1002/1521-3765(20000703)6:13<2435::aid-chem2435>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 13.Legault CY, Charette AB. J Am Chem Soc. 2005;127:8966–8967. doi: 10.1021/ja0525298. [DOI] [PubMed] [Google Scholar]
- 14.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem, Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 15.(a) Mohr JT, Nishimata T, Behenna DC, Stoltz BM. J Am Chem Soc. 2006;128:11348–11349. doi: 10.1021/ja063335a. [DOI] [PubMed] [Google Scholar]; (b) Marinescu SM, Nishimata TN, Mohr JT, Stoltz BM. Org Lett. 2008;10:1039–1042. doi: 10.1021/ol702821j. [DOI] [PMC free article] [PubMed] [Google Scholar]