

Abstract
Sulindac displays promising antineoplastic activity, but toxicities from cyclooxygenase (COX) inhibition limit its use for chemoprevention. Previous reports suggest that its anticancer properties may be attributed to a COX-independent mechanism, although alternative targets have not been well defined. Here we show that sulindac sulfide (SS) induces apoptosis and inhibits the growth of human breast tumor cells with IC50 values of 60-85 μM. Within the same concentration range, SS inhibited cGMP hydrolysis in tumor cell lysates, but did not affect cAMP hydrolysis. SS did not induce apoptosis of normal human mammary epithelial cells (HMEC), nor did it inhibit PDE activity in HMEC lysates. SS increased intracellular cGMP levels and activated protein kinase G in breast tumor cells, but not HMEC. The guanylyl cyclase (GC) activator, NOR-3, and cGMP PDE inhibitors, trequinsin and MY5445, displayed similar growth inhibitory activity as SS, but the adenylyl cyclase activator, forskolin, and other PDE inhibitors had no effect. Moreover, GC activation increased the sensitivity of tumor cells to SS, while GC inhibition reduced sensitivity. By comparing PDE isozyme profiles in breast tumor cells with HMEC and determining the sensitivity of recombinant PDE isozymes to SS, PDE5 was found to be overexpressed in breast tumor cells and selectively inhibited by SS. The mechanism of SS binding to the catalytic domain of PDE5 was revealed by molecular modeling. These data suggest that PDE5 inhibition is responsible for the breast tumor cell growth inhibitory and apoptosis inducing activity of SS and may contribute to the chemopreventive properties of sulindac.
Keywords: phosphodiesterase, cyclic GMP, breast cancer, chemoprevention, sulindac, NSAIDs, trequinsin, protein kinase G
Introduction
Breast cancer remains the most commonly diagnosed cancer and the second leading cause of cancer-related deaths for women in the United States (1). Despite ongoing efforts to develop novel therapeutics, mortality rates for breast cancer have only recently begun to decline, but only slightly and likely as a result of early detection of disease. Chemoprevention is widely believed to be an effective strategy for reducing cancer-related mortalities. However, with the exception of estrogen receptor antagonists and aromatase inhibitors, which have limited efficacy and potentially severe toxicities, no other drugs have received FDA approval for breast cancer chemoprevention.
Epidemiological studies have demonstrated that nonsteroidal anti-inflammatory drugs (NSAIDs) display promising breast cancer chemopreventive efficacy. For example, the Women's Health Initiative, a large observational study that followed more than 80,000 post-menopausal women for more than eight years, found that long-term regular use of any NSAID reduced breast cancer risk by 28%. This study also demonstrated that non-aspirin NSAIDs display stronger chemopreventive efficacy because aspirin use was associated with a 21% reduction of breast cancer risk, while ibuprofen use was associated with a 49% reduction of breast cancer risk (2). In addition to preventing primary occurrence of breast tumors, NSAIDs have also been shown to reduce the recurrence of breast cancer (3).
NSAIDs are a chemically diverse family of drugs used to treat a variety of inflammatory conditions and chronic pain associated with arthritis. The pharmacological basis for their activity involves inhibition of the cyclooxygenase (COX) enzymes. Inhibition of COX blocks the conversion of arachidonic acid to prostaglandins, prostacyclins, and thromboxanes that play an important role in inflammation and other physiological processes including renal function, clot formation, and gastrointestinal (GI) protection (4). Unfortunately, the depletion of physiologically important prostaglandins by NSAIDs and COX-2 inhibitors can result in sometimes fatal GI, renal, and cardiovascular toxicities that preclude their use for chemoprevention (4-6).
Several different lines of evidence suggest that a COX-independent mechanism may be fully or partially responsible for the antineoplastic activities of NSAIDs and COX-2 selective inhibitors (7-12). Most notably, much higher doses are required to inhibit tumor growth in vitro and in vivo compared with dosages required to inhibit COX-1 or COX-2 (13-14), which suggest that a low affinity, off-target effect may be responsible for their chemopreventive activity. In support of this possibility, the non-COX inhibitory sulfone metabolite of the NSAID sulindac has been shown to inhibit tumor cell growth and induce apoptosis in vitro (8-9) and prevent chemically induced tumor formation in several animal models including mammary tumorigenesis in the rat (15-21). Other studies have shown that sulindac sulfone can inhibit cyclic guanosine monophosphate phosphodiesterase (cGMP PDE) (18, 22-23). This effect may be shared by COX inhibitors based on a previous report showing that several chemically distinct NSAIDs, such as indomethacin and meclofenamic acid, as well as the COX-2 selective inhibitor, celecoxib, also inhibit cGMP PDE (12), although the specific isozyme(s) involved have not been identified.
Cyclic nucleotide PDEs are an important enzyme superfamily responsible for regulating second messenger signaling by hydrolyzing the 3′,5′-phosphodiester bond in the cyclic nucleotides 3′,5′-cyclic guanosine monophosphate (cGMP) and/or 3′,5′-cyclic adenosine monophosphate (cAMP). There are eleven PDE families with different substrate specificity, regulatory properties, tissue localization, and inhibitor sensitivity. Due to the expression of multiple genes, alternative mRNA splicing, and post-translational protein modifications, it is estimated that humans can express more than 100 distinct PDE isoforms or splice variants (24). Depending on the PDE isozyme content of the cell and the chemical selectivity of the inhibitor, PDE inhibition can increase the magnitude and/or the duration of the cAMP and/or cGMP signal(s). Increasing cyclic nucleotide levels activates specific signaling pathways, which, in the case of cGMP, can lead to activation of cGMP dependent protein kinase (PKG), cyclic nucleotide gated ion channels, or certain cGMP binding PDEs, resulting in protein phosphorylation, ion fluxes, or cyclic nucleotide hydrolysis to impact gene expression or other aspects of cellular activity (25).
Both cAMP and cGMP have been shown to have antiproliferative and pro-apoptotic effects (26-27). In addition, altered expression of one or more PDE isozymes has been reported in various carcinomas and hematological malignancies (18, 23, 28-33). However, little is known about whether cyclic nucleotides regulate proliferation and/or survival of breast tumor cells or which PDE isozymes are expressed. Here we show that cGMP elevation can inhibit growth of breast tumor cells, that this pathway is activated by the COX inhibitor, sulindac sulfide (SS); and that SS preferentially inhibits PDE5, which is overexpressed in breast tumor cells.
Materials and Methods
Drugs and Reagents
Sulindac sulfide, trequinsin, and forskolin were purchased from Sigma-Aldrich (St. Louis, MO). NOR-3 was purchased from BioMol (Plymouth Meeting, PA). LY83583 was purchased from Cayman Chemical (Ann Arbor, MI). Sildenafil was a generous gift from Pfizer. Recombinant PDE isozymes were purchased from BPS Biosciences (San Diego, CA). The family-specific anti-PDE antibodies were purchased from GeneTex (San Antonio, TX), anti-VASP antibody from BD Transduction Laboratories (San Jose, CA), and anti-phospho-VASP-Ser239 from Cell Signaling Technologies (Beverly, MA). Anti-rabbit and anti-mouse horseradish peroxidase-conjugated secondary antibodies were also obtained from Cell Signaling Technologies. All compounds were solubilized in DMSO and diluted to a final concentration of 1% in enzyme- and 0.1% in cell-based experiments, which did not interfere with the assays. Unless otherwise specified, all other reagents were purchased from Sigma-Aldrich.
Cells and Cell Culture
The human breast cancer cell lines MDA-MB-231 and SK-BR-3 were obtained from ATCC and grown under standard cell culture conditions in RPMI 1640 medium containing 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere with 5% CO2. Assays were performed using the same growth conditions except serum content was decreased to 1.5% FBS. Human mammary epithelial cells (HMEC) were obtained from Lonza (Basil, Switzerland) and were grown according to the specifications of the supplier in MEGM complete growth medium. Cell counts and viability were determined by trypan blue exclusion followed by hemacytometry. Only cultures displaying >95% viability were used for experiments.
Growth Assays
Tissue culture microtiter 96-well plates were seeded at a density of 5,000 cells per well. Cells were incubated for 18 to 24 hours, treated with the specified compound or vehicle control, and incubated an additional 72 hours. The inhibition of cell growth caused by treatment was determined using the luminescent Cell Titer Glo Assay (Promega), which measures viable cells based on ATP content. The assay was performed according to the manufacturer's specifications with a maximum DMSO concentration of 0.2%. Luminescence was measured using a VictorV (PerkinElmer) plate reader.
Caspase Assays
Tissue culture microtiter 96-well plates were seeded at a density of 10,000 cells per well. Cells were incubated for 18 to 24 hours, treated with the specified compound or vehicle control, and incubated an additional 6 hours. The induction of apoptosis caused by treatment was determined using the luminescent Caspase3/7 Glo Assay (Promega) which measures cleavage of a substrate for caspases 3 and 7. The assay was performed according to the manufacturer's specifications with a maximum DMSO concentration of 0.2%. Luminescence was measured using a VictorV (PerkinElmer) plate reader.
Cell Lysis
Cells were harvested, vortexed in ice cold lysis buffer (20mM TrisAcetate, 5mM Magnesium Acetate, 1mM EGTA, 0.8% Triton X-100, 50mM NaF, and protease inhibitor cocktail at pH 7.4), and clarified by centrifugation at 10,000g for 10 minutes at 4°C. Protein content was determined using the BCA Protein Assay (Pierce) following the manufacturer's specifications.
PDE Assays
PDE activity in cell lysates was measured using the IMAP fluorescence polarization (FP) phosphodiesterase assay (Molecular Devices) in which binding of hydrolyzed fluorescent cyclic nucleotide substrate to the IMAP reagent increases fluorescence polarization. The assay was modified to use fluorescein (Fl)-cAMP and tetramethylrhodamine (TAMRA)-cGMP as substrates, allowing for simultaneous measurement of cAMP and cGMP hydrolysis. Each well of a 96-well non-binding plate contained 0.25 mg/mL of whole cell lysate or recombinant enzyme preparations. Enzymes were incubated with SS for 30 minutes at 30°C prior to the addition of a substrate mixture containing 25 nM of each Fl-cAMP and TAMRA-cGMP. After 90 minutes of incubation at 30°C, the reaction was terminated by the addition of binding reagent. The maximum DMSO concentration for each experiment was 2%. FP was measured using a Synergy4 (BioTek) plate reader.
cGMP Assay
Cells were seeded at a density of 1×106 cells per 10 cm tissue culture dish, incubated for 48 hours, and treated with the specified compound or vehicle control. After 30 minutes of treatment, cells were lysed and assayed for cGMP content using the cGMP Direct Biotrak EIA kit (GE Biosciences). The assay was performed according to the manufacturer's specifications. Optical density was measured at 630 nm using a Synergy4 (BioTek) plate reader.
Western Blots
Whole cell lysates (30 μg protein) were separated by SDS-PAGE in a 12% polyacrylamide gel followed by electrophoretic transfer to a nitrocellulose membrane. The membranes were blocked with 5% BSA in TBS containing 0.05% Tween-20. Incubation of membranes in primary and secondary antibodies was performed according to the manufacturers' specifications. Protein bands were visualized on HyBlot CL (Denville Scientific) autoradiography film using Super Signal West Pico Enhanced Chemiluminescence Reagent (Pierce).
Molecular Modeling
Molecular modeling was performed using the Schrödinger Suite 2008 (Schrödinger, LLC). The PDE5 protein structure was obtained from the protein databank (PDB ID: 1UDT PDE5). The Induced Fit Docking (IFD) protocol, which takes into consideration of the ligand-induced receptor conformational change, was used for all docking studies. Residues within 5 Å from the ligand were allowed to be flexible. The docking results were scored using the extra-precision (XP) mode of Glide® version 4.5 (Schrödinger, LLC). The IDF docking protocol and parameters were first validated by docking sildenafil in the PDE5 catalytic site. The docking result excellently reproduced the sildenafil-PDE5 crystal complex conformation. The same protocol and parameters were then used to study the docking of SS to PDE5.
Experimental Design and Data Analysis
Drug effects on cell growth and PDE activity were measured and the potency expressed as an IC50 value, which is the concentration resulting in 50% inhibition when compared to the vehicle control. For growth assays, the IC50 value was determined by testing a range of 8 concentrations with a minimum of 4 replicates per dose. For the PDE assays, the IC50 value was determined by testing a range of 10 concentrations with a minimum of 2 replicates per dose. Dose response curves were constructed using Prism5 software (Graphpad), which calculates IC50 values using a four parameter logistic equation. All experiments were repeated a minimum of twice in order to determine the reproducibility of the results. All values represent a comparison between drug treatment at the specified concentration and vehicle treated controls. All error bars represent standard error of the mean (SEM). Calculation of p values was done by comparing the specified treatment group to vehicle treated controls using a student's t test.
Results
SS inhibition of breast tumor cell growth and apoptosis induction is associated with cGMP PDE inhibition, intracellular cGMP elevation, and PKG activation
SS inhibited the growth of the human SK-BR-3, ZR75-1, and MDA-MB-231 breast tumor cell lines with IC50 values of 59, 76 and 84 μM, respectively (Figure 1A). By comparison, primary cultures of normal human mammary epithelial cells (HMEC) were appreciably less sensitive with an IC50 value of 163 μM. Tumor cell growth inhibition by SS was associated with the induction of apoptosis as measured by the activation of effector caspases 3 and 7, which are specific biochemical markers of apoptotic cell death. As shown in Figure 1B, treatment with SS (100 μM, 5 hours) resulted in a 2-8 fold increase in the activity of caspases 3 and 7 when compared to vehicle treatment in MDA-MB-231, SKBR3, and ZR75-1 cells. HMEC were resistant to SS-induced activation of caspases, but were capable of undergoing apoptosis as evident by increased caspase activity following treatment with the non-selective apoptosis inducing agent, staurosporine. These results suggest that the reduced sensitivity of HMEC to growth inhibition by SS is attributed to the inability of SS to induce apoptosis. SS also increased TUNEL labeling of breast tumor cells, although did not have a significant effect on cell cycle distribution under the conditions used for the growth assays (data not shown).
Figure 1.
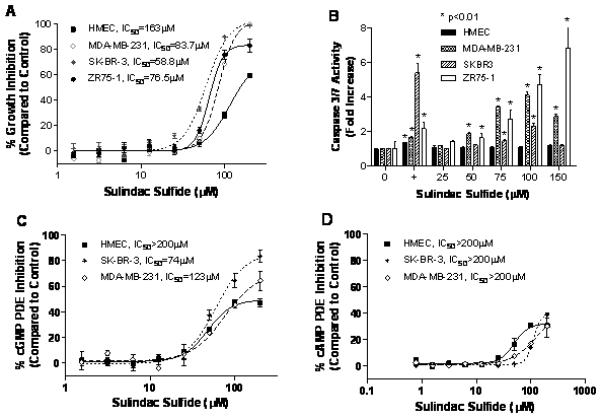
Inhibition of growth, induction of apoptosis, and inhibition of PDE activity by SS in human breast cells. A, dose-dependent growth inhibitory activity of SS in human MDA-MB-231, SK-BR-3, and ZR75-1 breast tumor cells and normal mammary epithelial cells (HMEC) after 72 hours of treatment. B, dose-dependent induction of apoptosis by SS in MDA-MB-231, SK-BR-3, ZR75-1, and HMEC as measured by activation of caspases 3 and 7 after 5 hours of treatment. “+” represents 1μM staurosporine positive control. C and D, dose-dependent inhibition of cGMP (C) and cAMP (D) PDE activity in whole cell lysates of HMEC, MDA-MB-231, and SK-BR-3 cells.
A fluorescence polarization assay that we developed to simultaneously measure cGMP and cAMP hydrolysis was used to determine if SS can inhibit cyclic nucleotide PDE activity. As shown in Figure 1C, SS inhibited cGMP hydrolysis in lysates from SK-BR-3 and MDA-MB-231 cells with IC50 values within the same range as concentrations that inhibited tumor cell growth and induced apoptosis, but did not significantly affect cAMP hydrolysis in breast tumor cell lysates (Figure 1D). Additionally, SS did not affect cGMP or cAMP hydrolysis in lysates from HMEC. These observations suggest that SS can preferentially inhibit cGMP degrading PDE isozymes and that there may be differences in the expression of such isozymes in normal and neoplastic cells.
To determine if the inhibition of cGMP degradation by SS can increase intracellular cGMP concentrations, cell associated cGMP levels were measured following 30 minutes of treatment with SS. As shown in Figure 2A, SS increased cGMP levels by 2-3 fold in MDA-MB-231 cells over a broad concentration range with levels peaking at 100 μM, which is comparable to the concentration range necessary for growth inhibition and caspase activation in this cell line. Higher concentrations of SS were less effective and may be attributed to necrotic cell death as reported previously (8), which resulted in the leakage of cGMP from the cells prior to the assay. Although basal levels of cGMP in MDA-MB-231 tumor cells and HMEC were comparable, SS failed to induce cGMP levels in HMEC.
Figure 2.
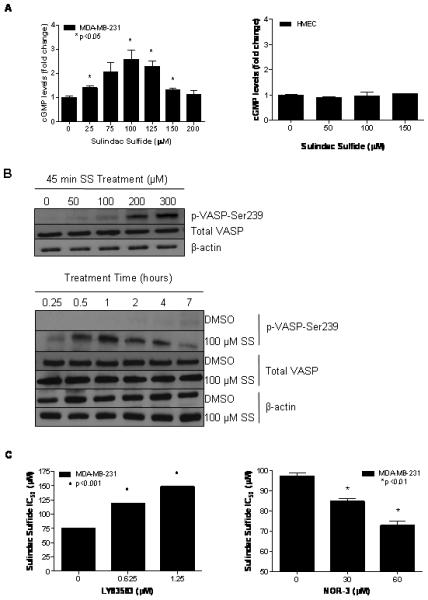
SS induced activation of cGMP signaling in MDA-MB-231 human breast tumor cells. A, dose-dependent increase in intracellular cGMP levels after 30 minutes of SS treatment in MDA-MB-231 (left) and HMEC (right). B, time- (top) and dose-dependent (bottom) increases in VASP phosphorylation at serine 239 after SS treatment. C, dose-dependent decrease in growth-inhibitory potency of 48 hours of SS treatment after 1 hour of pretreatment with the guanylyl cyclase inhibitor, LY83583 (left) and dose-dependent increase in growth-inhibitory potency of 72 hours of SS treatment after 1 hour of pretreatment with the guanylyl cyclase activator NOR-3 (right).
Treatment effects of SS on the phosphorylation of vasoactivator-stimulated phosphoprotein (VASP), a known substrate for phosphorylation by the cGMP-activated protein kinase, PKG (34), were measured to determine if the elevation of intracellular cGMP levels by SS is sufficient to activate cGMP signaling in breast tumor cells. As described previously, VASP is preferentially phosphorylated at the serine 239 residue by PKG, thereby allowing the measurement of phosphorylation at this residue to serve as an indicator of PKG activity and cGMP signaling within cells (34). Treatment of MDA-MB-231 cells with SS increased the levels of phosphorylated VASP (p-VASP) in a dose- (Figure 2B) and time-dependent (Figure 2C) manner without affecting the expression of total VASP. After 30 minutes of treatment, increases in VASP phosphorylation were apparent with 50 μM of SS. With 100 μM SS treatment, the level of VASP phosphorylation peaked after 1 hour of treatment. Importantly, the concentrations and time required for SS to increase VASP phosphorylation paralleled those required for cGMP elevation.
To determine if cGMP elevation is necessary for the tumor cell growth inhibitory activity of SS, the effects of modulating basal cGMP levels were studied using guanylyl cyclase (GC) activators and inhibitors. By pre-treating with the GC inhibitor, LY83583, significantly higher concentrations of SS were required to suppress the growth of MDA-MB-231 breast tumor cells as evident by almost a two-fold increase in the IC50 value (Figure 2D, left). As a control, LY83583 was tested for its ability to affect the sensitivity of the MDA-MB-231 cells to growth inhibition by the non-selective cytotoxic agent, doxorubicin, and was found to be ineffective (data not shown). In another series of experiments, cells were pre-treated with the NO donor and GC activator, NOR-3. NOR-3 significantly increased the sensitivity of breast tumor cells to the growth inhibitory activity of SS as evident by a significant reduction in the IC50 value. (Figure 2D, right) These experiments provide evidence that cGMP elevation is necessary for SS to inhibit the growth of human breast tumor cells.
Activation of cGMP signaling is a growth inhibitory signal in human breast tumor cells
To determine if cGMP or cAMP elevation is sufficient to inhibit the growth of human breast tumor cells, a number of known activators of cGMP or cAMP signaling were evaluated for effects on the growth of breast tumor cells. As shown in Figure 3A, NOR-3 inhibited the growth of SK-BR-3 and MDA-MB-231 breast tumor cell lines. By comparison, the adenylyl cyclase activator, forskolin, had no significant growth inhibitory activity (Figure 3B). In addition, a number of known PDE inhibitors were also tested for tumor cell growth inhibitory activity, including 8-methoxymethyl-IBMX (PDE1), EHNA (PDE2), milrinone (PDE3), rolipram (PDE4), sildenafil (PDE5), zaprinast (PDE5, 6, 9 and 11), dipyridamole (PDE7, 8, 10, 11), and the non-selective PDE inhibitor, IBMX, but none were found to be active (data not shown), with the exception of two inhibitors, trequinsin and MY5445, as shown in Figure 3C and 3D, respectively. Although the PDE isozyme specificity of trequinsin and MY5445 has not been well characterized, both inhibitors were found to selectively inhibit cGMP hydrolysis as described below (Table 1).
Figure 3.
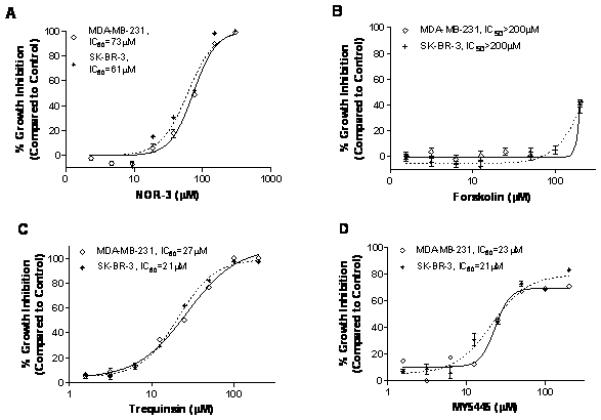
Growth inhibitory activities of cAMP and cGMP signaling inducers in MDA-MB-231, and SK-BR-3 human breast cells after 72 hours of treatment. A-D, growth inhibition induced by the NO donor and guanylyl cyclase activator NOR-3 (A), by the adenylyl cyclase activator forskolin (B), by the cGMP PDE inhibitor trequinsin (C), and by the cGMP PDE inhibitor MY5445 (D).
Table 1.
Substrate selectivity and sensitivity of PDE isozymes for inhibition by SS, trequinsin, or MY5445. Substrate selectivity is indicated by +/−, while sensitivity to inhibitors is indicated by an IC50 value (μM). SS was tested at concentrations up to 200 μM, trequinsin up to 50 μM, and MY5445 up to 100 μM. A dash (−) represents no detectable enzyme activity. A plus sign (+) represents detectable enzyme activity, with the number of plus signs corresponding to relative level of activity. If the level of inhibition was not significant and an IC50 value was undeterminable, “inactive” is indicated. If a drug was not evaluated , “n.d.” is indicated.
| Purified PDE Isoform |
Substrate Selectivity |
SS Sensitivity | Trequinsin Sensitivity | MY5445 Sensitivity | ||||
|---|---|---|---|---|---|---|---|---|
| cAMP | cGMP | cAMP | cGMP | cAMP | cGMP | cAMP | cGMP | |
| 1A | + | ++ | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 1B | + | ++ | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 1C | + | + | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| 2A | + | + | Inactive | 97 | 2 | 1.3 | Inactive | Inactive |
| 3A | +++ | + | Inactive | 84 | 0.00023 | 0.000067 | Inactive | Inactive |
| 4A | + | − | Inactive | - | n.d. | - | n.d. | - |
| 5A | − | + | - | 38 | - | 3.4 | - | 14.3 |
| 6C | − | + | - | Inactive | - | n.d. | - | n.d. |
| 7A | + | − | Inactive | - | n.d. | - | n.d. | - |
| 8A | + | − | Inactive | - | n.d. | - | n.d. | - |
| 9A | − | + | - | Inactive | - | n.d. | - | n.d. |
| 10A | ++ | + | Inactive | Inactive | n.d. | n.d. | n.d. | n.d. |
| 11A | ++ | + | Inactive | Inactive | n.d. | n.d. | n.d. | n.d. |
Normal and neoplastic breast cells display differential expression of cGMP PDE isozymes
The ability of SS to selectively inhibit growth and induce apoptosis of human breast tumor cells may be attributed to differential expression of SS-sensitive cGMP degrading PDE isozymes in breast tumor cells compared with HMEC. To study this possibility, we compared the expression of the cGMP-hydrolyzing enzymes PDE1A, 1B, 1C, 2A, 5A, and 9A in breast tumor cells and HMEC by western blotting using PDE family-specific antibodies. As shown in Figure 4, HMEC expressed a variety of PDE isozymes capable of cGMP hydrolysis, including two isoforms each of PDE1A, 5A and 9A and a single isoform each of PDE1B and 1C. By comparison, breast tumor cells expressed fewer isozymes, with PDE1B and PDE5A being the major isozymes detected. PDE1B was expressed at a comparable level in all three cell lines. On the other hand, a high molecular weight isoform of PDE5A, which corresponds to the size of PDE5A1, was overexpressed in breast tumor cells compared with HMEC. Interestingly, the lower molecular weight isoform of PDE5A, which corresponds to PDE5A3 displayed decreased expression in breast tumor cells compared with HMEC. None of the cell lines expressed detectable levels of PDE2A.
Figure 4.
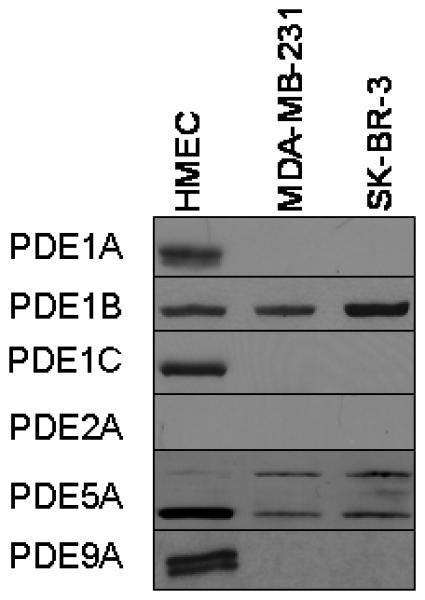
Expression levels of cGMP PDE isozymes in HMEC, MDA-MB-231, and SK-BR-3 human breast cells.
SS inhibits PDE5
To determine PDE isozyme selectivity of SS, the sensitivity of catalytically active preparations of recombinant PDE isozymes from each of the eleven PDE isozyme families to inhibition by SS, trequinsin, and MY5445 were determined. Initially, the recombinant enzymes were characterized for substrate selectivity. As summarized in Table 1 and consistent with the known substrate specificity of PDE isozyme families (24), PDE1, 2, 3, 10, and 11 were capable of hydrolyzing both cAMP and cGMP, while PDE4, 7, and 8 displayed cAMP specificity, and PDE5, 6, and 9 displayed cGMP specificity. Among all isozymes evaluated, PDE5 was found to be the most sensitive to inhibition by SS, which resulted in an IC50 value of 38 μM and is consistent with the ability of SS to selectively inhibit cGMP hydrolysis in tumor cell lysates. SS also inhibited cGMP hydrolysis by PDE2 and 3 with IC50 values of 97 and 84 μM, respectively, yet this activity appears to be irrelevant to its growth inhibitory activity because PDE2 was not expressed in the breast tumor cells and PDE3 displayed strong selectivity for cAMP hydrolysis. PDE1, 4, 6-11 were insensitive to SS at concentrations up to 200 μM. Trequinsin also inhibited PDE2, 3, and 5, but unlike SS, was appreciably more selective for inhibiting PDE3. By comparison, MY5445 was highly selective for PDE5. These results demonstrate that PDE5 is a common isozyme target of each of the cGMP PDE inhibitors that were capable of inhibiting breast tumor cell growth.
Molecular modeling studies
Molecular modeling studies were performed to determine the mode of SS binding to PDE5 and to compare it with the highly potent inhibitor, sildenafil. Induced fit docking was used to mimic the conformational change of the enzyme in response to drug binding. Figure 5 shows the crystal structure of the sildenafil-PDE5 complex (A) and SS-docked PDE5 structure (B). Both drugs were found to occupy essentially the same central core formed by Val782, Leu765, Tyr612, Phe786 and Phe820. The face-to-face π-π interaction with the phenyl ring of Phe820 was also well maintained. The hydrogen bonding between sildenafil and Gln817 was substituted with a halogen bonding in SS (DistF-H: 2.46 Â). The methylthiophenyl of SS aligned with the sulfonyl-piperazinyl group of sildenafil and extended into the so-called ‘lid’ region composed of Met816, Phe664, Ala823 and Gly819 (35). The carboxylic acid moiety of SS extended into the metal-binding site and directly interacted with the Zn ion (DistZn-O:2.10 Â), which further stabilized the SS binding to PDE5. These results demonstrate the ability of SS to bind the catalytic region of PDE5 in a manner similar to sildenafil.
Figure 5.
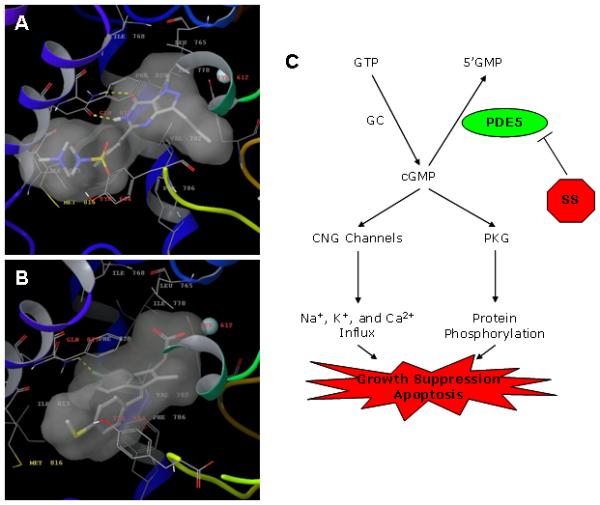
Molecular modeling of sildenafil and SS bound to PDE5. A, crystal structure of sildenafil-PDE5 complex. B, docked SS-PDE5 structure. The protein is represented with ribbons, while the residues involved in drug binding are shown as sticks. The zinc ion is represented in cyan. The transparent surface illustrates the drug occupied space within the binding pocket. C, mechanistic model of apoptosis induction by SS in human breast tumor cells. CNG = cyclic nucleotide gated ion channel.
Discussion
NSAIDs have shown promising antineoplastic activity in experimental models but their use for cancer chemoprevention in humans is not practical because of gastrointestinal, renal, and cardiovascular toxicities that result from COX-1 and/or COX-2 inhibition and the depletion of physiologically important prostaglandins. Previous studies have concluded that a COX-independent mechanism may be responsible for their tumor cell growth inhibitory and apoptosis inducing activities, which suggests the feasibility of developing safer and more efficacious drugs for cancer chemoprevention by targeting such mechanisms. The non-COX inhibitory sulfone metabolite of sulindac has been previously shown to inhibit cGMP PDE, although the specific isozyme(s) involved have not been identified, nor has this putative mechanism been studied with regard to the COX inhibitory sulfide metabolite of sulindac. Here we show for the first time that the COX-inhibitory sulfide metabolite of sulindac can selectively inhibit cGMP PDE activity in breast tumor cells, leading to the elevation of intracellular cGMP levels and activation of PKG. SS did not inhibit cGMP PDE activity in lysates from HMEC, nor did it increase cGMP levels in HMEC. By comparing the expression of cGMP degrading isozymes in breast tumor cells with HMEC and determining the sensitivity of PDE isozyme family members to SS, we conclude that PDE5 is overexpressed in human breast tumor cells and selectively inhibited by SS. This conclusion is supported by studies from other investigators showing antiproliferative and pro-apoptotic effects of PDE5 antisense in colon tumor cells (36).
Initial evidence for the involvement of a cGMP specific isozyme was suggested by experiments showing a high degree of selectivity for SS to inhibit cGMP hydrolysis compared with cAMP hydrolysis in the breast tumor cell lysates. Consistent with these observations, SS did not affect the activity of cAMP specific PDE enzymes (PDE4, 7, and 8), but selectively inhibited the cGMP specific PDE5 isozyme. The biological relevance of SS to inhibit PDE5 was confirmed by experiments involving intact cells, which showed that SS treatment increased intracellular cGMP levels in human breast tumor cells within the same concentration range as required for cGMP PDE inhibition in cell-free assays. Other experiments demonstrated that the magnitude of cGMP elevation by SS was sufficient to activate PKG, as measured by phosphorylation of VASP, a known PKG substrate, which occurred within the same concentration range and time period as required for cGMP elevation in breast tumor cells.
A mechanistic model for the tumor cell growth inhibitory and apoptosis inducing activity of SS involving PDE5 inhibition, cGMP elevation, and PKG activation is illustrated in Figure 5C. While PKG activation appears to play an important role in mediating the apoptosis inducing activity of SS, the specific substrates that are phosphorylated by PKG and downstream signaling events responsible for apoptosis induction, as well as the potential involvement of other cGMP-dependent proteins, such as cyclic nucleotide gated ion channels or cGMP binding PDEs, requires further study.
We also demonstrated that disrupting cGMP synthesis by GC inhibition could desensitize breast tumor cells to the growth inhibitory activity of SS, while GC activation enhanced sensitivity to SS. Together, these experiments support a mechanism of growth inhibition that involves cGMP PDE inhibition. In addition, GC activators and the cGMP PDE inhibitors trequinsin and MY5445 inhibited breast tumor cell growth. By contrast, compounds known to activate cAMP signaling, such as the adenylyl cyclase activator, forskolin, and cAMP PDE inhibitors had no significant effect on growth of breast tumor cells. These data demonstrate that the elevation of cGMP is both necessary and sufficient for SS-mediated growth inhibition in human breast cells and that cGMP plays an exclusive role in breast tumor cell survival compared with cAMP. Supporting a role of cGMP in chemoprevention, other investigators have demonstrated that GC deficient mice display increased susceptibility to colon tumorigenesis (37). In addition, GC agonists such as uroguanylin have been shown to have chemopreventive activity in the APCmin mouse model of colorectal tumorigenesis (38), which is highly sensitive model to sulindac as well.
SS inhibited PDE5 with an IC50 value of 38 μM, which is significantly higher compared with IC50 values of 2 and 6 μM to inhibit COX-1 and COX-2, respectively (39). Nonetheless, the higher concentration range of SS to inhibit PDE5 is consistent with the requirement of higher concentrations to inhibit breast tumor cell growth and induce apoptosis compared with COX-1 or COX-2. However, further studies are necessary to determine if clinically relevant dosages of sulindac cause PDE5 inhibition and cGMP elevation in vivo. In support of this possibility, pharmacokinetic studies in humans have shown that a 400 mg dosage of sulindac can achieve plasma concentrations of the sulfide that are comparable to those required for PDE5 inhibition and cGMP elevation as we report here and can cause adenoma regression in patients with familial adenomatous polyposis (40-41). Moreover, the sulfide metabolite can achieve higher levels in certain tissues compared with plasma (42) and is likely a reflection of the lipophilic properties of the drug.
As further evidence for direct binding of SS to PDE5, molecular modeling studies demonstrated the ability of SS to bind the catalytic domain of PDE5 in a manner similar to the highly potent and selective PDE5 inhibitor, sildenafil. These studies suggest a mechanism involving competitive inhibition of substrate binding. Molecular modeling also suggested a mechanism of selectivity to bind PDE5, but not other isozymes. For example, overlapping PDE5 with PDE1 showed that their catalytic pockets were structurally similar to each other except for the location of the “lid” region, which consisted of residues 661-680 in PDE5 and residues 271-290 in PDE1. However, using the same IDF docking protocol, SS resulted in very different binding configuration when docked to PDE1. Comparing with SS-PDE5 docking result, the docked SS-PDE1 complex showed a relatively unfavorable docking score of ~4 kcal/mol higher, which is equivalent to a ~800 fold higher dissociation constant and is consistent with results from enzyme assays. Previous studies have concluded that the closed catalytic pocket of PDE5 is responsible for the selectivity of sildenafil binding to PDE5 (35). The wider pocket of PDE1 may also explain the more favorable binding of SS to PDE5 compared with PDE1. The lack of the closed lid loop, especially the phenyl group of Phe664, to shield the methylthiophenyl group of SS from solvent may prevent SS from binding PDE1. In fact, such a lid covered pocket is quite unique for PDE5, while open pocket structures have been observed in other PDE isozymes including PDE2, PDE3 and PDE4 (PDB ID: 1Z1L, 1SO2 and 3D3P). This structural difference near the binding pocket might explain the selectivity of SS to bind PDE5.
While PDE5 appears to be the major cGMP degrading PDE isozyme expressed in breast tumor cells that is sensitive to SS, we cannot rule out the possibility that additional cGMP PDE isozymes or splice variants may be involved, especially since highly selective and potent PDE5 inhibitors (e.g. sildenafil) were unable to inhibit breast tumor cell growth. However, sildenafil was also unable to significantly increase intracellular cGMP levels or activate PKG in breast tumor cells, despite its ability to inhibit the majority of cGMP PDE activity in tumor cell lysates. This suggests that there may be another explanation as to why conventional PDE5 inhibitors do not have anticancer properties. For example, PDE5 in tumor cells may not be accessible to such drugs, possibly due to a restricted subcellular localization. Alternatively, such drugs may be effectively transported out of tumor cells by efflux mechanisms such as the multidrug resistance proteins (MRP), which are often upregulated in tumor cells.
We also show novel observations that the expression pattern of cGMP PDE isozymes is distinctly different between normal and neoplastic breast cells. With the exception of PDE5A1, HMEC display increased expression and greater diversity of all other cGMP degrading PDE isozymes when compared with breast tumor cells. The increased expression of PDE5A1 observed in breast tumor cells compared with HMEC is consistent with other studies reporting PDE5 overexpression in tumors from lung, bladder, and breast carcinomas (18, 23, 43). However, this is the first report showing that normal cells express a greater diversity of other cGMP PDE isozymes. Based on these observations, we speculate that the expression of cGMP PDE isozymes in HMEC, which are insensitive to SS, may provide a compensatory mechanism that allows normal cells to be refractory to SS.
From the data presented here, we conclude that PDE5 inhibition by SS leads to the intracellular accumulation of cGMP and activation of cGMP signaling to result in the selective apoptosis of breast tumor cells. The identification of more potent and selective compounds that can stimulate cGMP signaling in human breast tumor cells and thereby induce apoptosis could potentially yield a chemopreventive agent with sufficiently low toxicity and more complete efficacy for breast cancer chemoprevention compared with conventional NSAIDs and COX-2 inhibitors.
Acknowledgements
We are grateful to Dr. Graeme Bolger for critical reading of the article.
Financial Support: DoD CDMRP BCRP Grant W81XWH-07-1-0463, NCI Grant CA131378.
Abbreviations
- COX
cyclooxygenase
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- FP
fluorescence polarization
- GC
guanylyl cyclase
- IBMX
3-Isobutyl-1-methylxanthine
- MY5445
1-(3-chloroanilino)-4-phenylphthalazine
- NOR-3
(±)-(E)-Ethyl-2-((E)-hydroxyimino)-5-nitro-3-hexenamide
- NSAID
nonsteroidal anti-inflammatory drug
- PDE
phosphodiesterase
- PKG
protein kinase G
- SS
sulindac sulfide
- TUNEL
dUTP nick end labeling
- VASP
vasoactivator stimulated phosphoprotein
Footnotes
Potential Conflicts of Interest: There were no conflicts of interest among the authors.
References
- 1.Berry DA, Cronin KA, Plevritis SK, et al. Effect of screening and adjuvant therapy on mortality from breast cancer. N Engl J Med. 2005;353:1784–92. doi: 10.1056/NEJMoa050518. [DOI] [PubMed] [Google Scholar]
- 2.Harris RE, Chlebowski RT, Jackson RD, et al. Breast cancer and nonsteroidal anti-inflammatory drugs: prospective results from the Women's Health Initiative. Cancer Res. 2003;63:6096–101. [PubMed] [Google Scholar]
- 3.Kwan ML, Habel LA, Slattery ML, Caan B. NSAIDs and breast cancer recurrence in a prospective cohort study. Cancer Causes Control. 2007;18:613–20. doi: 10.1007/s10552-007-9003-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vane JR, Botting RM. Mechanism of action of antiinflammatory drugs. Int J Tissue React. 1998;20:3–15. [PubMed] [Google Scholar]
- 5.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee D. Selective cyclooxygenase-2 (COX-2) inhibitors and potential risk of cardiovascular events. Biochem Pharmacol. 2002;63:817–21. doi: 10.1016/s0006-2952(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 7.Alberts DS, Hixson L, Ahnen D, et al. Do NSAIDs exert their colon cancer chemoprevention activities through the inhibition of mucosal prostaglandin synthetase? J Cell Biochem Suppl. 1995;22:18–23. doi: 10.1002/jcb.240590804. [DOI] [PubMed] [Google Scholar]
- 8.Piazza GA, Rahm AL, Krutzsch M, et al. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995;55:3110–6. [PubMed] [Google Scholar]
- 9.Piazza GA, Rahm AK, Finn TS, et al. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction. Cancer Res. 1997;57:2452–9. [PubMed] [Google Scholar]
- 10.Elder DJ, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res. 1997;3:1679–83. [PubMed] [Google Scholar]
- 11.Hanif R, Pittas A, Feng Y, et al. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol. 1996;52:237–45. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 12.Soh JW, Kazi JU, Li H, Thompson WJ, Weinstein IB. Celecoxib-induced growth inhibition in SW480 colon cancer cells is associated with activation of protein kinase G. Mol Carcinog. 2008;47:519–25. doi: 10.1002/mc.20409. [DOI] [PubMed] [Google Scholar]
- 13.Raz A. Is inhibition of cyclooxygenase required for the anti-tumorigenic effects of nonsteroidal, anti-inflammatory drugs (NSAIDs)? In vitro versus in vivo results and the relevance for the prevention and treatment of cancer. Biochem Pharmacol. 2002;63:343–7. doi: 10.1016/s0006-2952(01)00857-7. [DOI] [PubMed] [Google Scholar]
- 14.Williams CS, Watson AJ, Sheng H, Helou R, Shao J, DuBois RN. Celecoxib prevents tumor growth in vivo without toxicity to normal gut: lack of correlation between in vitro and in vivo models. Cancer Res. 2000;60:6045–51. [PubMed] [Google Scholar]
- 15.Reddy BS, Kawamori T, Lubet RA, Steele VE, Kelloff GJ, Rao CV. Chemopreventive efficacy of sulindac sulfone against colon cancer depends on time of administration during carcinogenic process. Cancer Res. 1999;59:3387–91. [PubMed] [Google Scholar]
- 16.Piazza GA, Alberts DS, Hixson LJ, et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997;57:2909–15. [PubMed] [Google Scholar]
- 17.de Jong TA, Skinner SA, Malcontenti-Wilson C, et al. Inhibition of rat colon tumors by sulindac and sulindac sulfone is independent of K-ras (codon 12) mutation. Am J Physiol Gastrointest Liver Physiol. 2000;278:G266–72. doi: 10.1152/ajpgi.2000.278.2.G266. [DOI] [PubMed] [Google Scholar]
- 18.Piazza GA, Thompson WJ, Pamukcu R, et al. Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer Res. 2001;61:3961–8. [PubMed] [Google Scholar]
- 19.Malkinson AM, Koski KM, Dwyer-Nield LD, et al. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced mouse lung tumor formation by FGN-1 (sulindac sulfone) Carcinogenesis. 1998;19:1353–6. doi: 10.1093/carcin/19.8.1353. [DOI] [PubMed] [Google Scholar]
- 20.Thompson HJ, Briggs S, Paranka NS, et al. Inhibition of mammary carcinogenesis in rats by sulfone metabolite of sulindac. J Natl Cancer Inst. 1995;87:1259–60. [PubMed] [Google Scholar]
- 21.Thompson HJ, Jiang C, Lu J, et al. Sulfone metabolite of sulindac inhibits mammary carcinogenesis. Cancer Res. 1997;57:267–71. [PubMed] [Google Scholar]
- 22.Thompson WJ, Piazza GA, Li H, et al. Exisulind induction of apoptosis involves guanosine 3′,5′-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. 2000;60:3338–42. [PubMed] [Google Scholar]
- 23.Whitehead CM, Earle KA, Fetter J, et al. Exisulind-induced Apoptosis in a Non-Small Cell Lung Cancer Orthotopic Lung Tumor Model Augments Docetaxel Treatment and Contributes to Increased Survival. Mol Cancer Ther. 2003;2:479–88. [PubMed] [Google Scholar]
- 24.Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995;75:725–48. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- 25.Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. Faseb J. 1993;7:328–38. doi: 10.1096/fasebj.7.2.7680013. [DOI] [PubMed] [Google Scholar]
- 26.Wharton J, Strange JW, Moller GM, et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med. 2005;172:105–13. doi: 10.1164/rccm.200411-1587OC. [DOI] [PubMed] [Google Scholar]
- 27.Ahn YH, Jung JM, Hong SH. 8-Chloro-cyclic AMP-induced growth inhibition and apoptosis is mediated by p38 mitogen-activated protein kinase activation in HL60 cells. Cancer Res. 2005;65:4896–901. doi: 10.1158/0008-5472.CAN-04-3122. [DOI] [PubMed] [Google Scholar]
- 28.Drees M, Zimmermann R, Eisenbrand G. 3′,5′-Cyclic nucleotide phosphodiesterase in tumor cells as potential target for tumor growth inhibition. Cancer Res. 1993;53:3058–61. [PubMed] [Google Scholar]
- 29.Zhu B, Strada SJ. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr Top Med Chem. 2007;7:437–54. doi: 10.2174/156802607779941198. [DOI] [PubMed] [Google Scholar]
- 30.Zhu B, Strada S, Stevens T. Cyclic GMP-specific phosphodiesterase 5 regulates growth and apoptosis in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L196–206. doi: 10.1152/ajplung.00433.2004. [DOI] [PubMed] [Google Scholar]
- 31.Jiang X, Li J, Paskind M, Epstein PM. Inhibition of calmodulin-dependent phosphodiesterase induces apoptosis in human leukemic cells. Proc Natl Acad Sci U S A. 1996;93:11236–41. doi: 10.1073/pnas.93.20.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kloster MM, Hafte TT, Moltzau LR, et al. EBV infection renders B cells resistant to growth inhibition via adenylyl cyclase. Cell Signal. 2008;20:1169–78. doi: 10.1016/j.cellsig.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Lerner A, Epstein PM. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem J. 2006;393:21–41. doi: 10.1042/BJ20051368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deguchi A, Soh JW, Li H, Pamukcu R, Thompson WJ, Weinstein IB. Vasodilator-stimulated phosphoprotein (VASP) phosphorylation provides a biomarker for the action of exisulind and related agents that activate protein kinase G. Mol Cancer Ther. 2002;1:803–9. [PubMed] [Google Scholar]
- 35.Sung BJ, Hwang KY, Jeon YH, et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature. 2003;425:98–102. doi: 10.1038/nature01914. [DOI] [PubMed] [Google Scholar]
- 36.Zhu B, Vemavarapu L, Thompson WJ, Strada SJ. Suppression of cyclic GMP-specific phosphodiesterase 5 promotes apoptosis and inhibits growth in HT29 cells. J Cell Biochem. 2005;94:336–50. doi: 10.1002/jcb.20286. [DOI] [PubMed] [Google Scholar]
- 37.Li P, Schulz S, Bombonati A, et al. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology. 2007;133:599–607. doi: 10.1053/j.gastro.2007.05.052. [DOI] [PubMed] [Google Scholar]
- 38.Shailubhai K, Yu HH, Karunanandaa K, et al. Uroguanylin treatment suppresses polyp formation in the Apc(Min/+) mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res. 2000;60:5151–7. [PubMed] [Google Scholar]
- 39.Piazza GA, Keeton AB, Tinsley HN, et al. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev Res (Phila Pa) 2009;2:572–80. doi: 10.1158/1940-6207.CAPR-09-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies NM, Watson MS. Clinical pharmacokinetics of sulindac. A dynamic old drug. Clin Pharmacokinet. 1997;32:437–59. doi: 10.2165/00003088-199732060-00002. [DOI] [PubMed] [Google Scholar]
- 41.Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–6. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 42.Duggan DE, Hooke KF, Hwang SS. Kinetics of the tissue distributions of sulindac and metabolites. Relevance to sites and rates of bioactivation. Drug Metab Dispos. 1980;8:241–6. [PubMed] [Google Scholar]
- 43.Pusztai L, Zhen JH, Arun B, et al. Phase I and II study of exisulind in combination with capecitabine in patients with metastatic breast cancer. J Clin Oncol. 2003;21:3454–61. doi: 10.1200/JCO.2003.02.114. [DOI] [PubMed] [Google Scholar]