

Abstract
Although the mechanisms underlying striatal neurodegeneration are poorly understood, we have shown that striatal pathogenesis may be initiated by high synaptic levels of extracellular dopamine (DA). Here we investigated in rat striatal primary neurons the mobilization of the mitogen activated protein kinase (MAPK) signaling pathways after treatment with DA. Instead of observing an elevation of the archetypical pro-cytotoxic MAPKs, p-JNK and p-p38 MAPK, we found that DA, acting through D1 DA receptors, induced a sustained stimulation of the phosphorylated form of extracellular signal-regulated kinase (p-ERK) via a cAMP/PKA/Rap1/B-Raf/MEK pathway. Blockade of D2 DA receptors, β-adrenergic receptors or NMDA receptors with receptor-specific antagonists had no significant effect on this process. Activation of D1 DA receptors and PKA by DA caused phosphorylation and inactivation of the striatal–enriched tyrosine phosphatase (STEP), an important phosphatase for the dephosphorylation and subsequent inactivation of p-ERK in striatum. Interestingly p-ERK was primarily retained in the cytoplasm, with only low amounts translocated to the nucleus. The scaffold protein β-arrestin2 interacted with both p-ERK and D1 DA receptor, triggering the cytosolic retention of p-ERK and inducing striatal neuronal apoptotic death. These data provide unique insight into a novel role of p-ERK in striatal neurodegeneration.
Keywords: multiple system atrophy, D1 DA receptors, MAPKs, ERK, neurodegeneration, Parkinson’s disease
Introduction
The striatum plays a crucial role in motor control, memory and emotion regulation and is involved in the pathophysiology of several neuropathies. Striatal neurodegeneration is linked to several diseases, such as multiple system atrophy (MSA), L-DOPA-unresponsive parkinsonism subtype of multiple system atrophy (MSA-P subtype) (Ghorayeb I et al., 2000), methamphetamine-induced neurotoxicity (Stephans S and Yamamoto B, 1996; LaVoie MJ and Hastings TG, 1999), and secondary DA dysfunction in Huntington’s disease (Drago J et al., 1998), in which dopaminergic transmission is interrupted by progressive loss of the striatal neurons (Hantraye P, 1998). A characteristic feature of these neuropathies is the excessive accumulation and prolonged retention of DA in the nigrostriatal synapse (Calabresi P et al., 2000; Ghorayeb I et al., 2000; Nagatsu T, 2002). Normally, released DA is rapidly cleared and recycled into the presynaptic neurons by the DA transporter (Nelson N, 1998). In methamphetamine addictive states, synaptic DA accumulates through both blockade of DA transporter re-uptake and depletion of DA from presynaptic storage vesicles (Wang YM et al., 1997; Jones SR et al., 1998). Moreover, in DA transporter knock-out mice, sustained levels of extracellular DA in the striatal synapse causes selective degeneration of striatal neurons (Caron MG, 2003; Cyr M et al., 2003), highlighting the involvement of extracellular DA in striatal neurotoxicity.
In striatum, DA stimulates D1 and D2-like DA receptors on postsynaptic neurons. Physiological levels of DA promotes neuronal plasticity and exerts a neuroprotective role though receptor-dependent mechanism (Schapira, 2002; Yao et al., 2008a). For example, DA protects striatal neurons against glutamate-induced cell death, possibly through D1 DA receptor-mediated signaling (Amano et al., 1994), and apomorphine, a dopamine D1/D2 DA receptor agonist, protects neuronal cells against oxidative stress-induced apoptosis(Fornai et al., 2001; Hara et al., 2006). Further studies suggest that D2 DA receptors, more than D1 DA receptors, may mediate the protective action of DA (Bozzi and Borrelli, 2006). Indeed, activation of D2 DA receptors decreases glutamate-induced cytotoxicity in striatal neurons and the blockage of D2 DA receptors induces apoptosis in rat striatum and substantia nigra neurons (Cepeda et al., 1998; Mitchell et al., 2002). However, in pathological conditions, high level of DA, retained in the synapse, causes neurotoxicity through both activation of D1 DA receptors and its autoxidation to toxic products (Wersinger et al., 2004; Bozzi and Borrelli, 2006). The D1 DA receptors have a central role in the genesis and/or maintenance of Parkinson’s Disease [PD], as well as in hypersensitive responses to L-DOPA. Accumulating evidence in primates indicate that blockade of D1 DA receptors with antagonists has a strong neuroprotective and anti-Parkinsonian effect (Andringa G et al., 1999; Cools AR et al., 2002). Other studies have shown that inhibition of D1 DA receptors with its antagonist, SCH 23390, attenuates methamphetamine-induced neurotoxicity (Sonsalla PK et al., 1986; O’Dell SJ et al., 1993; Bronstein DM and Hong JS, 1995; Takaki M et al., 2001). In both rat striatal primary cultures (Wersinger C et al., 2004) and in SK-N-MC neuroblastoma cells (Chen J et al., 2003), which endogenously express D1 DA receptors, chronic treatment of these cells with DA induces cell death through oxidative and nitrative stress-dependent mechanisms. The neurotoxic effects of DA occur through two distinct pathways: the direct activation of the D1 DA receptor and the autoxidation of DA to toxic metabolites (Chen J et al., 2003). Accumulation of cAMP in SK-N-MC cells via activation of the D1 DA receptor preceded changes in ionic gradient, as measured by intracellular potassium concentration and leakage of cytochrome c into the cytosol, suggesting possible toxic events as a result of activation of the receptor (Moussa CE et al., 2006).
Mitogen-activated protein kinases (MAPKs), important mediators of signal transduction from the cell surface to the nucleus, have been implicated in a wide variety of cellular processes such as proliferation, differentiation, and apoptotic cell death (reviewed in Chang L and Karin M, 2001; Pearson G et al., 2001). In SK-N-MC neuroblastoma cells, we have found that DA promoted cytotoxicity through stimulation of D1 DA receptor and activation of the ERK-linked signaling cascade (Chen J et al., 2004). However, SK-N-MC cell is a transformed cell line, with survival mechanisms that may be not exist in the more complex milieu of primary striatal neurons. Current studies were undertaken in primary striatal neurons to analyze MAPKs and the upstream and down-stream components of the ERK signaling cascade. Our data demonstrates that DA, acting through D1 DA receptors, induces a sustained stimulation of ERK via a cAMP/PKA/Rap1/B-Raf/MEK pathway. In addition, the majority of p-ERK was retained in the cytoplasm, and not the nucleus, triggering striatal neuronal apoptotic death. This study suggests a novel mechanism of pERK activation in the genesis of striatal neurodegeneration after accumulation of extracellular DA.
EXPERIMENTAL METHODS
Materials
DA, hydrogen peroxide (H2O2), sodium metabisulfite (SMBS), SKF R-38393, SCH23390, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Brain-Derived Neurotrophic Factor (BDNF), spiperone, propranolol and MK801 were purchased from Sigma (St. Louis, MO). Wortmannin, genistein, mPKCi, KT5720, U0126 and SB203580 were from Calbiochem (San Diego, CA) and SB600125 was from Biomol Research Laboratories Inc. (Plymounth Meeting, PA).
Animals
Animal protocols were approved by the Georgetown University Animal Care and Use Committee. Female Sprague–Dawley rats were obtained from Harlan (Indianapolis, IN) and housed 2–3 per cage, fed and watered ad libitum, in the Georgetown Animal Care Facility until use. Female rats, gestation day 18, were decapitated, and embryos removed and used for the isolation of primary cultures.
Rat Primary Striatal Neuronal Culture and Treatment
Striata from 18 day old rat embryos were isolated, mechanically dissociated by gentle repetitive pipeting through a glass Pasteur pipette, and grown for 7 days as described previously by our group (Wersinger C et al., 2004). Neuronal cultures were exposed for the indicated time and appropriate concentrations of drugs, as described in the results section and legend to figures. Control neurons were treated with an equal concentration of solvent (0.2% H2O or DMSO).
Protein Analysis and Western Blotting
After appropriate treatments, neuronal cells were lysed in lysis buffer (20 mM Tris buffer pH 7.5 containing 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4) with protease inhibitor mixture (Roche Applied Science, Penzberg, Germany). Protein concentrations were determined by the Bradford method (Bradford MM, 1976). Immunoblot analysis was performed essentially as described previously (Wersinger C et al., 2004), using 20 μg/lane of protein loaded onto 10% SDS-PAGE gels. The following antibodies were used for Western blotting: p-ERK1/2(E-4) mouse monoclonal antibody (sc-7383), LaminB1 (C-20) goat polyclonal antibody (sc-6216), B-Raf (C-19) rabbit polyclonal antibody (sc-166), ERK2 (C-16) rabbit polyclonal antibody (sc-154) and Actin (H-196) rabbit polyclonal antibody (sc-7210) all from Santa Cruz Biotechnology (Santa Cruz, CA). JNK (#9252), p-JNK (Thr183/Tyr185) (81E11) (#4668) rabbit polyclonal antibody, p38 (#9212), p-p38 MAPK (Thr180/Tyr182) (3D7) (#9215) rabbit polyclonal antibody, MEK1/2 (#9122), p-MEK1/2 (Ser217/221) (#9121) rabbit polyclonal antibody (active from) and p-Elk-1 rabbit polyclonal antibody were bought from Cell Signaling Technology (Beverly, MA). STEP (23E5) mouse monoclonal antibody was from Upstate (Lake Placid, NY). Densitometric quantification of the immunoblots was performed using Scion Image (Scion Corporation, Frederick, MD).
Subcellular Fractionation
Cytosolic and nuclear extracts were prepared as described before (Chen et al., 2004). Briefly, After appropriate treatments, neurons were washed twice with ice-cold PBS and centrifuged at 1500 × g for 5 min. Cell pellets were resuspended in 200 μl of ice-cold lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.4% Nonidet P-40, 0.5 mM dithiothreitol, 0.5 mM PMSF, 1 mM sodium vanadate, 1 mM sodium fluoride), by gently pipetting up and down 10 times, followed by incubation on ice for 5 min. The lysate was centrifuged at 500 × g for 5 min to separate crude nuclei that were further purified as described below. The supernatant was transferred to anew tube. For cytosol preparation, the supernatant was centrifuged at 16,000 × g for 15 min. The crude nuclei were washed with 500 μl of lysis buffer and resuspended in 200 μl of nuclear extraction buffer(20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM dithiothreitol, 0.5 mM PMSF, 1 mM sodium vanadate, 1 mM sodium fluoride), vigorously shaken at 4 °C for 15 min, centrifuged at 16,000 × g for 15 min, and the supernatant (nuclear extracts) was transferred to a new tube. The purity of the nuclear and cytoplasmic extracts was assessed by immunoblotting the control cell extracts with the nuclear Lamin B and cytoplasmic HSP90 Abs.
Measurement of ERK Kinase Activity
ERK activity was measured by using the kinase assay kit from Cell Signaling Technology (Beverly, MA). Briefly, clarified neuronal cell lysates (200 μg), prepared as described above, were incubated overnight at 4°C on an orbital shaker with immobilized p-ERK monoclonal antibody (Cell Signaling Technology, Beverly, MA) to selectively immunoprecipitate pERK. Immobilized immune complexes were pelleted and washed twice with lysis buffer and kinase buffer, according to the manufacturer’s protocol. The kinase reaction was carried out at 30°C for 30 min in kinase buffer containing 200 μM ATP and 2 μg of GST-Elk-1 fusion protein, a specific p-ERK substrate. The reaction was terminated by the addition of SDS-sample buffer and boiling for 5 min, and analyzed by immunoblotting with phospho-specific Elk-1 (Ser383) antibody (Cell Signaling Technology, Beverly, MA) and densitometric quantification of the immunoblots was performed using Scion Image (Scion Corporation, Frederick, MD).
Cell Viability and Apoptosis Detection
Neuronal cell viability was measured by MTT assay following standard methods (Hussain RF et al., 1993). DNA fragmentation was detected by using Suicide-Track™ DNA ladder isolation Kit (Calbiochem, San Diego, CA). Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) staining was performed using a TACS apoptosis detection kit (Trevigen, Gaithersburg, MD). Briefly, striatal neurons grown on glass coverslips were fixed with 3% paraformaldehyde and permeabilized with 0.1 % Triton X-100, and processed for TUNEL staining (green). Hoechst (1:10000, Sigma) was added to stain neuron nuclei. Photomicrographs from 4–6 different fields in each coverslip were captured. Typically, ~300 cells were analyzed for the number of TUNEL-positive (apoptotic) cells. Total numbers of neurons were counted by Hoechst (Blue) staining. Apoptotic cell numbers were presented as a percentage of TUNEL-positive cells in relation to total cell numbers.
Assays of Rap1 activation and B-Raf Kinase activity
Rap1 activation was determined by using a Rap1 activation assay kit (Upstate, Lake Placid, NY). This assay uses a GST-fusion protein containing the Rap1 binding domain of human Ra1GDS to affinity precipitate active Rap1 (GTP-Rap1) from cell lysates. The pulled-down active or GTP-Rap1 is detected by Western blot analysis using a specific Rap1 antibody. Briefly, striatal neurons were treated, lysed and incubated with Rapl GDS-RBD pre-coupled to glutathione–agarose beads. GTP-bound Rap1 was eluted from beads analyzed by Western blotting. In addition, 20 μg of cell lysates were immunbloted for the total amount of Rap1. Kinase activities of B-Raf were measured by using B-Raf Kinase assay kit (Upstate, Lake Placid, NY). In brief, B-Raf was immunoprecipitated by B-Raf (C-19) rabbit polyclonal antibody (sc-166) (Santa Cruz Biotechnology) and incubated with Magnesium/ATP cocktail and GST-MEK-1 fusion protein at 30 °C for 30 min in a kinase reaction buffer. The samples were immunobloted with p-MEK rabbit polyclonal antibody ((#9121, Cell Signaling Technology), followed by reprobing with anti-B-Raf. The experiments demonstrated were repeated at three times with similar results.
Immunoprecipitation
Striatal neuronal cells were lysed and solubilized with 1% sodium cholate (Sigma), as described previously (Sidhu A et al., 1998). To the soluble extracts (200–400 μl/assay tube, 1–1.5 mg/ml protein), either of the following antiserum was added (1:100): Anti-Phosphoserine (AB1603) rabbit polyclonal (Chemicon, Temecula, CA) or p-ERK1/2 (Thr202/Tyr204) rabbit monoclonal (#9101)) (Cell Signaling Technology, Inc. Beverly, MA) or pan-β-arrestin rabbit polyclonal (PA1-732) (Affinity BioReagents, Golgen, CO) or D1 DA receptors mouse monoclonal (Luedtke et al., 1999). After overnight incubation, immune complexes were precipitated with protein A-Sepharose beads (CL-4B, Amersham Biosciences, Piscataway, NJ). Pellets were washed five times and subjected to 10% SDS-PAGE gels for Western blotting. Blots were probed with antibodies (1:1000) for either STEP (23E5) mouse monoclonal (05-730) (Upstate, Lake Placid, NY) or p-ERK1/2 mouse monoclonal (sc- sc-7383) (Santa Cruz Biotechnology) or β-arrestin2 (H-9) mouse monoclonal (sc-13140) (Santa Cruz Biotechnology) or D1 DA receptors mouse monoclonal. Proteins were visualized by using peroxidase-conjugated secondary antibodies (1:5000; Santa Cruz Biotechnology) and enhanced chemiluminescence (Amersham, Arlington Heights, IL).
Immunofluorescent Microscopy
Striatal cultures grown on glass coverslips were fixed with 3% paraformaldehyde and permeabilized with 0.1 % Triton X-100 in D-PBS, as described previously (Wersinger C et al., 2004). Cells were incubated overnight at 4°C with gentle shaking with 1:300 dilutions (in DPBS containing 0.1 % Triton X-100 and 1 % BSA) of rabbit polyclonal antisera against phospho-ERK1/2 (#9101) (Cell Signaling Technology, Beverly, MA), and for 2 h at room temperature with Alexa® Fluor® 568-conjugated goat anti-rabbit secondary antibodies (A-11036) (1:500; Molecular Probes, Eugene, OR). Cells were also stained with the DNA-specific fluorescent dye bisbenzamide (Hoechst) (1:10000, Sigma) to label nuclei and for counting of total cells. Cells were analyzed with a Nikon Eclipse E800 microscope equipped with a Nikon DXM1200 digital camera. Images from five independent fields for each experiment (n = 3 experiments for each treatment) were obtained using 20 or 40× objective with numerical aperture 0.5 and 0.75 respectively. The percentage of phospho-ERK1/2-positive cells was calculated by comparison with total Hoechst-stained cells.
Analysis of Data
All statistical analyses values were accomplished using Instat Statistical Software (Graphpad, Sorrento Valley, CA, U.S.A). Statistical comparisons were performed by using Student’s t test. Values of p<0.05 were considered statistically significant.
Results
DA Induces Phosphorylation of MAP kinases in rat striatal neuron
Striatal neurons were incubated for 20 minutes with increasing concentrations of DA, H2O2 or SKF R-38393, a D1 DA receptor agonist, and expression patterns of phosphorylated MAP kinases, p-ERK1/2, p-JNK and p-P38 MAPK, were examined. Both DA and SKF R-38393 caused a strong increase in p-ERK1/2 in a dose-dependent manner (Fig. 1A), and a substantial increase of 361 ± 45.2 % or 243 ± 37.4 %, respectively, was observed with 10 μM of DA or SKF R-38393. By contrast, H2O2 enhancement of p-ERK1/2 was weak (Fig. 1A), and at 10 μM of this compound, a modest increase of only 78% was seen. On the other hand, the increase in p-JNK levels by DA and H2O2 was equivalent (241% and 225%, respectively), while SKF R-38393 failed to increase p-JNK expression (Fig. 1B). DA and H2O2 also activated p-p38 MAPK to similar levels (126% and 142%, respectively), while SKF R-38393 was without any effect (Fig. 1C). These data suggest that direct stimulation of D1 DA receptors, through SKF R-38393 and partly via DA, causes selective activation of p-ERK, while H2O2 induces phosphorylation of p-38 MAPK and JNK.
Fig. 1. DA, H2O2 or SKF R-38393 induced phosphorylation of MAPKs in striatal neurons.


Neurons were treated for 20 min with 0–10 μM of DA, H2O2 or SKF R-38393, and Western blot analyses were conducted to probe for p-ERK1/2 [A], p-JNK [B] and p-p38 MAPK [C]. D. Time-course of p-ERK1/2 expression in striatal neurons treated with 10 μM of either DA or SKF R-38393. E. Time-course of p-ERK1/2 kinase activity in striatal neurons treated with 10 μM of either DA or SKF R-38393. F. Time course of p-MEK expression in striatal neurons treated with 10 μM of either DA or SKF R-38393. Equal levels of ERK2 expression were confirmed by Western blot (Data not shown). Blots were scanned and values expressed as the average ± S.E.M of three experiments (n=3). p<0.05 (*), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding DA treatment group; p<0.05 (#) and p<0.01 (##), represent the result of paired Student’s t test with 4 degrees of freedom for the corresponding vehicle control group.
To further examine the activation of p-ERK1/2 by D1 DA receptors in striatal neurons, the time-course of p-ERK1/2 production was examined. Increases in p-ERK1/2 levels were detected 5 min after addition of DA or SKF R-38393, reaching maximal levels after 20 min (326% and 268% for DA and SKF R-38393, respectively, p<0.01), and this increase was sustained for several hours (Fig. 1D). In general, DA-induced increase in p-ERK1/2 was higher than that seen with SKF R-38393, and it is likely due to the fact that the latter is a partial D1 DA receptor agonist (Sidhu A and Fishman PH, 1990). We next investigated whether the increased p-ERK1/2 was functionally active by measuring its ability to phosphorylate its substrate, Elk-1. Both DA and SKF R-38393 caused a time-dependent increase in p-Elk-1 levels (Fig. 1E), consistent with activate p-ERK1/2 (Fig. 1D). Since phosphorylation of ERK1/2 is exclusively regulated by MEK1/2, its upstream dual specificity kinase, we measured p-MEK1/2 levels, and found a time-dependent increase in p-MEK levels, reaching maximal levels after 20 min (307% and 214% for DA and SKF R-38393, respectively, p<0.01), and gradually subsiding after that (Fig. 1F), paralleling the increase and decrease in p-ERK1/2 expression (Fig. 1D).
The intracellular pathway for activating ERK1/2 upon DA treated striatal neurons
To investigate the mechanism involved in DA activation of ERK1/2, we treated neurons with either DA or SKF R-38393, in the presence or absence of the D1 DA receptor antagonist, SCH 23390; the antioxidant, SMBS; the PKA inhibitor, KT5720; and the MEK1/2 inhibitor, U0126. The increase in p-ERK1/2 levels induced by DA was completely blocked upon co-incubation with SCH 23390, but not with SMBS; SCH 23390 or SMBS alone had no effect on p-ERK1/2 expression levels (Fig. 2A). SKF R-38393-induced increase of p-ERK1/2 levels was also blocked by SCH 23390, but not by SMBS (Fig. 2B). These data further support the involvement of D1 DA receptors in mediating p-ERK1/2 activation in striatal neurons. Moreover, both KT5720 and U0126 blocked the DA-induced activation of ERK, attesting to the requirement for PKA and MEK kinases in this process (Fig. 2B).
Fig. 2. DA induced the ERK activation via D1 DA receptor and PKA dependent signaling pathway.

Striatal neurons were pretreated for 15 min with vehicle or with the following compounds: SCH 23390 (1 μM), SMBS (20 μM), KT5720 (1 μM) or U0126 (1 μM), and then treated with 10 μM of either DA [A] or SKF R-38393 [B] for 20 min. Immunoblot analysis of p-ERK1/2 expression was conducted; data shown are the average ± SEM of three independent experiments (n=3). In bar graph, P<0.05 (*), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding agonist (DA or SKF R-38393) alone-treated group; p<0.05 (#), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding control group. C. Immunocytochemistry of p-ERK1/2 labeling in striatal neurons up treatment with DA, H2O2 or SKF R-38393. Neuronal cultures were pretreated for 15 min with 1 μM each of either SCH 23390, KT5720 or U0126, or 20 μM of SMBS, prior to the addition of 10 μM of either DA, H2O2, SKF R-38393, or vehicle (0.2 % H2O) for 20 min. Immunocytochemical studies were conducted as described in Experimental Procedures. Scale bar, 100 μm.
Immunofluorescent studies confirmed our Western blot results. Treatment with 10μM DA significantly increased p-ERK1/2 immunolabelling in neurons (57.8±9.9%, p<0.05), which was substantially reduced in the presence of either SCH 23390 (12.2±5.1%, p<0.05), KT5720 (10.2±5.7%, p<0.05) or U0126 (14.5±5.9%, p<0.05), but not after pretreatment with the antioxidant SMBS (50.1±7.4%, p<0.05) (Fig. 2C, upper lane). Treatment with 10 μM H2O2 caused only a small increase in p-ERK1/2 expression (17.2±5.9%), which was not significantly different from p-ERK1/2 immunoreactivity in untreated neuronal cultures (7.2±4.3%) (Fig. 2C, middle lane). Incubation with 10 μM SKF R-38393 substantially increased p-ERK1/2 immunoreactivity in striatal neurons (48.7±8.9%, p<0.05), which was reversed by pretreatment with SCH 23390 (13.6±6.7, p<0.05), KT5720 (to 16.5±7.1%, p<0.05), or U0126 (to 9.2±6.1, p<0.05). By contrast, SMBS was devoid of any effect on SKF R-38393-induced increase in p-ERK1/2 staining, as 44.2±9.2% of total neurons were immunopositive for p-ERK1/2 in SKF R-38393 + SMBS treated neurons (Fig. 2C, lower lane). Taken together, these data provide further support for the participation of D1 DA receptors, PKA and MEK kinases in the DA-induced activation of p-ERK in striatal neurons.
ERK1/2 activation requires phosphorylation by the dual-specific kinase, MEK1/2, which isphosphorylated and activated by the Raf kinases(Houslay MD and Kolch W, 2000). In mammals, the Raf family is comprised of three members: Raf-1, A-Raf and B-Raf. Compared to other Raf proteins, B-Raf is a stronger activator of MEK1/2, and is highly expressed in neurons (Mercer KE and Pritchard CA, 2003). It has been reported that Rap1, a Ras-like small G protein, activates the ERK pathwayby activating B-Raf, while Rap1 in turn is activated by membrane depolarization in a cAMP/PKA dependent manner (reviewed in (Grewal SS et al., 2000). To test the possibility that Rap1/B-Raf participates in this signaling pathway, we investigated the activation of these proteins. Stimulationof striatal neurons with DA or SKF R-38393 for 10 min induced a large increase in the amount of Rap1-GTP (217% and 156% for 10 μM of DA and SKF R-38393, respectively, p<0.05) (Fig. 3A) and in B-Raf kinase activity (258% and 187% for 10 μM of DA and SKF R-38393, respectively, p<0.05) (Fig. 3B). Next we investigated B-Raf kinase activity in the presence of specific inhibitors. SKF-induced B-Raf kinase activity was significantly blocked by SCH23390 and KT5720 (p<0.05), but not by SMBS; SCH23390, KT5720 and SMBS alone had no effect on B-Raf kinase activity (Fig. 3C). These data demonstrate that in rat striatal neurons Rap1 and B-Raf are activated upon stimulation of D1 DA receptor in a PKA-dependent manner.
Fig. 3. Rap1 and B-Raf are activated in DA and SKF R-38393-treated strital neurons via D1 receptor and PKA pathway.

Neurons were treated for 10 min with 10 μM and 50 μM of DA or SKF R-38393. Rap1 activation [A] and B-Raf kinase activity [B] were conducted as described in Experimental Procedures. C. Striatal neurons were pretreated for 15 min with vehicle or with the following compounds: SCH 23390 (1 μM), SMBS (20 μM), KT5720 (1 μM), then treated with 10 μM of SKF R-38393 for 10 min. B-Raf kinase activity analyses were conducted. In bar graph, blots were scanned and values expressed as the average ± S.E.M of three experiments (n=3). p<0.05 (#), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding control group.
Striatal neurons express D2-like DA receptors, other catecholamine receptors and NMDA receptors which may also be activated by high levels of DA (reviewed in (Onn SP et al., 2000). To investigate whether other receptors also participated in D1 DA receptor induced p-ERK activation, we used different receptor-selective antagonists in the presence of DA or SKF R-38393. The increase in p-ERK1/2 levels by either DA or SKF R-38393 was not blocked upon co-incubation with D2-like receptor-selective antagonist, spiperone, β-adrenergic receptor antagonist, propranolol or NMDA receptor antagonist, MK801 (Fig. 4, A and B), and the inhibitors alone have no effect on p-ERK1/2 expression (Fig. 4, A and B).
Fig. 4. D2-like DA receptors, β-adrenergic receptors and NMDA receptors are not required in DA and SKF R-38393-induced ERK activation in striatal neurons.
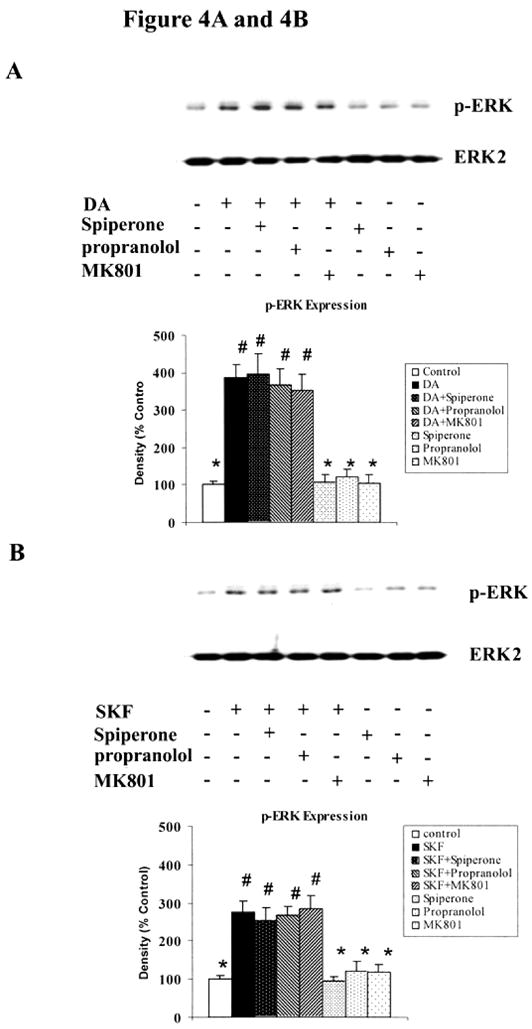
Striatal neurons were pretreated for 15 min with vehicle or with the following compounds: spiperone (10 μM), propranolol (10 μM) or MK801 (10 μM), and then treated with 10 μM of either DA [A] or SKF R-38393 [B] for 20 min. Immunoblot analysis of p-ERK1/2 expression was conducted; data shown are the average ± SEM of three independent experiments (n=3). In bar graph, P<0.05 (*), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding agonist (DA or SKF R-38393) alone-treated group; p<0.05 (#), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding control group.
In striatal neurons, p-ERK is rapidly inactivated by several phosphatases, including the striatal–enriched protein tyrosine phosphatase (STEP) (Paul S et al., 2000). Treatment of striatal neurons with DA or SKF R-38393 for 15 min substantially increased the phosphorylation of STEP61 (Fig. 5, A and B); phosphorylation of STEP61 causes a small upward mobility shift (Paul S et al., 2003). SKF R-38393-induced phosphorylation of STEP61 was significantly prevented by pretreatment with SCH 23390 or KT5720 (p<0.05), but not upon pretreatment with SMBS, the PKC inhibitor mPKCi, the PI3 kinase inhibitor wortmannin or the protein tyrosine kinase inhibitor genistein (Fig. 5B). Immunoprecipitation assays with phosphorylated serine antibody showed that STEP61 was phosphorylated on serine residues in DA or SKF R-38393-treated neurons (Fig. 5C). These data confirm previous findings showing that DA stimulation leads to the phosphorylation and inactivation of STEP (Paul S et al., 2000), a signaling event that reduces the dephosphorylation of p-ERK and prolongs its activation.
Fig. 5. DA and SKF R-38393 caused phosphorylation of STEP via D1 DA receptor and PKA dependent pathway.

A. Neurons were treated for 15 min with 10 μM and 50 μM of DA or SKF R-38393. STEP isoforms were immunobloted as described in Experimental Procedures with a monoclonal antibody against STEP, STEP(23E5). Phosphorylation of STEP (p-STEP) is indicated by the upward shift of the 61-KDa isoform (STEP61). B. Striatal neurons were pretreated for 15 min with vehicle or with the following compounds: mPKCi (1 μM), Wortmannin (1 μM), SCH23390 (1 μM), SMBS (20 μM), KT5720 (1 μM) or Genistein (10 μM) and then treated with 10 μM of SKF R-38393 for 15 min. Immunoblot analysis was conducted as described in Experimental Procedures. C. Neurons were treated for 15 min with 5 μM and 10 μM of DA or SKF R-38393. Immunoprecipitation was conducted by using phosphorylated serine (p-Serine) antibody and p-STEP in immunopellets was assessed by Western blots. In bar graph, blots were scanned for density. Data are expressed relative to control and represented as the average ± S.E.M of three experiments (n=3). In bar graph of B, left lane, p<0.05 (*), represents the result of paired Student’s t test with 4 degrees of freedom for SKF R-38393-treated group; p<0.05 (#), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding control group.
Activation of MEK/ERK kinase by DA promotes striatal neuronal cytotoxicity
Recent evidence has suggested that direct activation of D1 receptors accelerated cell death through oxidative/nitrative mechanisms in both SK-N-MC cells and striatal neurons (Chen J et al., 2003; Wersinger C et al., 2004). To assess the participation of the MAPKs in this process, striatal neurons were treated for 24 h with 10 μM of either DA or SKF R-38393, in the presence or absence of selective inhibitors of the various MAPKs, and cell viability and apoptotic cell counts were determined by the MTT assay, DNA ladder and TUNEL staining [see Methods]. Cells were also treated with H2O2 (10 μM) for 24 h to assess separately the involvement of the MAPKs in the oxidative pathway that is elicited by DA in our system. Chronic (24 h) treatment with 10 μM of DA, SKF R-38393, or H2O2 caused apoptotic striatal neuronal death (Fig. 6A, B and C). Incubation of striatal neurons with 10 μM of DA resulted in 27.6% neuronal loss (Fig. 6A), consistent with 26.7% TUNEL staining (apoptotic cells) (Fig. 6C). Chronic SKF R-38393 treatment also caused significant neuronal cell death (p<0.05), that was comparable in magnitude to that obtained with H2O2 treatment (~17% and 15% for H2O2 and SKF R-38393, respectively) (Fig. 6A), but was less than that obtained with DA treatment, consistent with the less DNA fragmentation (Fig. 6B) and TUNEL staining (Fig. 6C).
Fig. 6. MAPKs mediate DA-induced neuronal apoptotic death in striatal neurons.


A. Exposure of striatal neurons to either 10μM of DA, H2O2 or SKF R-38393 for 24 h. Cell viability (A), DNA fragmentation (B) and TUNEL staining (C) was conducted as described in Experimental Procedures. Values shown are the average ± S.E.M (n=5). p<0.05 (*), represents the result of paired Student’s t test with 8 degrees of freedom for the corresponding control group. In panels D, E, F and G, striatal neurons were exposed to 10 μM of DA, H2O2 or SKF R-38393 for 24 h, occurred in the presence (+) or absence (−) of either the selective p38 MAPK inhibitor SB203580 (D), or the selective JNK inhibitor SB600125 (E), or the selective MEK1/2 inhibitor U0126 (F), or the selective PKA inhibitor KT5720 (G). Neuronal viability was measured by the MTT cell viability assay. Kinase inhibitors (1 μM each) were added 15 min before treating the neurons. In panel H. Striatal neurons were pretreated for 15 min with vehicle or with 1 μM of SB203580, SB600125 or U0126, then treated with 10 μM of either DA, H2O2 or SKF R-38393 for 24 h. TUNEL staining was conducted. In bar graph, data are expressed relative to control, and represent as the average ± S.E.M of three independent experiments (n=3) for each treatment. p<0.05 (*), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding DA, H2O2 or SKF R-38393 treatment group.
SB203580 (1 μM), a selective inhibitor of the p38 MAPK, significantly reduced neuronal apoptotic death induced by both DA and H2O2 (p<0.05) (Fig. 6, D and H). In contrast, SB203580 caused no significant change in SKF R-38393-induced loss of neuronal viability (Fig. 6, D and H). Similarly, inhibition of p-JNK activity by its selective inhibitor, SB600125 (1 μM), had no significant effect on SKF R-38393-mediated neuronal apoptotic death, while significantly reducing neurotoxicity induced by either DA or H2O2 (Fig. 6, E and H).
Both U0126 or KT5720 (1 μM each) significantly (p<0.05) ablated DA or SKF R-38393-mediated neuronal cytotoxicity (Fig. 6, F, G and H), and restored neuronal cell viability after SKF R-38393 treatment to near control levels (Fig. 6, F and G). By contrast, H2O2-induced apoptotic neuronal death was not affected by the presence of the MEK1/2 or PKA inhibitors (Fig. 6, F, G and H). Taken together, these data suggest that the observed DA-mediated neurotoxicity is at least in part due to the direct stimulation of D1 receptors which activates the MEK/ERK signaling cascade via PKA.
Retention of p-ERK1/2 in the cytoplasm in DA-treated striatal neurons
Although activation of ERK pathway is widely believed to induce mitogenic responses, such as proliferation and differentiation, emerging data show that sustained stimulation of ERK signaling pathway is a toxic event promoting cell death (Oh-hashi K et al., 1999; Satoh T et al., 2000; Stanciu M et al., 2000; Kulich SM and Chu CT, 2001, 2003), as we have observed above. The precise cause of neurotoxicity by p-ERK is not clearly understood, but studies suggest that may be related to an abnormal cellular retention of pERK (reviewed in (Chu CT et al., 2004). Normally, p-ERK is rapidly translocated into the nucleus after activation, where it phosphorylates nuclear transcription factors, such as Elk-1, which induce cell cycle progression (reviewed in (Pouyssegur J et al., 2003).
We next explored the subcellular localization of p-ERK in DA and SKF R-38393 treated striatal neurons. Immunofluorescence of neurons showed only faint staining of p-ERK1/2 in control, untreated cells (Fig. 7A). Upon 20 min treatment with 10 μM of either DA or SKF R-38393, p-ERK1/2 immunoreactivity was found primarily in the cytoplasm of neurons, including neuritic processes, with modest levels of p-ERK1/2 seen in the nucleus, which was stained with Hoechst (Fig. 7A). To confirm these immunocytochemistry results, cell fractionation studies were conducted to quantify the amount of p-ERK1/2 present in cytoplasm and nuclei. After treatment with DA (Fig. 7B) or SKF R-38393 (Fig. 7C) for 20 min, total neuronal lysates were separated into cytoplasmic and nuclear fractions and p-ERK1/2 levels were assessed in both fractions. Although p-ERK1/2 levels were dose-dependently increased in both cytoplasm and nucleus after DA or SKF R-38393 treatment (Fig. 7, B and C), in all instances, a majority (~70%) of p-ERK1/2 was located in the cytoplasmic, with only modest level of p-ERK1/2 located in the nuclear fraction (Fig. 7, B and C), consistent with our immunofluorescence results.
Fig. 7. The subcellular distribution of p-ERK1/2 after treatment with DA and SKF R-38393.


A. Striatal neurons were treated for 20 min with 10 μM of either DA or SKF R-38393, and co-stained for p-ERK (red) and Hoechst (blue) immunoreactivity as described in Experimental Procedures. In panel B and C, p-ERK1/2 expression in cytoplasmic fraction and nuclear fraction was assessed after treatment with 0–10 μM of either DA [B] or SKF R-38393 [C]. In the bar graph, data are expressed relative to control, and represent the average ± S.E.M of three independent experiments (n=3) for each treatment. p<0.05 (*) and p<0.01 (**), represent the result of paired Student’s t test with 4 degrees of freedom for the corresponding cytoplasmic group. Panels D and E show the time-course of p-ERK1/2 expression in cytoplasmic fraction and nuclear fraction after 10 μM of DA [D] or SKF R-38393 [E] treatment. F. Time-course of p-ERK1/2 expression of cytoplasmic fraction and nuclear fraction after 50 ng/ml of BDNF treatment. Cytoplasmic and nuclear extracts were reblotted with anti-LaminB1 antibody (left lane) to demonstrate separation of the nuclear and cytosolic fractions.
To investigate the localization of p-ERK1/2 as a function of time, we conducted a time-course study and assessed p-ERK1/2 levels in cytoplasmic and nuclear fractions. DA or SKF R-38393 (10 μM) caused a time-dependent increase in p-ERK1/2 in both cytoplasmic and nuclear fractions, reaching maximal levels in 20 min (Fig. 7, D and E). The relative increase of p-ERK1/2 in the cytoplasm was significantly higher than that seen in the nuclei at all times (p<0.05) (Fig. 7, D and E). Cytoplasm p-ERK levels remained elevated for up to 2 h and then diminished gradually to control levels. To verify the ability of p-ERK to be appropriately translocated into the nucleus in these neurons, striatal neuronal cultures were treated with BDNF (50 ng/ml) for varying times and p-ERK1/2 levels in the cytoplasm and nucleus were examined. In contrast to our findings with DA and SKF R-38393, in BDNF-treated neurons the majority (67%) of p-ERK1/2 was rapidly translocated to the nuclei and only low levels (33%) of p-ERK1/2 remained in the cytoplasm (Fig. 7F). Moreover, this translocation reached maximal levels at 10 min, decreasing to control levels within 30 min. Cytoplasmic and nuclear extracts were reblotted with anti-Lamin B1 antibody (Fig. 7F, right panel), a nuclear protein, to demonstrate the appropriate cellular separation and lack of cross-contamination of the nuclear and cytoplasmic fractions.
p-ERK coupled with scaffolding protein β-arrestin
Upon activation, GPCRs are phosphorylated and then coupled to β-arrestin, resulting in endocytosis of the p-GPCR. Once internalized, β-arrestin acts as a scaffolding protein, binding to p-ERK to form a stable complex, leading to the cytosolic retention of the p-ERK (Oakley RH et al., 2000; Tohgo A et al., 2002; Tohgo A et al., 2003). Previously, it has been reported that the D1 receptor interacts with β-arrestin2 (Zhang J et al., 1999; Oakley RH et al., 2000). We analyzed whether stimulation of D1 receptors in striatal neurons also results in the scaffold of D1 DA receptor, β-arrestin2 and p-ERK. Striatal neurons were treated with DA (10 and 50 μM) or SKF R-38393 (10 and 50 μM) for 30 min, and co-immunoprecipitations (co-IP) were performed on cell lysates with antibodies against pan-β-arrestin (Fig. 8A), p-ERK (Fig. 8B) or the D1 DA receptors (Fig. 8C). In pan-β-arrestin immunopellets, both D1 receptors and p-ERK were detected in response to agonist treatment in a dose-dependent manner; there was an increase of ~70% and 40%, respectively, in D1 receptor co-immunoprecipitation (Fig. 8A, upper lane), and ~150% and 100% increase, respectively, in p-ERK1/2 co-immunoprecipitation (Fig. 8A, middle lane). In these immunopellets, β-arrestin2 was detected at similar levels in all fractions (Fig. 8A, lower lane), indicating that there were no differences in the amount of protein present in the various immunopellets. These data suggest that in striatal neurons, β-arrestin2 directly couples to both D1 DA receptors and p-ERK1/2 in response to DA or SKF R-38393 treatment. In pERK immunoprecipitated extracts, β-arrestin2 was detected in a dose-dependent manner (Fig. 8B), with an increase in protein levels of ~120% (β-arrestin2) and ~90% (p-ERK1/2) after DA (10 μM) treatment. An increase of ~90% (β-arrestin2) and ~70% (p-ERK1/2) was seen in SKF R-38393 (10 μM) treated neuronal extracts. However, only a modest amount of D1 receptors was observed in these immunopellets (~35% and 25% for DA and SKF R-38393 treatments, respectively. Finally, in co-IPs using D1 receptor antibodies, increased levels of β-arrestin2 (~90% and 60%, respectively, after DA and SKF R-38393 treatment, respectively) were detected in the immunopellets in a dose-dependent manner (Fig. 8C). Only modest levels of p-ERK were observed (~30% and 25% after DA and SKF R-38393 treatments, respectively), These results indicate that upon stimulation of D1 receptors with DA or SKF R-38393, β-arrestin2 coupled with D1 DA receptors and p-ERK1/2 in striatal neurons.
Fig. 8. p-ERK is retained in the cytoplasm in a heterotrimeric complex.

Striatal neurons treated with DA (10 and 50 μM) or SKF R-38393 (10 and 50 μM) for 30 min. Co-Immunoprecipitation were conducted on cell lysates with antibodies against pan-β-arrestin (A), p-ERK (B) or the D1 DA receptor (C), then immunobloted with β-arrestin2, p-ERK or the D1 receptor antibody. In bar graph, blots were scanned for density. Data are expressed relative to control and represented as the average ± S.E.M of three experiments (n=3). p<0.05 (#), represents the result of paired Student’s t test with 4 degrees of freedom for the corresponding control group.
Discussion
The results presented here provide novel insight into the mechanisms of striatal neurotoxicity induced upon stimulation of the D1 DA receptor, while providing the first direct evidence linking D1 DA receptors to apoptotic cell death. Indirect evidence that D1 receptors may cause apoptosis was recently demonstrated in vivo when SCH 23390 was found to alleviate caspase-3 expression after perinatal asphyxia or prenatal cocaine exposure (Mitchell ES and Snyder-Keller A, 2003). More recently, SCH 23390 or the D2-like DA receptor antagonist, raclopride, blocked methamphetamine-induced apoptotic striatal cell death (Xu W et al., 2005). Our data shows that direct stimulation of the D1 DA receptors at pathological levels of DA, will cause substantial apoptosis in striatal neuronal cultures.
In addition, these studies also provide evidence for a lesser-known role of ERK in the early events underlying neuronal degeneration. Previously, we had shown in SK-N-MC cells that DA accelerated cell death through two distinct pathways: its autoxidation to free radicals and through the direct activation of D1 DA receptors (Chen et al., 2003; Chen et al., 2004). Consistent with this, and similar to the effects elicited with H2O2, the autoxidation of DA caused activation of both p-JNK and p-P38 MAPK, which was blocked by the antioxidant SMBS. The failure of SMBS to alter DA-induced activation of p-ERK1/2, and the ability of SCH 23390 to completely suppress DA-mediated activation of p-ERK1/2, indicates that activation of D1 DA receptors induces toxicity via the p-ERK1/2 signaling cascade, an event also mimicked by SKF R-38393 (Chen et al., 2004). However, in SK-N-MC cells, we could not eliminate a possible participation of D2-like and/or catecholaminergic receptors, since SK-N-MC cells do not express such receptors. Thus, our findings in neurons, rather than SK-N-MC cells, are likely to more accurately reflect the pathophysiology in vivo, and highlight the limited information gleaned from cultured cell lines, while underscoring the necessity of conducting studies in neuronal cultures.
The concentrations of DA used in this study more closely mimic those seen in pathological conditions, where concentrations of striatal DA have been found to be in the 1–10 μM range (Chen and Reith, 1998; Jones et al., 1998; Spielewoy et al., 2000), which is much higher than physiological levels of DA (~0.1 μM)(Spielewoy et al., 2000; Robinson et al., 2002). The physiological function of DA is involved in cognition, motivation and motor control (Schapira, 2002; Yao et al., 2008a). Stimulation of D1 DA receptor is involved in neuronal protection and neuronal plasticity (Amano et al., 1994; Yao et al., 2008b), which is mediated via the interaction of D1 receptors and NMDA receptors(Scott et al., 2002; Cepeda and Levine, 2006). However, under pathological conditions, high levels of DA accumlates in the synapse, causing neurotoxicity through the activation of D1 DA receptors and its own autoxidation (Wersinger et al., 2004; Bozzi and Borrelli, 2006). Indeed, it is now widely accepted that blockade of D1 DA receptors reduces methamphetamine-induced neurotoxicity (Bronstein DM and Hong JS, 1995; Cadet JL et al., 1998; Takaki M et al., 2001), but the underlying mechanisms remain unknown.
Activation of the ERK signaling cascade by DA receptors is a complex process that may involve multiple mechanisms (Zhen X et al., 2001; Brami-Cherrier K et al., 2002; Gerfen CR et al., 2002; Liu JC et al., 2002). D1 DA receptors exert their actions by stimulating cellular cAMP/PKA second messenger-activated protein kinase via Gs/olf protein coupled receptor pathway (Sidhu A and Niznik HB, 2000; Corvol JC et al., 2001). Previous studies show that activation of ERK1/2 occurs via a Gs/cAMP/PKA-dependent pathway (Vossler MR et al., 1997; Schmitt JM and Stork PJ, 2002). In primary neuronal cells or a neuronal model, PC12 cells, cAMP induces ERK phosphorylation via the small GTPase, Rap1, which is activated by PKA and interacts with B-Raf, a strong activator for MEK, which then causes sustained ERK phosphorylation (Vossler MR et al., 1997; Dugan LL et al., 1999). The fact that inhibition of PKA completely blocked D1 receptor-induced ERK activation and neurotoxicity in straital neurons indicates the participation of the cAMP/PKA-dependent pathway in these processes. In addition, the stimulation of D1 receptors caused substantial activation of Rap1 and B-Raf in striatal neurons, leading to MEK phosphorylation. Furthermore, these activations were blocked by D1 receptor antagonist and PKA inhibitor, suggesting the participation of the D1/cAMP/PKA cascade in this process. Thus, following DA or SKF R-38393 treatment, the subsequent sequence of events presumably take place: DA or SKF R-38393 stimulates D1 receptors, triggering the activation of cAMP/PKA, inducing Rap1 activation, which then causes B-Raf/MEK phosphorylation, leading to the phosphorylation of ERK. Previous studies suggest that activation of D2-like DA receptors also induce ERK phosphorylation though RAS/C-Raf/MEK pathway(Luo et al., 1998; Oak et al., 2001). It is possible that DA-induced ERK phosphorylation is mediated, at least in part, by D2 receptors. However, the failure of antagonists of D2-like DA receptors, β-adrenergic receptors and NMDA receptors to alter DA-induced ERK phosphorylation indicated that DA-induced ERK phosphorylation was restricted to D1 DA receptors. This may be due to the fact that B-Raf is a much stronger activator for MEK than C-Raf (Mercer KE and Pritchard CA, 2003; Mizuchi D et al., 2005), and after activation, B-Raf competes with C-Raf in the activation of the MEK/ERK pathway (Mercer KE and Pritchard CA, 2003). Thus, a strong activation of B-Raf signaling by D1/PKA/Rap1 cascade will inhibit C-Raf signaling, such that even if D2 receptors were activated by DA, there would be no overall effect by these receptors on ERK phosphorylation.
In the striatum, p-ERK is rapidly inactivated by several phosphatases, including STEP; STEP is a striatal–enriched protein tyrosine phosphatase, which is phosphorylated on serine residues via a DA/D1 receptor/cAMP-dependent PKA pathway (Paul S et al., 2000). When phosphorylated, STEP lose its ability to dephosphorylate ERK within its ERK-binding domain (Paul S et al., 2000). In this study, we examined phosphorylation of STEP in striatal neurons treated with DA or D1 receptor agonist. Since phosphorylation of STEP results in a small upward electromobility shift (Paul S et al., 2003), we first detected p-STEP by Western blots. Both DA and SKF R-38393 caused a robust increase in phosphorylated STEP. We also found that DA and SKF R-38393 induced an increase in total STEP expression, which may be a compensation for the robust increment of p-ERK. Because the electromobility shift of p-STEP was sometimes indistinguishable from unphosphorylated STEP in some western blots, such as control cells, we further investigated p-STEP though immunoprecipitation by using phosphorylated serine antibodies to immunoprecipitate, and STEP61 antibody to detect p-STEP61. Consistent with Western blotting results, immunoprecipitation data indicated that both DA and SKF R-38393 induced a robust phosphorylation of STEP61. These data confirm previous findings showing that DA stimulation leads to the phosphorylation and inactivation of STEP (Paul S et al., 2000), a signaling event that reduces the dephosphorylation of p-ERK and prolongs its activation.
ERK is involved in neuronal development, synaptic plasticity, survival and adaptation (Xia Z et al., 1995; English JD and Sweatt JD, 1996; Segal RA and Greenberg ME, 1996; Atkins CM et al., 1998; Davis RJ, 2000; Sweatt JD, 2001). In resting cells, ERK is retained in the cytoplasm by the microtubule cytoskeleton, which serves as a major docking matrix for up to 35% of cellular ERK1 and ERK2 (Reszka AA et al., 1995). Other putative cytosol-anchoring proteins of ERK are the MAP kinase phosphatases (MKPs), in particular MKP3 (Camps M et al., 1998; Brunet A et al., 1999). ERK is also retained in the cytoplasm by its association with MEK1/2 (Fukuda M et al., 1997). After its phosphorylation by MEK1/2, p-ERK1/2 detaches from its cytosolic anchor and rapidly translocates into the nucleus (Chen RH et al., 1992). Once in the nucleus, activated ERK can phosphorylate several nuclear transcription factors, such as Elk-1 (Gille H et al., 1995), thereby transmitting signals originally received from cell surface receptors to the nucleus. The rapid nuclear translocation of activated ERK is essential for promoting cell cycle progression (reviewed in Pouyssegur J et al., 2003).
There is increasing evidence to suggest that the activation of the ERK signaling cascade also induces neurotoxicity (Alessandrini A et al., 1999; Oh-hashi K et al., 1999; Perry G et al., 1999; Stanciu M et al., 2000; Kulich SM and Chu CT, 2001; Namura S et al., 2001; Stanciu M and DeFranco DB, 2002; Kulich SM and Chu CT, 2003). A possible mechanism of activated ERK causing neurotoxicity, rather than neuroprotection, may result from its sustained activation and abnormal subcellular location (reviewed in (Grewal SS et al., 1999; Colucci-D’Amato L et al., 2003; Chu CT et al., 2004).
Previous in vivo studies indicate an important role of D1 receptor-mediated ERK activation in striatal neurons. Administration of various abuse drugs, such as cocaine or amphetamine, causes transient and rapid nuclear translocation of p-ERK. The cocaine-induced ERK activation is totally blocked by the D1 antagonist SCH23390 (Valjent et al., 2000). While cocaine treatment causes the increase of DA in the synapse, which then stimulates D1 receptors, recent studies demonstrate that activation of ERK by drugs of abuse is mediated by an interaction between D1 and NMDA receptors(Valjent et al., 2005), suggesting that this rapid (after 5–10 min of cocaine administration) and transient (less than 1h) ERK activation may have a different mechanism compared to our study. Since activation of both D1 receptor and NMDA receptors are necessary for drug-induced ERK activation (Cepeda and Levine, 2006; Lu et al., 2006), and transactivation of NMDA receptors can induce transient and rapid nuclear translocation of p-ERK (Paul S et al., 2003), these data indicate that NMDA receptors may have a central role in cocaine-induced activation of ERK.
In the current study, we found that DA, acting through D1 DA receptors, caused sustained activation of ERK and that the majority of activated ERK was retained in the cytoplasm, with only modest amounts translocated into the nucleus. The failure of p-ERK to translocate into the nucleus, coupled with its sustained activation, provides a possible mechanism by which ERK activation may trigger a neurotoxic, rather than a survival response upon activation of the D1 DA receptor. If abnormally retained in the cytoplasm, p-ERK1/2 can phosphorylate multiple plasma membrane, cytoplasmic and cytoskeletal substrates (Pearson G et al., 2001), resulting in altered, and perhaps inappropriate, series of cellular events. Although it is unclear as to which of these substrates of ERK are mediating neurotoxicity, recently studies conducted in brain cells such as astrocytes (Brambilla R et al., 2002), glia (Ferrer I et al., 2001b; Canals S et al., 2003) and neurons (Ferrer I et al., 2001a; Levinthal DJ and DeFranco DB, 2004) consistently found that p-ERK1/2 activation was linked to enhanced cell death, suggesting an alternate role for p-ERK1/2 in mediating neurotoxicity in the brain. Clinically, several studies have demonstrated increased p-ERK1/2 levels associated with early tau deposition in Alzheimer’s and Picks disease, and in progressive supranuclear palsy and corticobasal degeneration (Ferrer I et al., 2001b), as well as in familial taupathy with progressive supranuclear palsy (Ferrer I et al., 2003). Increased cytosolic p-ERK1/2 activity may also have a role in neurotoxins associated with AIDS dementia (Pulliam L et al., 2001). Finally, p-ERK1/2, but not p-p38 MAPK or p-JNK, were found in Lewy bodies in Parkinson’s disease and in dementia with Lewy bodies (Ferrer I et al., 2001b). Clearly, elucidation of the precise mechanisms that cause neurotoxicity of p-ERK1/2 is likely to provide important insights into the etiology of certain neurodegenerative diseases.
Acknowledgments
This study was supported in part by grants from the National Institutes of Health [NS45326, MH52711, MH01527, and AG028108]. We thank Ms. Haala Khatija Rokadia for assistance in preparation of Western samples.
Abbreviations
- DA
dopamine
- MAPKs
mitogen activated protein kinases
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- JNK
c-Jun N-terminal kinase
- p38 MAPK
38 kDa mitogen-activated protein kinase
- MEK1/2
MAPK/ERK kinase 1 and 2
- PKA
protein kinase A
- MSA
multiple system atrophy
- STEP
striatal–enriched tyrosine phosphatase
Contributor Information
Jun Chen, Department of Pediatrics, Georgetown University, Washington D.C. 20007, USA.
Milan Rusnak, Department of Pediatrics, Georgetown University, Washington D.C. 20007, USA.
Paul J. Lombroso, The Child Study Center, Yale University School of Medicine, New Haven, Connecticut, 06520
Anita Sidhu, Department of Pediatrics, Georgetown University, Washington D.C. 20007, USA.
References
- Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci U S A. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano T, Ujihara H, Matsubayashi H, Sasa M, Yokota T, Tamura Y, Akaike A. Dopamine-induced protection of striatal neurons against kainate receptor-mediated glutamate cytotoxicity in vitro. Brain Research. 1994;655:61–69. doi: 10.1016/0006-8993(94)91597-0. [DOI] [PubMed] [Google Scholar]
- Andringa G, Stoof JC, Cools AR. Sub-chronic administration of the dopamine D(1) antagonist SKF 83959 in bilaterally MPTP-treated rhesus monkeys: stable therapeutic effects and wearing-off dyskinesia. Psychopharmacology (Berl) 1999;146:328–334. doi: 10.1007/s002130051124. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Bozzi Y, Borrelli E. Dopamine in neurotoxicity and neuroprotection: what do D2 receptors have to do with it? Trends Neurosci. 2006;29:167–174. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Neary JT, Cattabeni F, Cottini L, D’Ippolito G, Schiller PC, Abbracchio MP. Induction of COX-2 and reactive gliosis by P2Y receptors in rat cortical astrocytes is dependent on ERK1/2 but independent of calcium signalling. J Neurochem. 2002;83:1285–1296. doi: 10.1046/j.1471-4159.2002.01239.x. [DOI] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Garcia M, Pages C, Hipskind RA, Caboche J. Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: a new route to cAMP response element-binding protein phosphorylation. J Neurosci. 2002;22:8911–8921. doi: 10.1523/JNEUROSCI.22-20-08911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein DM, Hong JS. Effects of sulpiride and SCH 23390 on methamphetamine-induced changes in body temperature and lethality. J Pharmacol Exp Ther. 1995;274:943–950. [PubMed] [Google Scholar]
- Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouyssegur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet JL, Ladenheim B, Hirata H. Effects of toxic doses of methamphetamine (METH) on dopamine D1 receptors in the mouse brain. Brain Res. 1998;786:240–242. doi: 10.1016/s0006-8993(97)01432-7. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Marfia GA, Pisani A, Sancesario G, G B. Synaptic transmission in the striatum: from plasticity to neurodegeneration. Prog Neurobiol. 2000;61:231–265. doi: 10.1016/s0301-0082(99)00030-1. [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, Arkinstall S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998;280:1262–1265. doi: 10.1126/science.280.5367.1262. [DOI] [PubMed] [Google Scholar]
- Canals S, Casarejos MJ, de Bernardo S, Solano RM, Mena MA. Selective and persistent activation of extracellular signal-regulated protein kinase by nitric oxide in glial cells induces neuronal degeneration in glutathione-depleted midbrain cultures. Mol Cell Neurosci. 2003;24:1012–1026. doi: 10.1016/j.mcn.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Caron MG. Transporter mediated regulation of neural communication. J Neurochem. 2003;85(Suppl):2. [Google Scholar]
- Cepeda C, Levine MS. Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE. 2006;333:pe20. doi: 10.1126/stke.3332006pe20. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Gruen E, Levine MS. Dopaminergic modulation of early signs of excitotoxicity in visualized rat neostriatal neurons. Eur J Neurosci. 1998;10:3491–3497. doi: 10.1046/j.1460-9568.1998.00357.x. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Chen J, Wersinger C, Sidhu A. Chronic stimulation of D1 dopamine receptors in human SK-N-MC neuroblastoma cells induces nitric-oxide synthase activation and cytotoxicity. J Biol Chem. 2003;278:28089–28100. doi: 10.1074/jbc.M303094200. [DOI] [PubMed] [Google Scholar]
- Chen J, Rusnak M, Luedtke RR, Sidhu A. D1 dopamine receptor mediates dopamine-induced cytotoxicity via the ERK signal cascade. J Biol Chem. 2004;279:39317–39330. doi: 10.1074/jbc.M403891200. [DOI] [PubMed] [Google Scholar]
- Chen NH, Reith ME. In vivo microdialysis for measurement of extracellular monoamine levels following inhibition of monoamine transporters. Methods Enzymol. 1998;296:719–730. doi: 10.1016/s0076-6879(98)96051-7. [DOI] [PubMed] [Google Scholar]
- Chen RH, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem. 2004;271:2060–2066. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci-D’Amato L, Perrone-Capano C, di Porzio U. Chronic activation of ERK and neurodegenerative diseases. Bioessays. 2003;25:1085–1095. doi: 10.1002/bies.10355. [DOI] [PubMed] [Google Scholar]
- Cools AR, Lubbers L, van Oosten RV, Andringa G. SKF 83959 is an antagonist of dopamine D1-like receptors in the prefrontal cortex and nucleus accumbens: a key to its antiparkinsonian effect in animals? Neuropharmacology. 2002;42:237–245. doi: 10.1016/s0028-3908(01)00169-1. [DOI] [PubMed] [Google Scholar]
- Corvol JC, Studler JM, Schonn JS, Girault JA, Herve D. Galpha(olf) is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J Neurochem. 2001;76:1585–1588. doi: 10.1046/j.1471-4159.2001.00201.x. [DOI] [PubMed] [Google Scholar]
- Cyr M, Beaulieu JM, Laakso A, Sotnikova TD, Yao WD, Bohn LM, Gainetdinov RR, Caron MG. Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc Natl Acad Sci U S A. 2003;100:11035–11040. doi: 10.1073/pnas.1831768100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 2000;268:14553–14556. [PubMed] [Google Scholar]
- Drago J, Padungchaichot P, Wong JY, Lawrence AJ, McManus JF, Sumarsono SH, Natoli AL, Lakso M, Wreford N, Westphal H, Kola I, DIF Targeted expression of a toxin gene to D1 dopamine receptor neurons by cre-mediated site-specific recombination. J Neurosci. 1998;18:9845–9857. doi: 10.1523/JNEUROSCI.18-23-09845.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan LL, Kim JS, Zhang Y, Bart RD, Sun Y, Holtzman DM, Gutmann DH. Differential effects of cAMP in neurons and astrocytes. Role of B-raf. J Biol Chem. 1999;274:25842–25848. doi: 10.1074/jbc.274.36.25842. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Puig B, Barrachina M, Gomez C, Ambrosio S. Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson’s disease and Dementia with Lewy bodies. J Neural Transm. 2001a;108:1383–1396. doi: 10.1007/s007020100015. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Hernandez I, Boada M, Llorente A, Rey MJ, Cardozo A, Ezquerra M, Puig B. Primary progressive aphasia as the initial manifestation of corticobasal degeneration and unusual tauopathies. Acta Neuropathol (Berl) 2003;106:419–435. doi: 10.1007/s00401-003-0756-4. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, Rey MJ, Cardozo A, Vinals F, Ribalta T. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 2001b;11:144–158. doi: 10.1111/j.1750-3639.2001.tb00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornai F, Battaglia G, Gesi M, Orzi F, Nicoletti F, Ruggieri S. Dose-dependent protective effects of apomorphine against methamphetamine-induced nigrostriatal damage. Brain Res. 2001;898:27–35. doi: 10.1016/s0006-8993(01)02125-4. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Gotoh Y, Nishida E. Interaction of MAP kinase with MAP kinase kinase: its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J. 1997;16:1901–1908. doi: 10.1093/emboj/16.8.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Miyachi S, Paletzki R, Brown P. D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci. 2002;22:5042–5054. doi: 10.1523/JNEUROSCI.22-12-05042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorayeb I, Fernagut PO, Aubert I, Bezard E, Poewe W, Wenning GK, Tison F. Toward a primate model of L-dopa-unresponsive parkinsonism mimicking striatonigral degeneration. Mov Disord. 2000;15:531–536. doi: 10.1002/1531-8257(200005)15:3<531::AID-MDS1017>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH, Shaw PE. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951–962. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Grewal SS, Horgan AM, York RD, Withers GS, Banker GA, Stork PJ. Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase. J Biol Chem. 2000;275:3722–3728. doi: 10.1074/jbc.275.5.3722. [DOI] [PubMed] [Google Scholar]
- Hantraye P. Modeling dopamine system dysfunction in experimental animals. Nucl Med Biol. 1998;25:721–728. doi: 10.1016/s0969-8051(98)00054-7. [DOI] [PubMed] [Google Scholar]
- Hara H, Ohta M, Adachi T. Apomorphine protects against 6-hydroxydopamine-induced neuronal cell death through activation of the Nrf2-ARE pathway. J Neurosci Res. 2006;84:860–866. doi: 10.1002/jnr.20974. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Kolch W. Cell-type specific integration of cross-talk between extracellular signal-regulated kinase and cAMP signaling. Mol Pharmacol. 2000;58:659–668. [PubMed] [Google Scholar]
- Hussain RF, Nouri AM, Oliver RT. A new approach for measurement of cytotoxicity using colorimetric assay. J Immunol Methods. 1993;160:89–96. doi: 10.1016/0022-1759(93)90012-v. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulich SM, Chu CT. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: implications for Parkinson’s disease. J Neurochem. 2001;77:1058–1066. doi: 10.1046/j.1471-4159.2001.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulich SM, Chu CT. Role of reactive oxygen species in extracellular signal-regulated protein kinase phosphorylation and 6-hydroxydopamine cytotoxicity. J Biosci. 2003;28:83–89. doi: 10.1007/BF02970136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinthal DJ, DeFranco DB. Transient phosphatidylinositol 3-kinase inhibition protects immature primary cortical neurons from oxidative toxicity via suppression of extracellular signal-regulated kinase activation. J Biol Chem. 2004;279:11206–11213. doi: 10.1074/jbc.M314261200. [DOI] [PubMed] [Google Scholar]
- Liu JC, Baker RE, Sun C, Sundmark VC, Elsholtz HP. Activation of Go-coupled dopamine D2 receptors inhibits ERK1/ERK2 in pituitary cells. A key step in the transcriptional suppression of the prolactin gene. J Biol Chem. 2002;277:35819–35825. doi: 10.1074/jbc.M202920200. [DOI] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006;29:695–703. doi: 10.1016/j.tins.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Luedtke RR, Griffin SA, Conroy SS, Jin X, Pinto A, Sesack SR. Immunoblot and immunohistochemical comparison of murine monoclonal antibodies specific for the rat D1a and D1b dopamine receptor subtypes. Journal of Neuroimmunology. 1999;101:170–187. doi: 10.1016/s0165-5728(99)00142-3. [DOI] [PubMed] [Google Scholar]
- Luo Y, Kokkonen GC, Wang X, Neve KA, Roth GS. D2 dopamine receptors stimulate mitogenesis through pertussis toxin-sensitive G proteins and Ras-involved ERK and SAP/JNK pathways in rat C6-D2L glioma cells. J Neurochem. 1998;71:980–990. doi: 10.1046/j.1471-4159.1998.71030980.x. [DOI] [PubMed] [Google Scholar]
- Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- Mitchell ES, Snyder-Keller A. Blockade of D1 dopaminergic transmission alleviates c-fos induction and cleaved caspase-3 expression in the brains of rat pups exposed to prenatal cocaine or perinatal asphyxia. Exp Neurol. 2003;182:64–74. doi: 10.1016/s0014-4886(03)00026-8. [DOI] [PubMed] [Google Scholar]
- Mitchell IJ, Cooper AC, Griffiths MR, Cooper AJ. Acute administration of haloperidol induces apoptosis of neurones in the striatum and substantia nigra in the rat. Neuroscience. 2002;109:89–99. doi: 10.1016/s0306-4522(01)00455-9. [DOI] [PubMed] [Google Scholar]
- Mizuchi D, Kurosu T, Kida A, Jin ZH, Jin A, Arai A, Miura O. BCR/ABL activates Rap1 and B-Raf to stimulate the MEK/Erk signaling pathway in hematopoietic cells. Biochem Biophys Res Commun. 2005;326:645–651. doi: 10.1016/j.bbrc.2004.11.086. [DOI] [PubMed] [Google Scholar]
- Moussa CE, Tomita Y, Sidhu A. Dopamine D1 receptor-mediated toxicity in human SK-N-MC neuroblastoma cells. Neurochem Int. 2006;48:226–234. doi: 10.1016/j.neuint.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Nagatsu T. Parkinson’s disease: changes in apoptosis-related factors suggesting possible gene therapy. J Neural Transm. 2002;109:732–745. doi: 10.1007/s007020200061. [DOI] [PubMed] [Google Scholar]
- Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson N. The family of Na+/Cl− neurotransmitter transporters. J Neurochem. 1998;71:1785–1803. doi: 10.1046/j.1471-4159.1998.71051785.x. [DOI] [PubMed] [Google Scholar]
- O’Dell SJ, Weihmuller FB, Marshall JF. Methamphetamine-induced dopamine overflow and injury to striatal dopamine terminals: attenuation by dopamine D1 or D2 antagonists. J Neurochem. 1993;60:1792–1799. doi: 10.1111/j.1471-4159.1993.tb13405.x. [DOI] [PubMed] [Google Scholar]
- Oak JN, Lavine N, Van Tol HH. Dopamine D(4) and D(2L) Receptor Stimulation of the Mitogen-Activated Protein Kinase Pathway Is Dependent on trans-Activation of the Platelet-Derived Growth Factor Receptor. Mol Pharmacol. 2001;60:92–103. doi: 10.1124/mol.60.1.92. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Oh-hashi K, Maruyama W, Yi H, Takahashi T, Naoi M, Isobe K. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun. 1999;263:504–509. doi: 10.1006/bbrc.1999.1237. [DOI] [PubMed] [Google Scholar]
- Onn SP, West AR, Grace AA. Dopamine-mediated regulation of striatal neuronal and network interactions. Trends Neurosci. 2000;23(10 Suppl):S48–56. doi: 10.1016/s1471-1931(00)00020-3. [DOI] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Paul S, Snyder GL, Yokakura H, Picciotto MR, Nairn AC, Lombroso PJ. The Dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. J Neurosci. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Perry G, Roder H, Nunomura A, Takeda A, Friedlich AL, Zhu X, Raina AK, Holbrook N, Siedlak SL, Harris PL, Smith MA. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport. 1999;10:2411–2415. doi: 10.1097/00001756-199908020-00035. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2003;64:755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- Pulliam L, Irwin I, Kusdra L, Rempel H, Flitter WD, Garland WA. CPI-1189 attenuates effects of suspected neurotoxins associated with AIDS dementia: a possible role for ERK activation. Brain Res. 2001;893:95–103. doi: 10.1016/s0006-8993(00)03293-5. [DOI] [PubMed] [Google Scholar]
- Reszka AA, Seger R, Diltz CD, Krebs EG, Fischer EH. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc Natl Acad Sci U S A. 1995;92:8881–8885. doi: 10.1073/pnas.92.19.8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DL, Heien ML, Wightman RM. Frequency of dopamine concentration transients increases in dorsal and ventral striatum of male rats during introduction of conspecifics. J Neurosci. 2002;22:10477–10486. doi: 10.1523/JNEUROSCI.22-23-10477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Nakatsuka D, Watanabe Y, Nagata I, Kikuchi H, Namura S. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci Lett. 2000;288:163–166. doi: 10.1016/s0304-3940(00)01229-5. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Neuroprotection and dopamine agonists. Neurology. 2002;58:S9–18. doi: 10.1212/wnl.58.suppl_1.s9. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Stork PJ. PKA phosphorylation of Src mediates cAMP’s inhibition of cell growth via Rap1. Mol Cell. 2002;9:85–94. doi: 10.1016/s1097-2765(01)00432-4. [DOI] [PubMed] [Google Scholar]
- Scott L, Kruse MS, Forssberg H, Brismar H, Greengard P, Aperia A. Selective up-regulation of dopamine D1 receptors in dendritic spines by NMDA receptor activation. Proc Natl Acad Sci U S A. 2002 Feb 5;99(3):1661–499. 1661–1664. doi: 10.1073/pnas.032654599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Fishman PH. Identification and characterization of functional D1 dopamine receptors in a human neuroblastoma cell line. Biochem Biophys Res Commun. 1990;166:574–579. doi: 10.1016/0006-291x(90)90847-g. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Niznik HB. Coupling of dopamine receptor subtypes to multiple and diverse G proteins. Int J Dev Neurosci. 2000;18:669–677. doi: 10.1016/s0736-5748(00)00033-2. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Kimura K, Uh M, White BH, Patel S. Multiple coupling of human D5 dopamine receptors to guanine nucleotide binding proteins Gs and Gz. J Neurochem. 1998;70:2459–2467. doi: 10.1046/j.1471-4159.1998.70062459.x. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Gibb JW, Hanson GR. Roles of D1 and D2 dopamine receptor subtypes in mediating the methamphetamine-induced changes in monoamine systems. J Pharmacol Exp Ther. 1986;238:932–937. [PubMed] [Google Scholar]
- Spielewoy C, Gonon F, Roubert C, Fauchey V, Jaber MCM, Roques BP, Hamon M, Betancur C, Maldonado R, Giros B. Increased rewarding properties of morphine in dopamine-transporter knockout mice. Eur J Neurosci. 2000;12:1827–1837. doi: 10.1046/j.1460-9568.2000.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J Biol Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Stephans S, Yamamoto B. Methamphetamines pretreatment and the vulnerability of the striatum to methamphetamine neurotoxicity. Neuroscience. 1996;72:593–600. doi: 10.1016/0306-4522(95)00587-0. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Takaki M, Ujike H, Kodama M, Takehisa Y, Nakata K, Kuroda S. Two kinds of mitogen-activated protein kinase phosphatases, MKP-1 and MKP-3, are differentially activated by acute and chronic methamphetamine treatment in the rat brain. J Neurochem. 2001;79:679–688. doi: 10.1046/j.1471-4159.2001.00615.x. [DOI] [PubMed] [Google Scholar]
- Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277:9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol J, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Herve D, Girault JA. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- Wersinger C, Chen J, Sidhu A. Bimodal induction of dopamine-mediated striatal neurotoxicity is mediated through both activition of D1 dopamine receptors and autoxidation. Mol Cell Neurosci. 2004;25:124–137. doi: 10.1016/j.mcn.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Xu W, Zhu JP, Angulo JA. Induction of striatal pre- and postsynaptic damage by methamphetamine requires the dopamine receptors. Synapse. 2005;58:110–121. doi: 10.1002/syn.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W-D, Spealman RD, Zhang J. Dopaminergic signaling in dendritic spines. Biochemical Pharmacology. 2008;75:2055–2069. doi: 10.1016/j.bcp.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Barak LS, Anborgh PH, Laporte SA, Caron MG, Ferguson SS. Cellular trafficking of G protein-coupled receptor/beta-arrestin endocytic complexes. J Biol Chem. 1999;274:10999–11006. doi: 10.1074/jbc.274.16.10999. [DOI] [PubMed] [Google Scholar]
- Zhen X, Zhang J, Johnson GP, Friedman E. D(4) dopamine receptor differentially regulates Akt/nuclear factor-kappa b and extracellular signal-regulated kinase pathways in D(4)MN9D cells. Mol Pharmacol. 2001;60:857–864. [PubMed] [Google Scholar]