

Abstract
Anthrax is an infectious disease caused by Bacillus anthracis, a Gram-positive, rod-shaped, anaerobic bacterium. The lethal factor (LF) enzyme is secreted by B. anthracis as part of a tripartite exotoxin and is chiefly responsible for anthrax-related cytotoxicity. As LF can remain in the system long after antibiotics have eradicated B. anthracis from the body, the preferred therapeutic modality would be the administration of antibiotics together with an effective LF inhibitor. Although LF has garnered a great deal of attention as an attractive target for rational drug design, relatively few published inhibitors have demonstrated activity in cell-based assays and, to date, no LF inhibitor is available as a therapeutic or preventive agent. Here we present a novel in silico high-throughput virtual screening protocol that successfully identified 5 non-hydroxamic acid small molecules as new, preliminary LF inhibitor scaffolds with low micromolar inhibition against that target, resulting in a 12.8% experimental hit rate. This protocol screened approximately thirty-five million non-redundant compounds for potential activity against LF and comprised topomeric searching, docking and scoring, and drug-like filtering. Among these 5 hit compounds, none of which has previously been identified as a LF inhibitor, three exhibited experimental IC50 values less than 100 µM. These three preliminary hits may potentially serve as scaffolds for lead optimization, as well as templates for probe compounds to be used in mechanistic studies. Notably, our docking simulations predicted that these novel hits are likely to engage in critical ligand-receptor interactions with nearby residues in at least two of the three (S1’, S1–S2 and S2’) subsites in the LF substrate binding area. Further experimental characterization of these compounds is in process. We found that micromolar-level LF inhibition can be attained by compounds with non-hydroxamate zinc-binding groups that exhibit monodentate zinc chelation, as long as key hydrophobic interactions with at least two LF subsites are retained.
Introduction
The Bacillus anthracis bacterium secretes an exotoxin comprising three proteins: a lethal factor (LF), a calmodulin-activated edema factor adenylate cyclase (EF), and a protective antigen (PA), produced by the pXO1 plasmid.1 Most critical for pathogenesis is LF, an 89-kDa Zn metalloprotease which combines with PA to form the anthrax lethal toxin.2 Once translocated by PA into the cytoplasm of host target cells, LF cleaves members of the mitogen-activated protein kinase kinase (MEK) family, including mitogen activated protein kinase kinases (MAPKKs) 1–3, in the proline-rich N-terminal area adjacent to the kinase domain,3,4 thereby interrupting MAPKK phosphorylation that, in turn, interferes with cellular immune/inflammatory defense mechanisms against pathogens.5–8 In subsequent stages of the disease, LF also targets endothelial cells and causes disruption of vascular barriers.4,9–11 The sole existing therapeutic modality for anthrax is antibiotic treatment, but early administration is crucial, as antibiotics have no effect on the exotoxin itself, and diagnosis is often inconclusive in the initial stages of the disease. Moreover, high levels of LF may remain in the system for days after B. anthracis has been cleared, and can produce fatal residual toxemia in the absence of viable bacteria. Since weaponized anthrax continues to pose a threat to society, there remains a critical need for small-molecule LF inhibitors that can be administered concurrent with antibiotics to increase the probability of host survival.
The LF enzyme consists of four domains: the N-terminal domain (I); the large central domain (II); a small helical domain (III); and the C-terminal catalytic domain (IV).12,13 Domains II–IV (1YQY.pdb)14 are illustrated in Figure 1. The C-terminal domain forms the LF active site, and has therefore been the primary focus of LF inhibition studies. This domain contains a catalytic Zn2+ coordinated to three active-site residues: His686, His690, and Glu735 (Figure 2). Two histidines are located on an α-helix near the bottom of the LF substrate binding site, and form part of the signature Zn metalloproteinase HEXXH consensus motif that is also present in most matrix metalloproteinases (MMPs).9,15 Glu735 is located on a separate, but closely adjacent, helix near the top of the active site. The binding cleft itself encompasses three general subsites: the deep, strongly hydrophobic, and sterically constrained S1’ subsite; the largely hydrophobic but less restricted S1–S2 region, which is an open-ended, partly solvent-exposed tunnel; and the less well characterized and somewhat more electrostatically complicated S2’ area (Figure 2).
Figure 1.

Anthrax toxin lethal factor domains II–IV (residues 297–809) (1YQY.pdb14), colored by residuum order (N-terminus=blue, C-terminus=red), with catalytic Zn2+ (yellow sphere).
Figure 2.

Substrate cleavage site of anthrax toxin lethal factor (1YQY.pdb14), with electrostatic potential mapping (red = positive, purple = negative); catalytic Zn2+ (pink sphere); Zn-chelating residues His686, His690, and Glu735; and illustrating three key subsite regions: S1’, S1–S2, and S2’.
Many studies have been conducted toward the design of small molecules that target the LF active site.9,14–20 The first active LF inhibitors were, like the earliest matrix metalloproteinase (MMP) inhibitors, small peptide sequences designed to parallel the natural MAPKK substrate, with hydroxamic acid zinc-binding groups (ZBGs).4,21,22 However, while these early attempts offered valuable insight into important LF structural features and ligand-receptor interactions, they showed limited promise as therapeutics due to relatively poor bioavailability and lack of selectivity. Subsequent attempts to develop effective nonpeptidic LF inhibitors resulted in the discovery of sulfonamide hydroxamate compounds demonstrating high (~54 nM) potency against LF;9 but the therapeutic value of these compounds was also hindered by selectivity issues and the well-documented range of pharmacokinetic liabilities exhibited by hydroxamic acids.
Recent attention has therefore been strongly focused on the development of new LF inhibitor scaffolds that incorporate non-hydroxamate ZBGs.15–20,23–35 Many of these investigations involved small- to medium-scale high-throughput screening (HTS) of compound collections using fluorescence resonance energy transfer (FRET) assays. Scaffolds investigated to date include cationic polyamines,17 aminoglycosides,24,34 pyrazolones,18 ECGC and related polyphenolics,31 tetracyclines,25 α-defensins,32 rhodanines,15,16,26,33 and hydroxypyrothiones.35 The majority of compounds identified in these studies are active in the micromolar range against LF, and some small molecules9,16,17 achieved nanomolar inhibition. However, there is still currently no effective therapeutic on the market that can counteract LF-mediated cell death. In the current paper, we present novel virtual screening methodologies, validated by experimental biological activity data, that are designed to cover an exceptionally wide range of non-hydroxamate structures and thereby identify previously uninvestigated LF inhibitor scaffolds. From an initial virtual screen of millions of compounds, we identified five novel non-hydroxamate small molecules with at least micromolar-level inhibition of LF. None of these compounds was previously identified as a LF inhibitor, and three of these small molecules show particular promise for further modification and optimization as potential drug and/or probe scaffolds.
Results and Discussion
Large-scale virtual screening
To explore current chemical space as broadly as possible for potential LF active-site lead/probe scaffolds, to investigate LF ligand-receptor interactions, and to select a small, structurally diverse library of previously unevaluated compounds for preliminary experimental assays, we screened approximately thirty-five million non-redundant compounds in silico for potential activity against the anthrax toxin lethal factor (Figure 3). These structures were obtained from seven small-molecule databases: DrugBank,36,37 LeadQuest,38 NIH Molecular Libraries Small Molecule Repository (MLSMR),39 GDB (26.4 million structures comprising organic molecules of up to 11 atoms containing C, N, O, and F),40–42 ZINC 7,43 NCI,44 and the University of Minnesota Institute for Therapeutics Discovery and Development (ITDD) in-house database. (For those compounds whose three-dimensional structures were not available in the databases, 3D configurations were generated using the SciTegic Pipeline Pilot data analysis and reporting platform (Accelrys, Inc.)). These databases comprise a diverse array of drug-like molecules, commercially available compounds, currently marketed drugs, probe molecules and natural products.
Figure 3.

Large-scale virtual screening protocol for small molecules against the anthrax toxin lethal factor.
In the first stage of this virtual screen, the LeadQuest, NIH MLSMR, GDB, ZINC 7, NCI and University of Minnesota Institute for Therapeutics Discovery and Development (ITDD) in-house vendor database compounds were subjected to a shape-based, “topomeric” searching technique developed by Cramer et al.45–53 as implemented in the Topomer Search module (SYBYL 8.0, Tripos, Inc.). In this method, one or more already-proven active compounds are used to search collections of molecules for “hits” that exhibit similar three-dimensional shapes, as defined by conformationally independent topomeric fields. This type of similarity searching has proven effective for pinpointing active compounds within datasets of varying size and compound diversity, and has been successfully used to select compounds for experimental screening, synthesis and optimization.45,46 Compounds identified as similar using topomeric searching are often true “lead-hops,” that is, they are often significantly dissimilar in terms of traditional two-dimensional structural fingerprints.45 Such structures are therefore more likely to reside in less extensively explored chemical space for a particular target than those identified by 2D similarity searching methodologies, and will often carry activity-enhancing structural features beyond those that have been previously published and/or investigated.45,46 Here, we selected (2R)-2-[(4-fluoro-3-methylphenyl)sulfonylamino]-N-hydroxy-2-(tetrahydro-2H-pyran-4-yl)acetamide (Figure 4, compound 40) developed by Xiong et al. 9 as our topomeric search template, as it demonstrated an IC50 of 54 nM in a LF enzyme assay and an IC50 of 210 nM in a macrophage cytotoxicity assay.9 Although this compound was strongly active against LF, it was not feasible as a therapeutic due to poor pharmacokinetics and lack of selectivity. The objective of our topomeric searching protocol here was to use a highly active but “compromised” compound as a template to “scaffold-hop” to new structures that exhibit similar three-dimensional shapes but different functional groups, in order to retain biological activity while avoiding potential pharmacokinetic impediments such as metabolic instability. Our topomeric search based on this strategy identified 22,133 preliminary hits (Figure 3). Broken down by database, with respective hit rates in parentheses, the topomeric search yielded 94 LeadQuest (0.18%), 2402 NIH MLSMR (1.02%), 206 NCI (0.08%), 5731 ITDD (0.3%), 10 GDB (3×10−5%), and 13690 ZINC 7 (0.23%) compounds. It is perhaps not surprising that the NIH MLSMR would exhibit the most favorable topomeric search hit rate, as this database comprises known bioactives including drugs and metabolites, natural products and their derivatives, and additional specific libraries targeting proteases. Similarly, one could expect the least favorable hit rate from the very large GDB database, which enumerates all possible organic molecules containing C, N, O and F within the 11-atom limit, resulting in compounds that are somewhat small (average MW = 153 ± 7) compared to the typical drug molecule (MW ≈ 340),40 and are less likely to match the shape descriptors of the topomeric search template (MW = 346.37).
Figure 4.

Potent sulfonamide hydroxamate LF inhibitor 40,9 used as the topomeric searching template.
All of these hit compounds were subsequently docked into the LF active site (1YQY.pdb) together with 3,462 structures from the DrugBank bio- and cheminformatics resource database that had been pre-filtered to exclude compounds with inorganic atoms. Docking was done using Surflex-Dock54–57 in the SYBYL 8.0 discovery suite (Tripos, Inc.). Docked poses were scored using the CScore consensus scoring package58 and were ranked by total Surflex-Dock score expressed as calculated −log(Kd). To check the accuracy of this docking procedure for LF, the 1YQY.pdb cocrystallized ligand (also compound 40) was docked back into the LF active site. The best docked ligand conformation differed from that of the X-ray structure by an RMSD of only 0.54 Å. Although this validation procedure does not guarantee that predicted binding modes for all our database compounds will be accurate, it does indicate that Surflex-Dock was able to accurately reproduce experiment for this system.
The most favorable docked pose of 40 displayed a total Surflex-Dock score of 9.91; therefore all structures with a total Surflex-Dock score of 10 or greater, corresponding to estimated Kd values of 0.1 nM or lower, were assembled to create dataset D1. This dataset comprised 643 non-redundant compounds: 213 topomeric searching hits and 430 high-scoring DrugBank structures. The topomeric searching hits consisted of 5 LeadQuest, 7 MLSMR, 137 ZINC 7, 7 NCI and 57 ITDD structures. While ten GDB compounds passed the initial topomeric filter, none of them achieved the Surflex-Dock cutoff value.
To refine this selection to a smaller subset of compounds for subsequent in vitro screening, dataset D1 was subjected to a series of four filters in the SciTegic Pipeline Pilot data analysis and reporting platform (Accelrys, Inc.) (Figure 3). The first filter retained structures that satisfied Lipinski’s Rule of Five.59 The SciTegic high-throughput screening (HTS) filter was then applied, eliminating molecules likely to be poor candidates for assays commonly used in HTS, including those compounds containing inorganic atoms and reactive substructures. A backup organic filter was implemented to “fail” any structures containing inorganic atoms that may have been missed by the HTS filter. Finally, in order to avoid selecting compounds residing in “well-explored” zinc metalloproteinase inhibitor territory, any molecules containing hydroxamate and/or sulfonyl functionalities were rejected. Applying this series of filters to D1 yielded 301 structures comprising a variety of scaffolds: 203 topomeric searching hits (2 LeadQuest, 3 MLSMR, 137 ZINC 7, 4 NCI and 57 ITDD structures), and 98 high-scoring DrugBank structures. Thirty-nine of these compounds were found to be commercially available, comprising focused dataset D2, and were selected for purchase and in vitro screening assays as described below.
MMPI screen for in vitro assay and docking/scoring validation
In order to assess the suitability of our experimental assay procedure and docking/scoring methodologies for the lethal factor, we first screened 19 nonselective MMP inhibitors against LF, both experimentally and in silico. This MMPI screening set was obtained from EMD-Calbiochem Inc. and included the potent peptide hydroxamate MMP and LF inhibitor ilomastat (GM6001/M364205, Table 1), which was cocrystallized with LF by Liddington and coworkers (1PWU.pdb).22 All nineteen compounds were evaluated for activity against recombinant LF by means of an in vitro FRET assay (see Experimental Section), using a MAPKKide consensus sequence peptide substrate (List Biological Laboratories). This assay correctly identified GM6001/M364205 as active against LF (IC50 value = 10.2 ± 0.7 µM), as well as M444264, a structurally similar peptide hydroxamate (Table 1). The other seventeen MMPIs screened did not demonstrate biological activity against LF; their structures and Surflex-Dock scores are provided in Supporting Information. In the MMPI docking study, GM6001/M364205 yielded a highly favorable Surflex-Dock score of 10.55, corresponding to a Kd of 0.03 nM. Most notably, the docked configuration of this compound (Figure 5) confirms that Surflex-Dock is able to accurately predict reported experimental binding modes for this system,22,60 where the hydroxamate moiety chelates the catalytic Zn (with the carbonyl oxygen located further from the metal center), the leucine mimetic partly fills the S1’ subsite, and the hydrophobic Trp sidechain is oriented towards the S2’ region.
Table 1.
Two matrix metalloproteinase inhibitors (MMPIs) found to be biologically active against the anthrax toxin lethal factor, with IC50 values from in vitro FRET assays reported with standard errors of measurement, and total Surflex-Dock scores.
| Compound | IC50 (µM) | Total Surflex-Dock Score (−log Kd) |
|---|---|---|
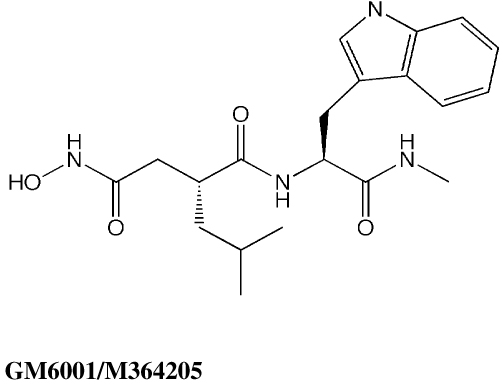 |
10.2 ± 0.72 | 10.55 |
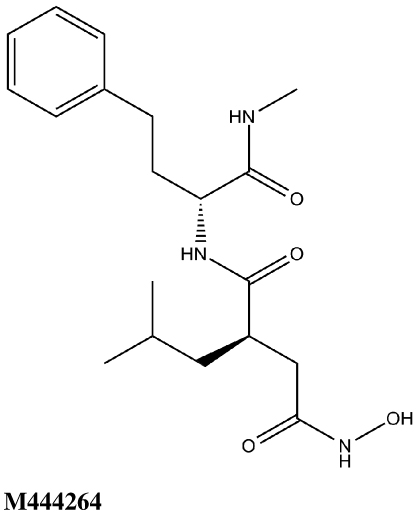 |
7.1 ± 1.28 | 12.31 |
Figure 5.

Ligand-receptor interaction diagram of GM6001/M364205 docked into the LF active site (1YQY.pdb14) (MOE 2007.09, Chemical Computing Group, Inc.) Green spheres = “greasy” residues; spheres with red outline = acidic residues; spheres with blue outline = basic residues; spheres with black outline = polar residues; blue background spheres = receptor exposure to solvent; blue spheres on ligand atoms = ligand exposure to solvent. Green dotted lines = sidechain donors/acceptors; blue dotted lines = backbone donors/acceptors; purple dotted line = metal contact; grey dotted line = proximity contour.
Experimental screening of the focused dataset
All thirty-nine of the commercially available compounds in dataset D2 were subjected to the aforementioned validated in vitro LF FRET assay, again using a consensus sequence peptide substrate, oAbz/Dnp-substrate (List Biological Laboratories). Screening dataset D2 resulted in five hit compounds with at least micromolar inhibition of LF (Table 2), four of which were dibenzylamine derivatives. Three of these closely related dibenzylamines demonstrated IC50 values less than 100 µM: compounds 5426202 (2-[(benzyl(ethyl)amino)methyl]-4,6-diiodophenol, IC50 = 49.5 ± 1.5 µM); 5421384 (2-[(benzyl(ethyl)amino)methyl]-4-chlorophenol, IC50 = 67.5 ± 2.0 µM); and 5428736 (2-[(benzyl(ethyl)amino)methyl]-4-bromophenol, IC50 = 73.9 ± 4.5 µM) (Table 2). All were topomeric searching hits from ITDD’s in-house vendor database. The hits were analyzed for identity and purity utilizing 1H NMR and LC-MS; the identity of all three compounds was confirmed and their purity established at >95%.
Table 2.
Five hit compounds from in vitro FRET screen of dataset D2, with experimental IC50 values against LF, reported with standard errors of measurement, and total Surflex-Dock scores.
| Compound | IC50 (µM) | Total Surflex-Dock Score (−log Kd) |
|---|---|---|
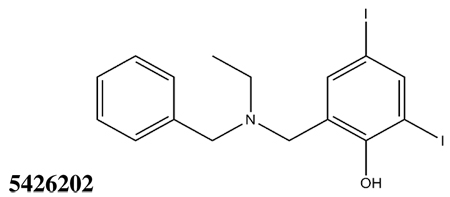 |
49.5 ± 1.5 | 10.07 |
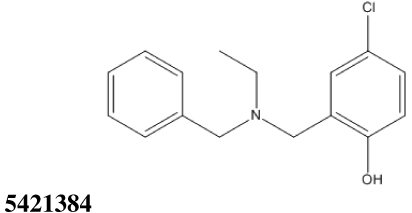 |
67.5 ± 2.0 | 10.44 |
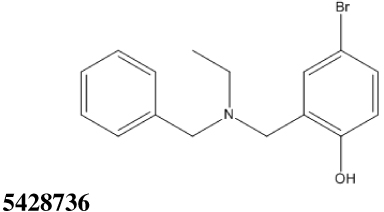 |
73.9 ± 4.5 | 10.48 |
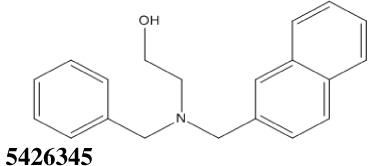 |
124.5 ± 3.5 | 10.14 |
 |
150.4 ± 6.7 | 10.49 |
The three hits exhibited total Surflex-Dock scores of 10.07, 10.44, and 10.48, respectively; three-dimensional renderings of docked configurations are illustrated in Figure 6. Best docked poses of these molecules predict very similar ligand-binding modes, which is not unexpected given their high structural similarity. A representative two-dimensional ligand-receptor interaction map for 5426202 is shown in Figure 7. In all three compounds, the phenol oxygen is predicted to coordinate the catalytic zinc, while the S1–S2 area is occupied by the halophenol moiety, with the two iodines in 5426202 (and the chloro and bromo functionalities in 5421384 and 5428736, respectively) partly solvent-exposed, and the S1’ subsite is targeted by the opposite benzyl group. Hydrophobic interactions appear to play a prominent role: the phenol aryl moiety interacts hydrophobically with Leu658 in the S1–S2 region, (see Figure 6), while the benzyl interacts with Val675 and Leu677 in the S1’ area. This benzyl also engages in π−π stacking with Tyr728. Interestingly, the N-ethyl grouping is oriented toward uncharged polar residues at or near the entrance to the S2’ area, including Gly657, Gly674, Gly683 and Ser655, and the backbone of Lys656. The only significant difference in the docked configurations of these top hits involves hydrogen bonding to the zinc-chelating oxygen. His686, His 690 and Glu687 are predicted to hydrogen bond with this oxygen in all three cases; however, Glu687 engages in two hydrogen bonds with the phenol oxygen in compound 5428736, while participating in only one hydrogen bond with that oxygen in compounds 5421384 and 5426202. This may be due to a slight but discernible shift in the positioning of the 5428736 bromophenol moiety in the S1–S2 subsite (Figure 6).
Figure 6.



Three-dimensional renderings of the top three in vitro hits (hydrogens undisplayed) docked into the LF active site (1YQY.pdb14), with catalytic Zn2+ (grey sphere): (a) 5426202; (b) 5421384; and (c) 5428736 (SYBYL 8.1, Tripos, Inc).
Figure 7.

Ligand-receptor interaction diagram of the top in vitro hit (5426202) docked into the LF active site (1YQY.pdb14) (MOE 2007.09, Chemical Computing Group, Inc.) Green spheres = “greasy” residues; spheres with red outline = acidic residues; spheres with blue outline = basic residues; spheres with black outline = polar residues; blue background spheres = receptor exposure to solvent; blue spheres on ligand atoms = ligand exposure to solvent. Green dotted lines = sidechain donors/acceptors; blue dotted lines = backbone donors/acceptors; purple dotted lines = metal contact; grey dotted line = proximity contour.
Concluding Remarks
In this paper, we have presented an original virtual and experimental screening strategy that was able to identify three non-hydroxamate, previously uninvestigated small molecules with biological activity against the anthrax toxin lethal factor in the low micromolar range, with an overall 12.8% experimental hit rate (5 hits out of 39 final prioritized compounds). These initial screening hits may serve as starting points toward lead optimization and eventual nanomolar inhibition. The topomeric searching portion of the virtual screen resulted in the selection of 22,133 topomerically similar yet structurally diverse compounds from an initial dataset of over thirty-five million structures. Notably, all three top hit compounds were identified in silico by means of topomeric searching. Each of these three hits demonstrates monodentate zinc coordination as predicted by docking and scoring; none exhibits the traditionally preferred bidentate zinc chelation. Hydrophobic Val and Leu residues in the S1’ area, Leu in the S1–S2 region, and uncharged polar residues including Gly and Ser in the S2’ region appear to play critical roles in ligand binding, as do two His residues which are also Zn chelators. While several docking validation runs were performed to check the ability of Surflex-Dock to reproduce bound ligand conformations in LF ligand-receptor crystal structures, it will be important to further validate our screening protocol by experimentally assessing binding modes of new hits via X-ray crystallography; this work is currently underway. The results of our screening strategy also confirm that micromolar-level LF inhibition can be achieved by small molecules with non-hydroxamate, monodentate ZBGs, as long as critical hydrophobic interactions with at least two LF subsites (in this case, S1–S2 and S1’) are maintained.
Experimental Section
1. Virtual Screening
Three-dimensional configurations were generated for each virtual compound using SciTegic Pipeline Pilot, using the “SD Reader,” “3D Coordinates,” “Add Hydrogens,” “Minimize Molecule,” and “SD Writer” components, in that order. The “3D Coordinates” module was used to calculate 3D atomic coordinates for each structure by breaking the compound into ring and chain fragments, generating 3D structures for each fragment, reassembling the compound, and conducting a brief geometry optimization on the reassembled structure. The “Minimize Molecule” component subsequently carried out a more thorough energy minimization on each compound after hydrogens were added, by means of the Clean force field.61 Topomeric searching was done using the Topomer Search module in SYBYL 8.0. In the Topomer Search input options, default computational parameters (Maximum Distance Considered Hit = 185) were used, and all weighting factors (steric, aromatic, positive/negative, donor/acceptor) were set to 1.000.
Docking and scoring calculations were carried out using Surflex-Dock and CScore in the SYBYL 8.0 discovery software suite (Tripos, Inc.). In Surflex-Dock, the 1YQY.pdb cocrystallized ligand (#40)9 was used to guide the protomol generation process. Default parameters of 0.5 and 0 were used for docking threshold and bloat, respectively. The maximum number of conformations per compound fragment and the maximum number of poses per ligand were both set to their default values of 20, and the maximum number of rotatable bonds per molecule was set to 100. Post-dock minimizations were done on each molecule to enhance the quality of results, and all four CScore consensus scoring functions were implemented.
Three-dimensional visualizations of small-molecule docked configurations were rendered in SYBYL 8.1 (Tripos, Inc.). All ligand-receptor interaction diagrams were obtained using MOE 2007.09 (Chemical Computing Group, Inc.) Additional visualizations were obtained using PyMOL (DeLano Scientific LLC), and the iMol Molecular Visualizer for Mac OS X.62 All SYBYL, MOE and PyMOL calculations and visualizations were done on Minnesota Supercomputing Institute (MSI) workstations running under the Suse Linux Enterprise Desktop 10.2 operating system. SciTegic Pipeline Pilot analyses were conducted on MSI workstations running under Microsoft Windows Server 2003. iMol visualizations were done in Mac OS X version 10.5.7.
2. LF Enzyme Expression
The anthrax toxin lethal factor was expressed in-house from the BH450 and BH450/pSJ115 attenuated strains of B. anthracis, following the protocols of Leppla and coworkers63 and Knaus and coworkers.64 A 1-liter culture of B. anthracis BH450/pSJ115 (provided by S. Leppla, NIAID) was grown at 37°C overnight with shaking in modified FA media. Solid ammonium sulfate (450 g per liter of culture) was slowly dissolved in clarified culture supernatant at 4°C. Precipitated LF was re-suspended in 50 mM Tris pH 8.0, 5 mM EDTA and AmSO4 was added to give a final concentration of 1.5M. This solution was loaded onto a 50 mL phenyl-sepharose column and eluted with a linear gradient to 0.0M AmSO4. Fractions containing LF were pooled, dialyzed into 50 mM Tris pH 8.0, 50 mM NaCl, 5 mM EDTA and loaded onto a 50 mL Q-sepharose column. Protein was eluted with a linear gradient of 50–300 mM NaCl. Fractions containing LF were pooled, concentrated and loaded onto a HiPrep 26/60 Sephacryl S-200 HR column (GE Healthcare) equilibrated with 50 mM Tris pH 8.0, 50 mM NaCl, 100 µM ZnSO4, and 20% glycerol. Fractions containing LF were concentrated to 24 mg/mL and frozen at −80°C. Final yield of purified LF was ~40 mg/L of culture.
3. In vitro Assays
High-throughput screening kinetic assays were performed on a SpectraMax M2e fluorescence microplate reader in a 384-well plate-based format, following modified procedures of Shoop and coworkers14 and Goldman and coworkers.17 10 mM stock solutions of each test compound were made in DMSO. To create the compound dose response assays, varying volumes of stock solutions were added to a Corning microplate (Cat. #3573) using a Labcyte Echo® 550 acoustic dispenser. For assay uniformity, appropriate volumes of DMSO were backfilled into assay wells in order to achieve a final 5% DMSO per well. 35 µL of a 50 nM solution of anthrax toxin lethal factor (University of Minnesota) in buffer was then added to the assay plate (final concentration 25 nM) using a MultiDrop (Thermo-Fisher), and the plate was pre-incubated at 37°C for 15 minutes prior to addition of substrate. The reaction was initiated by the addition of 35 µL of 60 µM oAbz/Dnp-substrate in water (final substrate concentration of 30 µM). The time-dependent increase in fluorescence intensity was monitored at 37° C every 60 seconds for 20 minutes. Excitation and emission wavelengths were set to 320 nm and 420 nm, respectively. Final IC50 values were obtained by dose-response measurements, using the 8-point dose response curve obtained from the assay as described above. Measurements were made in duplicate columns of a 384-well plate and the plate was repeated to obtain quadruplicate data points for analysis using SofMax Pro software, with the 100% and 0% values set by controls run in the same plate as the compounds (i.e., complete reaction mixture with no inhibitor as 100% positive control and reaction mixture less enzyme as 0% negative control). In addition, a dose response curve or single-point concentration for GM6001, a known inhibitor, was run as a control in all plates under identical conditions.
4. Identity and purity analysis
Compounds 5426202, 5421384 and 5428736 were analyzed for identity and purity using 1H NMR and LC-MS. NMR samples were prepared by weighing ~5 mg of each compound, and dissolving in 0.6 mL deuterated DMSO. Observed NMR spectra were compared to the corresponding spectra predicted by the CS ChemBioOffice 2008 software package.65 Purity determinations for each compound were done using LC-MS on a Waters uPLC instrument with PDA detector and a Waters ZQ mass spectrometer, with C8 BEH 1.7mm and 2.1 × 50mm columns. Column temperature was 25°C. The solvents used for the mobile phase gradient were 95:5 water:acetonitrile with 0.1% formic acid, and 95:5 acetonitrile:water with 0.1% formic acid. The PDA wavelength was set to 220 nm, with a mass range of 150–800 and ESI positive mode mass detection. All associated spectra, and the LC-MS solvent gradient table, are provided in Supporting Information.
Supplementary Material
Acknowledgements
The authors express their appreciation to Dr. Stephen Leppla for generously providing the BH450 and BH450/pSJ115 attenuated strains of Bacillus anthracis for in-house LF enzyme expression. The authors also gratefully acknowledge Jennifer Nguyen and Darlene Charboneau for valuable contributions. This work was supported in part by the National Institutes of Health (R01 AI083234-01 to E.A.A.), the University of Minnesota Department of Medicinal Chemistry, the University of Minnesota Institute for Therapeutics Discovery and Development, the University of Minnesota Supercomputing Institute for Advanced Computational Research, the University of Minnesota Undergraduate Research Opportunities Program, and the University of Minnesota Academic Health Center.
References
- 1.Pezard C, Berche P, Mock M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 1991;59:3472–3477. doi: 10.1128/iai.59.10.3472-3477.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chopra A, Boone S, Liang X, Duesbery N. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J. Biol. Chem. 2003;278:9402–9406. doi: 10.1074/jbc.M211262200. [DOI] [PubMed] [Google Scholar]
- 3.Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- 4.Turk B. Discovery and development of anthrax lethal factor metalloproteinase inhibitors. Curr. Pharm. Biotechnol. 2008;9:24–33. doi: 10.2174/138920108783497604. [DOI] [PubMed] [Google Scholar]
- 5.Gaddis B, Avramova L, Chmielewski J. Inhibitors of anthrax lethal factor. Bioorg. Med. Chem. Lett. 2007;17:4575–4578. doi: 10.1016/j.bmcl.2007.05.089. [DOI] [PubMed] [Google Scholar]
- 6.Biondi R, Nebreda A. Signaling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem. J. 2003;372:1–13. doi: 10.1042/BJ20021641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanoue T, Nishida N. Molecular recognitions in the MAP kinase cascades. Cell. Signalling. 2003;15:455–462. doi: 10.1016/s0898-6568(02)00112-2. [DOI] [PubMed] [Google Scholar]
- 8.Bardwell A, Abdollahi M, Bardwell L. Anthrax lethal factor-cleavage products of MAPK (mitogen-activated protein kinase) kinases exhibit reduced binding to their cognate MAPKs. Biochem. J. 2004;378:569–577. doi: 10.1042/BJ20031382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiong Y, Wiltsie J, Woods A, Guo J, Pivichny J, Tang W, Bansal A, Cummings R, Cunningham B, Friedlander A, Douglas C, Salowe S, Zaller D, Scolnick E, Schmatz D, Bartizal K, Hermes J, MacCoss M, Chapman K. The discovery of a potent and selective lethal factor inhibitor for adjunct therapy of anthrax infection. Bioorg. Med. Chem. Lett. 2006;16:964–968. doi: 10.1016/j.bmcl.2005.10.088. [DOI] [PubMed] [Google Scholar]
- 10.Moayeri M, Leppla S. The roles of anthrax toxin in pathogenesis. Curr. Opin. Microbiol. 2004;7:19–24. doi: 10.1016/j.mib.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Warfel J, Steele A, D'Agnillo F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005;166:1871–1881. doi: 10.1016/S0002-9440(10)62496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pannifer A, Wong T, Schwarzenbacher R, Renatus M, Petosa C, Bienkowska J, Lacy D, Collier R, Park S, Leppla S, Hanna P, Liddington R. Crystal structure of the anthrax lethal factor. Nature. 2001;414:229–233. doi: 10.1038/n35101998. [DOI] [PubMed] [Google Scholar]
- 13.Rawlings N, Tolle D, Barrett A. MEROPS: the peptidase database. Nucl. Acids Res. 2004;32:D160–D164. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shoop W, Xiong Y, Wiltsie J, Woods A, Guo J, Pivnichy J, Felcetto T, Michael B, Bansal A, Cummings R, Cunningham B, Friedlander A, Douglas C, Patel S, Wisniewski D, Scapin G, Salowe S, Zaller D, Chapman K, Scolnick E, Schmatz D, Bartizal K, MacCoss M, Hermes J. Anthrax lethal factor inhibition. Proc. Natl. Acad. Sci. USA. 2005;102:7958–7963. doi: 10.1073/pnas.0502159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whittaker M, Floyd C, Brown P, Gearing A. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- 16.Forino M, Johnson S, Wong T, Rozanov D, Savinov A, Li W, Fattorusso R, Becattini B, Orry A, Jung D, Abagyan R, Smith J, Alibek K, Liddington R, Strongin A, Pellecchia M. Efficient synthetic inhibitors of anthrax lethal factor. Proc. Natl. Acad. Sci. USA. 2005;102:9499–9504. doi: 10.1073/pnas.0502733102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson S, Jung D, Forino M, Chen Y, Satterthwait A, Rozanov D, Strongin A, Pellecchia M. Anthrax lethal protease inhibitors: synthesis, SAR, and structure-based QSAR studies. J. Med. Chem. 2006;49:27–30. doi: 10.1021/jm050892j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldman M, Cregar L, Nguyen D, Simo O, O'Malley S, Humphreys T. Cationic polyamines inhibit anthrax lethal factor protease. BMC Pharmacol. 2006;6:1–8. doi: 10.1186/1471-2210-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schepetkin I, Khlebnikov A, Kirpotina L, Quinn M. Novel small-molecule inhibitors of anthrax lethal factor identified by high-throughput screening. J. Med. Chem. 2006;49:5232–5244. doi: 10.1021/jm0605132. [DOI] [PubMed] [Google Scholar]
- 20.Karginov V, Nestorovich E, Moayeri M, Leppla S, Bezrukov S. Blocking anthrax lethal toxin at the protective antigen channel by using structure-inspired drug design. Proc. Natl. Acad. Sci. USA. 2005;102:15075–15080. doi: 10.1073/pnas.0507488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tonello F, Seveso M, Marin O, Mock M, Montecucco C. Screening inhibitors of anthrax lethal factor. Nature. 2002;418:386. doi: 10.1038/418386a. [DOI] [PubMed] [Google Scholar]
- 22.Turk B, Wong T, Schwarzenbacher R, Jarrell E, Leppla S, Collier R, Liddington R, Cantley L. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat. Struct. Mol. Biol. 2004;11:60–66. doi: 10.1038/nsmb708. [DOI] [PubMed] [Google Scholar]
- 23.Panchal R, Hermone A, Nguyen T, Wong T, Schwarzenbacher R, Schmidt J, Lane D, McGrath C, Turk B, Burnett J, Aman M, Little S, Sausville E, Zaharevitz D, Cantley L, Liddington R, Gussio R, Bavari S. Identification of small molecule inhibitors of anthrax lethal factor. Nat. Struct. Mol. Biol. 2004;11:67–72. doi: 10.1038/nsmb711. [DOI] [PubMed] [Google Scholar]
- 24.Lee L, Bower K, Liang F, Shi J, Wu D, Sucheck S, Vogt P, Wong C. Inhibition of the proteolytic activity of anthrax lethal factor by aminoglycosides. J. Am. Chem. Soc. 2004;126:4774–4775. doi: 10.1021/ja0495359. [DOI] [PubMed] [Google Scholar]
- 25.Kocer S, Walker S, Zerler B, Golub L, Simon S. Metalloproteinase inhibitors, nonantimicrobial chemically modified tetracyclines, and ilomastat block Bacillus anthracis lethal factor activity in viable cells. Infect. Immun. 2005;73:7548–7557. doi: 10.1128/IAI.73.11.7548-7557.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson S, Chem L, Harbach R, Sabet M, Savinov A, Cotton N, Strongin A, Guiney D, Pellecchia M. Rhodanine derivatives as selective protease inhibitors against bacterial toxins. Chem. Biol. Drug. Des. 2008;71:131–139. doi: 10.1111/j.1747-0285.2007.00617.x. [DOI] [PubMed] [Google Scholar]
- 27.Min D, Tang W, Mrksich M. Chemical screening by mass spectrometry to identify inhibitors of anthrax lethal factor. Nat. Biotechnol. 2004;22:717–723. doi: 10.1038/nbt973. [DOI] [PubMed] [Google Scholar]
- 28.Houseman B, Huh J, Kron S, Mrksich M. Peptide chips for the quantitative evaluation of protein kinase activity. Nat. Biotechnol. 2002;20:270–274. doi: 10.1038/nbt0302-270. [DOI] [PubMed] [Google Scholar]
- 29.Min D, Su J, Mrksich M. Profiling kinase activities by using a peptide chip and mass spectrometry. Angew. Chem. Int. Ed. 2004;43:5973–5977. doi: 10.1002/anie.200461061. [DOI] [PubMed] [Google Scholar]
- 30.Su J, Mrksich M. Using mass spectrometry to characterize self-assembled monolayers presenting peptides, proteins and carbohydrates. Angew. Chem. Int. Ed. 2002;41:4715–4718. doi: 10.1002/anie.200290026. [DOI] [PubMed] [Google Scholar]
- 31.Dell’Aica I, Dona M, Tonello F, Piris A, Mock M, Montecucco C, Garbisa S. Potent inhibitors of anthrax lethal factor from green tea. EMBO Rep. 2004;5:418–422. doi: 10.1038/sj.embor.7400118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim C, Gajendran N, Mittrucker H, Weiwad M, Song Y, Hurwitz R, Wilmanns M, Fischer G, Kaufmann S. Human alpha-defensins neutralize anthrax lethal toxin and protect against its fatal consequences. Proc. Natl. Acad. Sci. USA. 2005;102:4830–4835. doi: 10.1073/pnas.0500508102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson S, Chen L, Pellecchia M. A high-throughput screening approach to anthrax lethal factor inhibition. Bioorg. Chem. 2007;35:306–312. doi: 10.1016/j.bioorg.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dalkas G, Papakyriakou A, Vlamis-Gardikas A, Spyroulias G. Low molecular weight inhibitors of the protease anthrax lethal factor. Mini Rev. Med. Chem. 2008;8:290–306. doi: 10.2174/138955708783744083. [DOI] [PubMed] [Google Scholar]
- 35.Agrawal A, de Oliveira C, Cheng Y, Jacobsen A, McCammon JA, Cohen S. Thioamide hydroxypyrothiones supersede amide hydroxypyrothiones in potency against anthrax lethal factor. J. Med. Chem. 2009;52:1063–1074. doi: 10.1021/jm8013212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wishart D, Knox C, Guo A, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–D906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wishart D, Knox C, Guo A, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Exelgen Ltd., UK: 2008. [accessed 8/6/2008]. http://www.leadquest.com. [Google Scholar]
- 39.Austin C, Brady L, Insel T, Collins F. NIH molecular libraries initiative. [accessed 8/6/2008];Science. 2004 306:1138–1139. doi: 10.1126/science.1105511. http://www.ncbi.nlm.nih.gov/sites/entrez. [DOI] [PubMed]
- 40.Fink T, Reymond J. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model. 2007;47:342–353. doi: 10.1021/ci600423u. [DOI] [PubMed] [Google Scholar]
- 41.Fink T, Bruggesser H, Reymond J. Virtual exploration of the small-molecule chemical universe below 160 Daltons. Angew. Chem. Int. Ed. 2005;44:1504–1508. doi: 10.1002/anie.200462457. [DOI] [PubMed] [Google Scholar]
- 42.Amin E. Small-molecule databases for drug discovery and chemical space exploration. ChemTracts: Org. Chem. 2007:181–184. [Google Scholar]
- 43.Irwin J, Shoichet B. ZINC – a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. [accessed 8/6/2008]; http://cactus.nci.nih.gov/ncidb2/download.html.
- 45.Cramer R, Poss M, Hermsmeier M, Caulfield T, Kowala M, Valentine M. Prospective identification of biologically active structures by topomer shape similarity searching. J. Med. Chem. 1999;42:3919–3933. doi: 10.1021/jm990159q. [DOI] [PubMed] [Google Scholar]
- 46.Cramer R, Jilek R, Guessregen S, Clark S, Wendt B, Clark R. ‘Lead-Hopping.’ Validation of topomer similarity as a superior predictor of similar biological activities. J. Med. Chem. 2004;47:6777–6791. doi: 10.1021/jm049501b. [DOI] [PubMed] [Google Scholar]
- 47.Cramer R. Leadhopping – and beyond. Expert Opin. Drug Discov. 2006;1:311–321. doi: 10.1517/17460441.1.4.311. [DOI] [PubMed] [Google Scholar]
- 48.Cramer R, Soltanshahi F, Jilek R, Campbell B. Generating and searching ~1020 synthetically accessible structures. doi: 10.1007/s10822-006-9093-8. Abs. ACS, 231st mtg: CINF 38. [DOI] [PubMed] [Google Scholar]
- 49.Jilek R, Cramer R. Topomers: a validated protocol for their self-consistent generation. J. Chem. Inf. Comput. Sci. 2004;44:1221–1227. doi: 10.1021/ci049961d. [DOI] [PubMed] [Google Scholar]
- 50.Cramer R, Jilek R, Andrews K. dbtop: Topomer similarity searching of conventional databases. J. Mol. Graph. Model. 2002;20:447–462. doi: 10.1016/s1093-3263(01)00146-2. [DOI] [PubMed] [Google Scholar]
- 51.Andrews K, Cramer R. Toward general methods of targeted library design: topomer shape similarity searching with diverse structures as queries. J. Med. Chem. 2000;43:1723–1740. doi: 10.1021/jm000003m. [DOI] [PubMed] [Google Scholar]
- 52.Cramer R, Clark R. Computer-implemented method of generating and characterizing representative three-dimensional conformations of reactant molecules. No. 7,184,893. US Patent. 2007
- 53.Cramer R, et al. Method for searching heterogeneous compound databases using topomeric shape descriptors and pharmacophoric features. Application No. 0060982. US Patent. 2003
- 54.Jain A. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput.-Aided Mol. Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 55.Pham T, Jain A. Parameter estimation for scoring protein-ligand interactions using negative training data. J. Med. Chem. 2006;49:5856–5868. doi: 10.1021/jm050040j. [DOI] [PubMed] [Google Scholar]
- 56.Jain A. Virtual screening in lead discovery and optimization. Curr. Opin. Drug Discov. Devel. 2004;7:396–403. [PubMed] [Google Scholar]
- 57.Jain A. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 58.Meng C, Shoichet B, Kuntz I. Automated docking with grid-based energy evaluation. J. Comp. Chem. 1992;13:505–524. [Google Scholar]
- 59.Lipinski C, Lombardo F, Dominy B, Feeney P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 60.Gowravaram M, Tomczuk B, Johnson J, Delecki D, Cook E, Ghose A, Mathiowetz A, Spurlino J, Rubin B, Smith D, Pulvino T, Wahl R. Inhibition of matrix metalloproteinases by hydroxamates containing heteroatom-based modifications of the P1’ group. J. Med. Chem. 1995;38:2570–2581. doi: 10.1021/jm00014a010. [DOI] [PubMed] [Google Scholar]
- 61.Hahn M. Receptor surface models. 1. Definition and construction. J. Med. Chem. 1995;38:2080–2090. doi: 10.1021/jm00012a007. [DOI] [PubMed] [Google Scholar]
- 62. [accessed 5/19/2009]; http://www.pirx.com/iMol.
- 63.Park S, Leppla SH. Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr. Purif. 2000;18:293–302. doi: 10.1006/prep.2000.1208. [DOI] [PubMed] [Google Scholar]
- 64.Lehmann M, Noack D, Wood M, Perego M, Knaus UG. Lung epithelial injury by B. anthracis lethal toxin is caused by MKK-dependent loss of cytoskeletal integrity. PLoS ONE. 2009;4:e4755. doi: 10.1371/journal.pone.0004755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.CambridgeSoft Life Science Enterprise Solutions. 2008 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.