

Abstract
While Nature excels at performing selective modifications of complex polyfunctional molecules through the use of tailoring enzymes, synthetic chemistry has lagged behind in this regard. In prior work, we have applied a biomimetic approach to this problem, developing small peptides to achieve various group transfer reactions on polyol substrates with high enantio- or regioselectivity. The utility of sulfonates as synthetic building blocks and the scarcity of direct, selective methods for their preparation prompted our investigation into this area. In this article we report the development of a π-methyl histidine-based tetrameric peptide that effects the desymmetrization of meso-1,3-diols through enantioselective (mono)sulfonylation. The catalyst exhibits structural similarities to another catalyst found to be effective in orthogonal group transfers, but results in modification of the enantiotopic alcohol. The practical and mechanistic implications of this discovery may extend beyond synthetic considerations and provide analogies to the diverse roles of histidine in enzyme active sites.
Selective derivatization of single hydroxyl groups within frameworks containing several is a long-standing challenge in synthetic chemistry.i Work in our group has examined a variety of asymmetric group transfer reactions of alcohols (e.g., acylation,ii,iii phosphorylation,iv sulfinylation,v thiocarbonylationvi) predicated on putative nucleophilic catalysis by histidine-containing peptides (Figure 1). This concept has proven surprisingly general, furnishing additional powerful new methods and catalysts.vii,viii This strategy is at the heart of one approach to the systematic modification of complex polyol structures, such as those found in complex antibiotics like erythromycin A. Using this tactic, one can employ a unique catalyst library type, along with a range of electrophiles, to produce a set of natural product-derived analogs for biological evaluation.ix,x In order to expand the repertoire of analogs that can be accessed and to increase the general utility of these group transfer processes in organic synthesis, the addition of novel electrophiles is a consistent goal of this work. One underdeveloped sub-field in this area is the asymmetric transfer of sulfur-based electrophiles. Asymmetric sulfinylation may be the best-studied sub-class of S-group transfer.xi,xii Yet, despite the widespread use of sulfonates in complex molecule synthesis, approaches to asymmetric catalytic sulfonate transfer are rare.xiii,xiv,xv Herein, we report a tetrapeptide catalyst for the desymmetrization of select meso-1,3-diols through a direct enantioselective monosulfonylation reaction. Notably, the lead catalyst bears strong stereochemical analogy to scaffolds previously found to be effective for orthogonal group transfer reactions. Yet, detailed studies of the stereochemical aspects of these two group transfer processes (S-electrophile versus P-electrophile) reveal opposite enantioselectivties from a common catalyst scaffold for these processes.xvi Stereochemical divergence among related catalysts for orthogonal group transfers may reveal an as yet underappreciated mechanistic nuance for histidine-dependent catalysts. Moreover, from the functional point of view, these observations expand the utility of complementary group transfer processes, allowing access to complementary stereoisomeric products from a common catalyst scaffold.
Figure 1. π-Methyl-histidine-containing peptides for group transfer chemistry.

Previous work from the Miller group has resulted in selective transfer of acyl, phosphoryl, sulfinyl, and thiocarbonyl groups to alcohols using π-methyl-histidine-containing peptides. These reactions are thought to proceed through a N-methyl imidazolium intermediate generated by nucleophilic attack of the imidazole moiety on the electrophile. Careful design and optimization of the peptide catalysts can allow for the facile synthesis of a variety of analogs of complex polyols.
Results
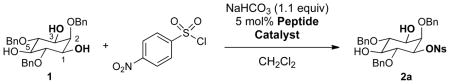
To achieve catalytic enantioselective synthesis of sulfonate esters by sulfonyl transfer, we chose 2,4,6-tribenzyl-myo-inositol (1) as a test-substrate and p-nitrobenzenesulfonyl chloride (p-NsCl) as the sulfonylating agent (Table 1). Derivatives of myo-inositol have proven valuable in earlier studies of enantioselective group transfer, in particular for analogous transfers of phosphorous(V)-based reagents such as diphenylphosphoryl chloride.xvii myo-Inositol derivative 1 presents the challenge of desymmetrization of the enantiotopic hydroxyl groups at the 1- and 3-positions, as well as the free hydroxyl group at the 5-position, along the mirror plane of the substrate. As hindered amine bases are often sufficient to promote the sulfonylation of alcohols in the absence of any catalyst, insoluble inorganic bases were first evaluated as the stoichiometric base. Among the bases screened (including MgO, K2CO3, and Na2CO3), NaHCO3 was identified as the optimal base, providing no observable sulfonate product in the absence of catalyst at 23 °C in 24 h, and good conversions in the presence of N-methyl imidazole (NMI) or peptide catalyst.
Table 1.
Selected data from sulfonylation catalyst screen.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Temp. | i+1 | i+2 | i+3 | i+4 | erb |
| 1 | 3 | 23 °C | Hyp(But) | Sp5 | Tyr(But) | Phe | 67.5:32.5 |
| 2 | 4 | 23 °C | Hyp(But) | Sp5 | Leu | Phe | 84.5:15.5 |
| 3 | 5 | 23 °C | Hyp(But) | Aib | Leu | Phe | 83.5:16.5 |
| 4 | 6 | 23 °C | Pro | Aib | Leu | Phe | 76.5:23.5 |
| 5 | 4 | 0 °C | Hyp(But) | Sp5 | Leu | Phe | 89.0:11.0 |
| 6 | 5 | 0 °C | Hyp(But) | Aib | Leu | Phe | 87.0:13.0 |
| 7 | 7 | 0 °C | Hyp(But) | D-Val | Leu | Phe | 92.5:7.5 |
| 8 | 8 | 0 °C | Hyp(But) | Val | Leu | Phe | 60.0:40.0 |
| 9 | 9 | 0 °C | Hyp(But) | Sp5 | Leu | D-Phe | 91.5:8.5 |
| 10 | 10 | 0 °C | Hyp(But) | Sp5 | Leu | ----- | 97.0:3.0 |
| 11 | 11 | 0 °C | Hyp(But) | D-Val | Leu | ----- | 95.5:4.5 |
Reactions were run with 1.0 equiv 2,4,6-tribenzyl-myo-inositol (1) and 1.0 equiv 4-nitrobenzenesulfonyl chloride. Reactions were run under a common set of conditions, and conversions were found to be approximately 10%, with some variation. The issue of secondary kinetic resolution was addressed subsequently (vide infra).
All enantiomer ratios were measured using chiral HPLC.
With appropriate conditions established, a series of π-methyl-histidine (Pmh)-containing peptides were evaluated based on their ability to influence the enantioselectivity of the reaction (Table 1). Catalyst screening began with peptide 3 due to its high enantiocontrol in the phosphorylation of inositol substrate 1.iv Although the enantiomer ratio (er) observed with peptide 3 was quite modest (67.5:32.5 er, entry 1), this result provided a starting point for catalyst optimization. Changing the amino acid in the i+3 position from an aromatic residue to leucine (Leu) dramatically increased the enantioselectivity to 84.5:15.5 er (catalyst 4, entry 2). The tert-butoxy group of the L-hydroxy proline (Hyp) residue was found to be important for high er’s as replacement with L-proline (Pro) resulted in a significant decrease in enantioselectivity (entry 3 vs. 4). The impact of the i+2 position on enantioselectivity was probed through the use of gem-disubstituted amino acids, L-valine (Val), and D-valine (D-Val) (entries 5–8). The use of 1-amino-isobutyric acid (Aib) resulted in a small decrease in enantioselectivity compared to the five-membered spirocyclic residue (Sp5). Incorporation of a D-amino acid improved the enantioselectivity (92.5:7.5 er, entry 7) while a L-amino acid led to reduced enantioselectivity (60.0:40.0 er, entry 8). It is possible that these changes influence β-turn stability.xviii The stereochemistry of the i+4 position was also probed through the use of D-Phe (catalyst 9). This catalyst resulted in a more selective reaction than the analogous L-Phe peptide (91.5:8.5 er for catalyst 9, and 89.0:11.0 er for catalyst 4). The small degree of influence, however, suggested that the peptide could be truncated and still retain its high enantioselectivity. In fact, tetrapeptide 10, showed the highest enantioselectivity to date (97.0:3.0 er, entry 10).
In addition to p-NsCl, a variety of other sulfonylating agents were assayed using catalyst 10 and triol 1 as the substrate (Figure 2). Usage of ortho-nitrosulfonyl chloride led to reduced enantioselectivity (97.0:3.0 er for 2a vs. 66.0:34.0 er for 2b) suggesting the reaction is sensitive to the steric effects. Nitro-substitution in the 3-position was less consequential (2c, 95.5:4.5 er). Additionally, the er of the reaction was found to be sensitive to the electronic properties of the arenesulfonyl chloride, with electron donating groups having a detrimental effect on enantioselectivity (2d is formed with an 85.5:14.5 er; 2e, 77.5:22.5 er).
Figure 2. The influence of the arene-sulfonyl chloride on the enantioselectivity and yield.

The reactions were run using various substituted arene-sulfonyl chlorides. Yields were low for all reactions except when p-nitrobenzenesulfonyl chloride was used (product 2a). Moving the nitro group to the meta-position had little effect on the enantiomer ratio (er) of the product, while moving the nitro group in the ortho-position dramatically decreased the er of the products. Additionally, a slight decrease in er of the products was observed when electron-rich para-substituents were used (X = p-Me or p-MeO, 2d and 2e, respectively).
Concurrent with catalyst and sulfonylation agent optimization, other reaction parameters were explored to maximize rate and enantioselectivity. A variety of solvents were evaluated, but none proved superior to CH2Cl2. To further increase rates, catalyst 10 was evaluated at 23 °C. Indeed, faster rates were observed, but the er was smaller (92.0:8.0). While high enantioselectivity was consistently observed when solid NaHCO3 was used as the base, the reaction rates were slow and inconsistent yields (25–75%) were recorded. As a result, two new sets of conditions were identified that furnish consistently good yields of 2a in shorter reaction times: condition A uses a biphasic system of saturated NaHCO3 (aq)/CH2Cl2 (1:2 v/v) to produce (mono)sulfonate 2a in 76% yield and 97.0:3.0 er;xix condition B uses 2,6-lutidine in CH2Cl2 to give 2a in 66% yield and 95.0:5.0 er (Table 2, entry 1).
Table 2.
Substrate scope for sulfonylation of meso-1,3-diols.a
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Cond.b | Yieldc | erd | Entry | Substrate | Cond.b | Yieldc | erd |
| 1 | 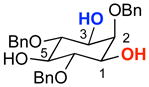 |
A | 76% | 97.0:3.0 | 6 |  |
A | 75% | 96.5:3.5 |
| B | 66% | 95.0:5.0 | B | 56% | 95.5:4.5 | ||||
| 2 | 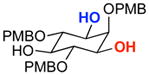 |
A | 75% | 97.0:3.0 | 7 | 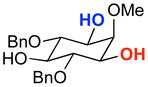 |
A | 57% | 88.0:12.0 |
| B | 58% | 95.0:5.0 | B | 42% | 82.5:17.5 | ||||
| 3 | 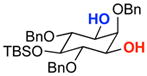 |
A | 62% | 95.5:4.5 | 8 | 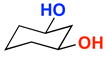 |
A | 12% | 50.0:50.0 |
| B | 60% | 95.0:5.0 | B | 61% | 50.0:50.0 | ||||
| 4 | 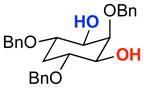 |
A | 58% | 95.5:4.5 | 9 | 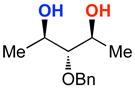 |
A | 74% | 89.0:11.0 |
| B | 53% | 91.5:8.5 | B | 65% | 86.0:14.0 | ||||
| 5 | 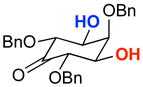 |
A | 76% | 96.0:4.0 | 10 | 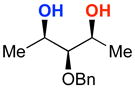 |
A | 16% | 62.0:38.0 |
| B | 76% | 94.5:5.5 | B | 40% | 69.0:31.0 | ||||
All reactions were run with 5 mol% peptide 10, 1.0 equiv substrate and 1.3 equiv 4-nitrobenzenesulfonyl chloride in for 5–24 hours.
Condition A uses 0.2 mL sat. aq. NaHCO3, condition B uses 1.5 equiv 2,6-lutidine.
Isolated yield of mono(sulfonate), an average of at least two trials.
All enantiomer ratios were measured using chiral HPLC and are an average of at least two trials.
The rapid identification of a catalyst for the desymmetrization of 1 bodes extremely well for the identification of highly effective catalysts for enantioselective sulfonylation of broad and diverse structural types.xx,xxi Moreover, the studies above establish simple tetrapeptide catalysts as fertile ground for study of a variety of stereochemical arrays. For studies of polyol modification, catalyst “generality” is much less significant than catalyst library “diversity.” Neverthless, given the simplicity of catalyst 10, and the highly practical nature of conditions A and B, we asked whether or not the processes identified for substrate 1 might be applicable to others. Notably, both conditions A and B enable desymmetrization of a number of other 1,3-diols (Table 2). In general, the biphasic conditions (A) provide higher yields and enantioselectivities. For example, the benzyl protecting groups can be exchanged for p-methoxy benzyl groups without loss of enantioselectivity (entry 2). Additionally, the reaction is tolerant of quite dramatic modifications of the 5-position of inositol (entries 3–6). Small changes at the 2-position of inositol lead to some decrease in enantioselectivity (88.0:12.0 er, entry 7). Furthermore, while cis-1,3-cyclohexane diol shows good reactivity, the reaction is not enantioselective (entry 8). We believe that condition A gives a low yield of product due to the high water solubility of the starting material. Interestingly, the acyclic 1,3-diol derived from adonitol (19) shows high enantioselectivity (89.0:11.0 er) and good reactivity (entry 9), while all-syn stereoisomer 20 reacts slowly and with depreciated enantioselectivity (62.0:38.0 er; entry 10).
From a mechanistic point of view, the observed desymmetrizations appear to be a result of enantiotopic group selection, rather than secondary kinetic resolution of the monofunctionalized product.xxii As shown in Figure 3, when peptide 10 was evaluated for kinetic resolution of racemic (±)-2a, the starting material was recovered (60% mass recovery) with only 57.0:43.0 er (krel = 1.7).
Figure 3. Kinetic resolution of racemic mono(sulfonate) 2a.

Mono(sulfonate) 2a was prepared in racemic form and subjected to the reaction conditions. After 40% conversion to the bis(nosylate) 21, the starting material was recovered with 57:43 er, corresponding to a relative rate constant ratio of the fast enantiomer to the slow enantiomer (krel) of 1.7. This low krel indicates that the high enantioselectivity observed for the desymmetrization of inositol-based 1,3-diols is primarily the result of enantiotopic group selection rather than kinetic resolution of the monofunctionalized product.
These initial mechanistic studies set the stage for a rather remarkable observation. The determination of the absolute configuration of the products derived from the mono(sulfonylation) reveals a stark contrast between peptide-catalyzed asymmetric sulfonylations and the equivalent peptide-catalyzed phosphorylations. We have shown on a number of occasions a strong correlation between the secondary structure of a given enantioselective histidine-based catalyst and the absolute sense of chirality transfer for various electrophiles. For example, we have shown that the enantioselectivity of acylation reactions mediated by diastereomeric peptides was strongly dependent on the type of β–turn nucleated by either a D-Proline residue or an L-Proline residue in the catalyst (Figure 4).xviii,xxiii Analogous observations consistently appear throughout our studies of asymmetric phosphorylation.xvii
Figure 4. Chirality of proline determines the absolute sense of asymmetric induction.

In previous work in the Miller group, the enantioselectivity of acylation reactions mediated by diastereomeric peptides was strongly dependent on whether the -turn of the peptide catalyst was nucleated by a D-Proline residue or an L-Proline residue.
On the other hand, we had not systematically compared the absolute sense of induction provided by related peptide-based catalysts when challenged with different electrophiles. In fact, naïvely, we had often assumed that although the mechanisms of acyl transfer, phosphoryl transfer and other group transfers were clearly nuanced and different, the details of catalyst-substrate recognition could likely be analogous, and the absolute senses of asymmetric induction would also be the same in most cases. Thus, in order to evaluate this issue, we synthesized the mono(nosylated), mono(phosphorylated) inositol 25 through two independent methods (Figure 5) and compared their HPLC retention times. From previous work, it is known that peptide 3 phosphorylates 1 with high levels of enantioselectivity (>99:1 er) at the 3-position of inositol to provide 24.xvii Upon mono(sulfonylation) with an achiral catalyst, 25 was then obtained. However, mono(sulfonate) 2a, obtained from reaction with peptide 10 (97.0:3.0 er), was then subjected to phosphorylation and surprisingly also gave compound 25. It should be noted that neither 24 nor 2a racemize under the sulfonylation or phosphorylation conditions. On the other hand, selective phosphorylation at the 1-position of 1,iv followed by nosylation gives the opposite enantiomer. Thus, peptides 3 and 10, which possess superficially similar peptide sequences, catalyze bond-forming reactions at enantiotopic hydroxyl groups when subjected to either phosphorylation or sulfonylation conditions, respectively. These results suggest disparate details of catalyst-substrate recognition in the respective group transfer reactions, depending on conditions and the nature of the electrophile. The mechanistic basis of these enantiodivergent reactions at present is elusive, and a myriad of mechanistic scenarios must be considered. Changes in mechanism as drastic as the difference between nucleophilic and general base catalysis represent but one possibility under consideration. Likewise, the differences in the nature of the charge distributions exhibited by various activated Lewis basic catalytic intermediates may have fundamentally different consequences for activated catalyst conformations.xxiv Elucidation of these mechanistic nuances requires extensive physical organic studies, and these are underway in our laboratory.
Figure 5. Determination of absolute configuration of sulfonates by comparison to phosphorylation.

(Mono(nosylated), mono(phosphorylated) inositol 25 was prepared through two independent methods, and the products were compared by HPLC retention times. From previous work, it was known that peptide 3 selectively phosphorylates inositol 1 at the 3-position to give diol 24. Sulfonylation of 24 using an achiral catalyst produces compound 25. Surprisingly, phosphorylation of mono(sulfonate) 2a obtained through nosylation mediated by peptide 10 also yielded 25. Therefore, despite the similarity between peptides 3 and 10, they act at enantiotopic alcohols for phosphorylation and sulfonylation.
The significance of these observations is both methodological and fundamental. On the one hand, these results expand the scope of asymmetric peptide-catalyzed group transfer reactions to include an important class of S-based electrophiles. The substrate scope exhibited by even a single catalyst is encouraging as expanded catalyst libraries may now be applied to more complex substrates with the goal of achieving disparate sites of reaction for the realization of analogs. In addition, our new findings point to intriguing mechanistic nuances when different electrophiles are employed. The notion that a unique amino acid side chain may exhibit distinct and unusual roles is well precedented in enzymes, in particular when diverse reactions are carried out by different enzymes. For example, the active site histidine in various histidine-dependent kinases is often demonstrated to serve as a catalytic nucleophile.xxv On the other hand, in lipases and proteases the active site histidines more often serve the role of a general base.xxvi While our experiments in no way imply so drastic a change in mechanism, the reversal of enantioselectivity in two superficially similar reactions support very different transition states for the two enantiomer determining steps. Highly selective catalysts that bear structural similarity, but exhibit their catalytic prowess by different mechanisms have been viewed by some as a hallmark of privileged catalysts in nature,xxvii or the laboratory.xxviii We hope to learn more about these issues as we pursue methodological goals with this intriguing family of peptide-based catalysts in parallel.
Methods
General procedure for sulfonylation using condition A (Table 2)
To an oven-dried 1 dram vial equipped with a magnetic stir bar was added diol substrate (0.10 mmol) and CH2Cl2 (0.4 mL). The peptide catalyst 10 (3.4 mg, 0.005 mmol) was then added and the reaction was cooled to 0 °C. 4-Nitrobenzenesulfonyl chloride (28.8 mg, 0.13 mmol) was added followed by 0.2 mL sat. aq. NaHCO3 solution. The reaction was monitored by TLC for the disappearance of the sulfonyl chloride and allowed to stir at 0 °C for 5–48 h. The biphasic reaction mixture was diluted with CH2Cl2 (2 mL) and sat. aq. NaHCO3, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 2 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting yellow oil was purified by silica gel flash chromatography.
General procedure for sulfonylation using condition B (Table 2)
To an oven-dried 1 dram vial equipped with a magnetic stir bar was added diol substrate (0.10 mmol) and CH2Cl2 (0.4 mL). The peptide catalyst 10 (3.4 mg, 0.005 mmol) was then added and the reaction was cooled to 0 °C. 4-Nitrobenzenesulfonyl chloride (28.8 mg, 0.13 mmol) was added followed by 2,6-lutidine (17.5 μL, 0.15 mmol). The reaction was monitored by TLC for the disappearance of the sulfonyl chloride and allowed to stir at 0 °C for 5–48 h. The reaction mixture was diluted with CH2Cl2 (2 mL), and 1M HCl (0.5 mL) was added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 2 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting yellow oil was purified by silica gel flash chromatography.
Supplementary Material
Figure 6.

Figure 7.

Acknowledgments
This work is supported by the NIH (GM-068649). K.W.F. would like to thank the NIH for a postdoctoral fellowship (1F32GM083622).
Footnotes
Author contributions. All authors conceived and designed the experiments and analyzed the data, K.W.F and A.L.A.P. performed the experiments, K.W.F and S.J.M. wrote the paper jointly, and all authors edited and commented on the manuscript.
SUPPORTING INFORMATION. Experimental procedures and characterization.
References
- i.Peddibhotla S, Dang Y, Liu JO, Romo D. Simultaneous arming and structure/activity studies of natural products employing O–H insertions: an expedient and versatile strategy for natural products-based chemical genetics. J Am Chem Soc. 2007;129:12222–12231. doi: 10.1021/ja0733686. [DOI] [PubMed] [Google Scholar]
- ii.Miller SJ, Copeland GT, Papaioannou N, Horstmann TE, Ruel EM. Kinetic resolution of alcohols catalyzed by tripeptides containing the N-alkylimidazole substructure. J Am Chem Soc. 1998;120:1629–1630. [Google Scholar]
- iii.Copeland GT, Miller SJ. Selection of enantioselective acyl transfer catalysts from a pooled peptide library through a fluorescence-based activity assay: an approach to kinetic resolution of secondary alcohols of broad structural scope. J Am Chem Soc. 2001;123:6496–6502. doi: 10.1021/ja0108584. [DOI] [PubMed] [Google Scholar]
- iv.Sculimbrene BR, Miller SJ. Discovery of a catalytic asymmetric phosphorylation through selection of a minimal kinase mimic: a concise total synthesis of D-myo-inositol-1-phosphate. J Am Chem Soc. 2001;123:10125–10126. doi: 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]
- v.Evans JW, Fierman MB, Miller SJ, Ellman JA. Catalytic enantioselective synthesis of sulfinate esters through the dynamic resolution of tert-butanesulfinyl chloride. J Am Chem Soc. 2004;126:8134–8135. doi: 10.1021/ja047845l. [DOI] [PubMed] [Google Scholar]
- vi.Sánchez-Roselló M, Puchlopek ALA, Morgan AJ, Miller SJ. Site-selective catalyis of phenyl thionoformate transfer as a tool for regioselective deoxygenation of polyols. J Org Chem. 2008;73:1774–1782. doi: 10.1021/jo702334z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Enantioselective silyl protection of alcohols catalysed by an amino-acid-based small molecule. Nature. 2006;443:67–70. doi: 10.1038/nature05102. [DOI] [PubMed] [Google Scholar]
- viii.Ishihara K, Kosugi Y, Umemura S, Sakakura A. Kinetic resolution of racemic carboxylic acids by an L-histidine-derived sulfonamide-induced enantioselective esterification reaction. Org Lett. 2008;10:3191–3194. doi: 10.1021/ol801007m. [DOI] [PubMed] [Google Scholar]
- ix.Lewis CA, Miller SJ. Site-selective derivatization and remodeling of erythromycin A by using simple peptide-based chiral catalysts. Angew Chem, Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- x.Lewis CA, Merkel J, Miller SJ. Catalytic site-selective synthesis and evaluation of a series of erythromycin analogs. Bioorg Med Chem Lett. 2008;18:6007–6011. doi: 10.1016/j.bmcl.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xi.Peltier HM, Evans JW, Ellman JA. Catalytic enantioselective sulfonyl transfer using cinchona alkaloid catalysts. Org Lett. 2005;7:1733–1736. doi: 10.1021/ol050275p. [DOI] [PubMed] [Google Scholar]
- xii.Shibata N, Matsunaga M, Fukuzumi T, Nakamura S, Toru T. Cinchona alkaloid-sulfinyl chloride combinations: catalytic enantioselective sulfinylation of alcohols. Synlett. 2005:1699–1702. [Google Scholar]
- xiii.Demizu Y, Matsumoto K, Onomura O, Matsumura Y. Copper complex catalyzed asymmetric monosulfonylation of meso-vic-diols. Tetrahedron Lett. 2007;48:7605–7609. [Google Scholar]
- xiv.Onomura O, Mitsuda M, Nguyen MTT, Demizu Y. Asymmetric tosylation of racemic 2-hydroxyalkanamides with chiral copper catalyst. Tetrahedron Lett. 2007;48:9080–9084. [Google Scholar]
- xv.Demizu Y, Kubo Y, Matsumura Y, Onomura O. Nonenzymatic kinetic resolution of 3-hydroxyalkanamides with chiral copper catalyst. Synlett. 2008:433–437. [Google Scholar]
- xvi.Sibi MP, Liu M. Reversal of stereochemistry in enantioselective transformations. Can they be planned or are they just accidental? Curr Org Chem. 2001;5:719–755. [Google Scholar]
- xvii.Sculimbrene BR, Morgan AJ, Miller SJ. Enantiodivergence in small molecule catalysis of asymmetric phosphorylation: concise total syntheses of the enantiomeric D-myo-inositol-1-phosphate and D-myo-inositol-3-phosphate. J Am Chem Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]
- xviii.Haque TS, Little JC, Gellman SH. Stereochemical requirements for β-hairpin formation: model studies with four-residue peptides and depsipeptides. J Am Chem Soc. 1996;118:6975–6985. [Google Scholar]
- xix.For a Schotten-Baumann-type sulfonylation method using N-alkylimidazole catalysts, see: Asano K, Matsubara S. Amphiphilic organocatalyst for Schotten-Baumann-type tosylation of alcohols under organic solvent free condition. Org Lett. 2009;11:1757–1759. doi: 10.1021/ol900125y.
- xx.Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. Remote desymmetrization at near-nanometer group separation catalyzed by a miniaturized enzyme mimic. J Am Chem Soc. 2006;128:16454–16455. doi: 10.1021/ja067840j. [DOI] [PubMed] [Google Scholar]
- xxi.Lewis CA, Gustafson JL, Chiu A, Balsells J, Pollard D, Murry J, Reamer RA, Hansen KB, Miller SJ. A case of remote asymmetric induction in the peptide-catalyzed desymmetrization of a bis(phenol) J Am Chem Soc. 2008;130:16358–16365. doi: 10.1021/ja807120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxii.Schreiber SL, Schreiber TS, Smith DB. Reactions that proceed with a combination of enantiotopic group and diastereotopic face selectivity can deliver products with very high enantiomeric excess: experimental support of a mathematical model. J Am Chem Soc. 1987;109:1525–1529. [Google Scholar]
- xxiii.Copeland GT, Jarvo ER, Miller SJ. Minimal acylase-like peptides. Conformational control of absolute stereospecificity. J Org Chem. 1998;63:6784–6785. doi: 10.1021/jo981642w. [DOI] [PubMed] [Google Scholar]
- xxiv.Denmark SE, Beutner GL. Lewis base catalysis in organic synthesis. Angew Chem, Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- xxv.Pirrung MC. Histidine kinases and two-component signal transduction systems. Chem Biol. 1999;6:R167–R175. doi: 10.1016/S1074-5521(99)80044-1. [DOI] [PubMed] [Google Scholar]
- xxvi.Jencks WP. Catalysis in Chemistry and Enzymology. Dover; 1975. pp. 218–226. [Google Scholar]
- xxvii.Anantharaman V, Aravind L, Koonin EV. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr Opin Chem Biol. 2003;7:12–20. doi: 10.1016/s1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- xxviii.Yoon TP, Jacobsen EN. Privileged chiral catalysts. Science. 2003;299:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.