

Abstract
Borrelia burgdorferi, the Lyme disease spirochete, adapts as it moves between the arthropod and mammalian hosts that it infects. We hypothesize that BosR serves as a global regulator in B. burgdorferi to modulate the oxidative stress response and adapt to mammalian hosts. To test this hypothesis, a bosR mutant in a low passage B. burgdorferi isolate was constructed. The resulting bosR::kanR strain was altered when grown microaerobically or anaerobically suggesting that BosR is required for optimal replication under both growth conditions. The absence of BosR increased the sensitivity of B. burgdorferi to hydrogen peroxide and reduced the synthesis of Cdr and NapA, proteins important for cellular redox balance and the oxidative stress response, respectively, suggesting an important role for BosR in borrelial oxidative homeostasis. For the bosR mutant, the production of RpoS was abrogated and resulted in the loss of OspC and DbpA, suggesting that BosR interfaces with the RpoS-RpoN-Rrp2 regulatory cascade. Consistent with the linkage to RpoS, cells lacking bosR were non-infectious in the mouse model of infection. These results indicate that BosR is required for resistance to oxidative stressors and provides a regulatory response that is necessary for B. burgdorferi pathogenesis.
INTRODUCTION
The etiological agent of Lyme disease is the spirochete Borrelia burgdorferi and is transmitted by the bite of an infected tick. Infection with B. burgdorferi results in a multisystem disorder characterized by a skin lesion known as erythema migrans that is accompanied by a flu-like illness. If treatment is not sought at this stage, the infected individual is at risk for developing later stage pathology including carditis, neurologic issues, and arthritic manifestations, which contribute to the profound morbidity associated with this infection (Nadelman and Wormser, 1998; Steere et al., 2004).
Given the recent advent of borrelial genetic techniques (reviewed in (Rosa et al., 2005), the ability to evaluate individual loci in the context of experimental Lyme infection is now possible, assuming that the gene in question is not essential for in vitro growth. We have a long-standing interest in the role of the BosR regulatory protein, a homologue of the Fur family of regulators (Boylan et al., 2003; Hyde et al., 2006; Katona et al., 2004; Seshu et al., 2004b), in the ability of B. burgdorferi to regulate the oxidative stress response in this pathogen. Within the Fur family, BosR is most similar to PerR, a repressor that regulates genes involved in the oxidative stress response in Bacillus spp. (Bsat et al., 1998b; Fuangthong et al., 2002; Herbig and Helmann, 2001a; Mongkolsuk and Helmann, 2002). In contrast to PerR, Boylan et al. demonstrated that BosR activates expression of target borrelial genes involved in the oxidative stress response including napA (dps) and a CoA disulfide reductase designated cdr (Boylan et al., 2003; Boylan et al., 2006). In this regard, the role of BosR resembles that of OxyR, an activator in E. coli that promotes the expression of genes required for oxidative stress detoxification (Georgiou, 2002; Storz and Imlay, 1999; Zheng et al., 1998). In biochemical assays, BosR binds to putative operator sequences upstream of napA (dps), cdr, superoxide dismutase (sodA), bosR, bb0646, and oppA-V, providing suggestive evidence that BosR functions as a global regulator to many unlinked genes within the B. burgdorferi genome (Boylan et al., 2003; Boylan et al., 2006; Katona et al., 2004; Medrano et al., 2007; Seshu et al., 2004b). To date, all genetic analyses related to bosR have been limited to non-infectious isolates of B. burgdorferi (Hyde et al., 2006; Seshu et al., 2004b); that is, all prior attempts to genetically inactivate bosR in low-passage isolates have been unsuccessful.
Using a previously described limiting dilution transformation method (Yang et al., 2004), we have isolated a genetic knockout of bosR in low-passage B. burgdorferi and have begun characterizing the importance of this regulator in the physiological and pathogenic properties of this spirochete. As expected, the bosR mutant in low-passage B. burgdorferi is impaired during in vitro growth. We also found that the bosR mutant is more sensitive to H2O2 and exhibits differential production of proteins involved in the oxidative stress response. Unexpectedly, the bosR mutant was incapable of synthesizing RpoS, which functions as a master switch for expression of a number of genes during mammalian infection (Burtnick et al., 2007; Caimano et al., 2004; Caimano et al., 2007; Fisher et al., 2005; Hubner et al., 2001; Yang et al., 2000b), including those encoding the well-characterized virulence determinants OspC and DbpA (Blevins et al., 2008; Caimano et al., 2004; Gilbert et al., 2007; Grimm et al., 2004a; Pal et al., 2004; Shi et al., 2008; Tilly et al., 2007; Weening et al., 2008; Yang et al., 2005). As such, BosR is not only involved in the response to reactive oxygen species (ROS), but also interfaces with the pathogenic potential of B. burgdorferi, suggesting that BosR plays a crucial role in both borrelial oxidative homeostasis and the adaptive response important for infectivity.
RESULTS
Inactivation of bosR in low-passage B. burgdorferi
All prior attempts to isolate a bosR mutant in infectious B. burgdorferi using BSK-II agarose plating were unsuccessful and, given our recent success isolating a dbpBA deletion strain using a liquid limiting dilution method (Weening et al., 2008; Yang et al., 2004), this approach was utilized for the inactivation of bosR in low-passage B. burgdorferi. B. burgdorferi strain B31 derivative ML23 was transformed with pJS167 that contains the bosR::kanR allele, used previously to inactivate bosR in non-infectious B. burgdorferi (Seshu et al., 2004b), to isolate strain JH300 (Fig. 1A). Transformants were obtained and the putative mutants screened for the bosR::kanR allele by both PCR (Fig. 1B) and Southern blot analysis (data not shown). Transformants were identified by the amplification of a 2.5-kb band representing bosR interrupted by a 2-kb stable transposon that confers resistance to kanamycin in B. burgdorferi (JH300, Fig. 1B). The same oligonucleotide primers amplify the approximate 0.5-kb native bosR gene (ML23, Fig. 1B). Those candidates that contained the desired bosR::kanR allele were then tested for their ability to produce BosR. Three independently isolated mutants were analyzed by Western immunoblot analysis under conditions where the parental strain produced BosR. All putative mutants did not produce BosR and one representative clone, designated JH300, was chosen for further study (Fig. 1C). The same compendium of plasmids was observed for JH300 as is seen for ML23 (i.e., missing cp9 and lp25; data not shown) (Labandeira-Rey and Skare, 2001a; Labandeira-Rey et al., 2003). Subsequently, JH300 was transformed with pCADDY, which contains the bbe22 (pncA) gene on a streptomycin resistant borrelial shuttle vector. This plasmid restores infectivity to the lp25 deficient strain ML23 (Purser et al., 2003) and its transformation into JH300 was required to determine how the inactivation of bosR affects borrelial pathogenesis.
FIG. 1.

Schematic of the bosR-containing operon and screening of bosR::kanR mutant, JH300, in B. burgdorferi. A. The schematic depicts the chromosomal region of B. burgdorferi containing bosR and the genetic disruption of bosR with the insertion of PflgB-kanR resulting in strain JH300. The arrows indicate the location of oligonucleotide primers utilized to screen clones for presence of kanR cassette. B. PCR screen of JH300 was performed using primers specific for bosR that amplified a 586 bp product in ML23 or an approximate 2.5-kb product in JH300, indicating the insertion of PflgB-kanR. Lane 1: ML23; Lane 2: JH300. Markers (in bp) are shown on the left. C. BosR is not produced in strain JH300. Cultures were grown to 5 × 107/ml and cell lysates were resolved by SDS-PAGE, immobilized on PVDF membranes, and probed with antisera specific for BosR.
The growth phenotype of strain JH300
To determine how the loss of bosR affects B. burgdorferi growth, JH300 was grown both microaerobically and anaerobically and compared with its isogenic parental strain ML23. Microaerobically grown JH300 showed no lag in growth but exhibited a significant decrease in growth rate (as seen by the decreased slope) and did not attain the maximum cell density relative to its parental strain ML23 (Fig. 2A). Anaerobically grown JH300 exhibited a longer lag period than did ML23 (approximately 3 days), but demonstrated a comparable exponential growth rate and achieved the same final cell density seen for ML23 (Fig. 2B). The difference in cell growth for the bosR mutant when grown microaerobically and anaerobically suggests that BosR is needed to mediate different adaptive responses under these different growth conditions. It is important to note that for anaerobically grown JH300, there is a significant difference in cell density when a given day of growth is compared; however, once the lag in growth is relieved, the normal rate of growth is observed.
FIG. 2.

The bosR::kanR mutant, JH300, displays a delayed growth phenotype relative to the parental, ML23 under microaerobic and anaerobic conditions. Independent cultures were grown in triplicate, cell densities averaged and standard error indicated by bars. A. ML23 (dark squares) and JH300 (open circles) were grown under microaerobic conditions (3.48 ppm dissolved O2, 1% CO2). B. ML23 (dark squares) and JH300 (open circles) were grown under anaerobic conditions (0.087 ppm dissolved O2, 5% CO2). Asterisks indicate significant differences in cell density for a given day (P < 0.001).
Sensitivity of the bosR mutant to hydrogen peroxide
Given the linkage of BosR to the oxidative stress response in B. burgdorferi (Boylan et al., 2003; Boylan et al., 2006; Seshu et al., 2004b), we were interested in determining how the bosR mutant compared with its parental strain in its resistance to ROS. To this end, B. burgdorferi strains JH300 and ML23 were treated with H2O2 in concentrations ranging from 1 mM to 50 mM for 4 hours and compared to samples not exposed to H2O2. The incorporation of propidium iodide was tracked to assess the toxicity of H2O2 treatment since cells with compromised/depolarized cytoplasmic membranes rapidly take up propidium iodide. The resulting samples were then scored by flow cytometry to obtain an unbiased evaluation of cell viability for the samples and treatment tested. Comparison of strain JH300 and its parent ML23 indicated that the cells making BosR (ML23) were more resistant to H2O2 than those that did not make BosR (JH300), with the exposure at 5 mM H2O2 yielding the most pronounced differential in cell viability (Fig. 3). Statistical analyses indicated that a significant difference is observed across all concentrations of H2O2 tested (logit analysis yielded a P < 0.001). It is also important to emphasize that each data point shown is based on approximately 40,000 borrelial cells for each strain and concentration of H2O2 tested, and summarizes the data set from two independent experiments.
FIG. 3.

BosR is required for optimal protection from H2O2. Two independent anaerobic cultures were grown to exponential phase and treated with various concentrations of H2O2 up to 50 mM each in triplicate. JH300 (open circles) is significantly more sensitive to killing by H2O2 relative to parental, ML23 (solid squares) with a P value across all H2O2 concentrations tested of less than 0.001. The lines shown represent the best fit to the logit regression model for the bosR mutant (JH300, top line) and the parental strain (ML23, bottom line) for the data obtained. Please note that the data points for the 1 mM H2O2 treatment are of equal value for both strains tested and thus overlap one another.
Inactivation of bosR alters the production of oxidative stress proteins
To determine how BosR levels affect the production of proteins involved in the oxidative stress response, we assessed the levels of BosR, NapA, Cdr, and SodA in strain JH300 when grown both anaerobically and microaerobically. BosR production was undetectable in JH300 under all tested growth conditions but was abundantly produced in strains MSK5 and ML23 grown anaerobically with CO2 present (Fig. 4) as previously reported (Hyde et al., 2007). The increase in BosR in MSK5 and ML23 under these conditions is not due to an increase in transcription since bosR transcripts were unchanged under both microaerobic and anaerobic conditions (Hyde et al., 2007), and instead reflects a post-transcriptional regulatory effect that is not yet characterized.
FIG. 4.

BosR is required for maximal expression of NapA and Cdr. Wild type infectious MSK5, parental strain ML23 and JH300 (ML23 bosR::kanR) were grown under both anaerobic (A) and microaerobic (M) growth conditions. Samples were probed with antibodies specific for the antigen indicated. Endoflagellum (EF) levels were used as a control to normalize for equivalent protein loading.
The production of NapA was increased in the low-passage parental strains ML23 and MSK5 (infectious B31 strain containing all known plasmids) relative to the bosR mutant JH300 (Fig. 4), when the parental strains were grown anaerobically. The increased production of NapA during anaerobiosis (Fig. 4) is consistent with the increased production of BosR under anaerobic conditions as previously described (Hyde et al., 2007). Although JH300 is capable of producing NapA, the absence of BosR prevents the increased production of NapA during anaerobic growth as is observed for ML23 and MSK5 (Fig. 4). A similar pattern was observed for Cdr (Fig. 4), a protein required for redox balance in the borrelial cytoplasm (Boylan et al., 2006). The results for NapA and Cdr provide further support to the prior contention that BosR functions as an activator for both napA and cdr (Boylan et al., 2003; Boylan et al., 2006). Given that both NapA and Cdr were produced at greater levels in ML23 and MSK5, it was surprising that SodA levels were not affected by the absence of BosR (Fig. 4). Nevertheless, based on the response seen for both NapA and Cdr, it is clear that BosR is required for optimal production of a subset of borrelial oxidative stress responsive proteins.
The loss of bosR abrogates RpoS-dependent regulation
When strain JH300 was grown anaerobically and compared with anaerobically grown ML23 and MSK5, it was apparent that JH300 did not synthesize an abundant 23-kDa protein species (Fig. 5A). Previous work demonstrated that OspC is induced under anaerobic growth (Hyde et al., 2007) and, given this similarity, we asked whether the bosR mutant was unable to make OspC and DbpA, as well as RpoS. The profiles observed indicate that the loss of bosR in JH300 abrogates the production of detectable levels of RpoS, OspC and DbpA when these cells are grown anaerobically (Fig. 5B). Thus, BosR appears to interface with the activation of rpoS, which is an alternative sigma factor needed for both ospC and dbpBA expression (Caimano et al., 2004; Caimano et al., 2007; Fisher et al., 2005; Gilbert et al., 2007; Hubner et al., 2001; Yang et al., 2005). Since both OspC and DbpA play important roles in borrelial pathogenesis, the linkage of BosR to their production implicates BosR in the pathogenic potential of B. burgdorferi. Since RpoS production is dependent on the Rrp2-RpoN system (Boardman et al., 2008; Burtnick et al., 2007; Caimano et al., 2004; Caimano et al., 2007; Fisher et al., 2005; Hubner et al., 2001; Yang et al., 2000b; Yang et al., 2003), the steady-state levels of Rrp2 and its cognate histidine Hk2 were evaluated. The absence of BosR had no effect on the production of either Rrp2 or Hk2 (data not shown for Rrp2 and Fig. 5B).
FIG. 5.

BosR alters the synthesis of RpoS and virulence determinants regulated by the Rrp2-RpoN-RpoS pathway. Wild type infectious MSK5, parental strain ML23, and JH300 (ML23 bosR::kanR) were grown under anaerobic (A) and microaerobic (M) growth conditions to 5×107 cells/ml. A. Coomassie-stained SDS-PAGE gel of cell lysates. The arrow indicates the presence of a 23-kDa protein in MSK5 and ML23 that is absent in JH300. B. Samples were probed with antibodies specific for the antigen indicated. As in Fig. 4, endoflagellum (EF) is used as a control to normalize for equivalent protein loading.
It is important to note that RpoS, OspC and DbpA are all present in microaerobically grown ML23 and MSK5 but at levels significantly below that seen for anaerobically grown B. burgdorferi. Furthermore, each lane is equivalently loaded as shown in the endoflagellum (EF) control immunoblot and Coomassie blue-stained gel (Fig. 5). Longer exposures did show the presence of RpoS, OspC and DbpA in microaerobically grown ML23 and MSK5 (not shown). However, even at these longer exposures, no detectable levels of RpoS, OspC or DbpA were visualized for JH300 grown either microaerobically or anaerobically.
Role of bb0646 in the JH300 phenotype
Another possible explanation for the growth phenotype and change in antigen production observed for strain JH300 could be the elimination of the gene downstream from bosR, bb0646, which is predicted to encode an exported lipase (Katona et al., 2004). To address this possibility, strain DS102 (ML23 bb0646::gentR) was compared with strains ML23 and JH300 for various phenotypic traits. Recent results indicated that DS102 exhibited no overt defect in growth rate relative to its parental strain (Hyde et al., 2010), suggesting that the growth defect seen for strain JH300 was restricted to the loss of bosR and not bb0646.
To assess the role of BB0646 in antigen production previously attributed to BosR-dependent mechanisms, we tested the ability of DS102 to generate RpoS and other RpoS-dependent proteins. The results indicated that DS102 makes unimpaired levels of BosR, and more importantly RpoS (Fig. 6) and OspC (data not shown). Furthermore, we have recently tested strain DS102 in the mouse model of experimental Lyme borreliosis and determined that strains lacking BB0646 retain infectivity (Shaw and Skare, unpublished observations). Taken together, these data support the notion that the phenotype observed for JH300 is associated with the absence of BosR and is not due to a polar effect involving the loss of bb0646.
FIG. 6.
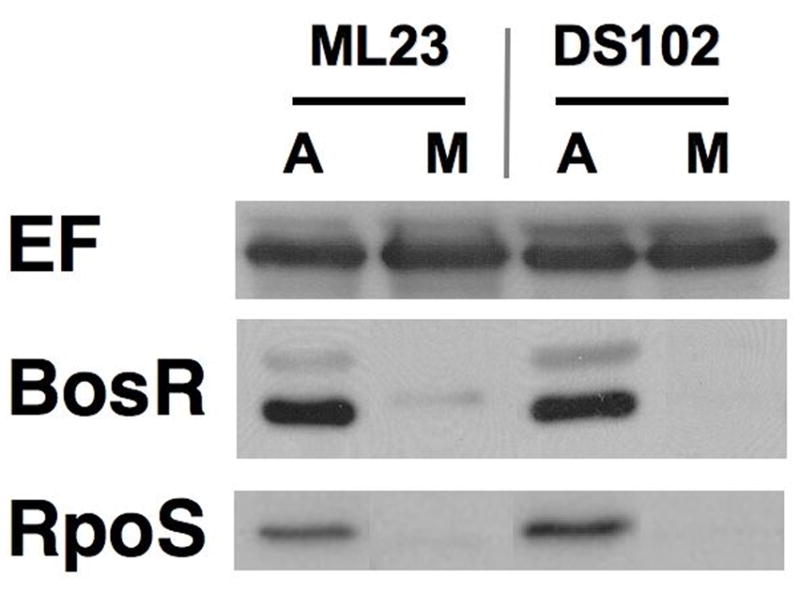
ML23 bb0646::gentR does not alter the synthesis of RpoS. The parental strain ML23 and DS102 (ML23 bb0646::gentR) were grown anaerobically and microaerobically, resolved by SDS-PAGE, immobilized on PVDF membrane and probed with antisera for BosR and RpoS. Endoflagellum (EF) was used as a control for equivalent protein loading.
Infectivity of B. burgdorferi lacking BosR
To assess the role of BosR in borrelial pathogenesis, we tested whether B. burgdorferi lacking bosR were able to infect mice. Although the inability of the bosR mutant to produce OspC and DbpA suggested that the strain would be significantly impaired in its ability to infect, it is possible that the effect seen is limited to an in vitro growth characteristic of bosR-deficient B. burgdorferi. When mouse infections were conducted with JH300 pCADDY and compared with its infectious parental strain ML23 pCADDY, we found that JH300 pCADDY supported no infectivity at either a 103 or a 105 inoculum dose (Table 3). This is in contrast to the parental strain ML23 pCADDY, which infected all mice at each inoculum dose tested. The largest limitation of this comparison was our inability to genetically complement the bosR mutant either in trans on a shuttle vector or via cis complementation at a heterologous site in the chromosome (Li et al., 2007). Despite this limitation, these infectivity results, coupled with the linkage of BosR to RpoS production, strongly suggest that BosR plays an important role in the pathogenic potential of B. burgdorferi.
Table 3.
Infectivity data for C3H/HeN mice infected with B. burgdorferi ML23 pCADDY and JH300 pCADDY.
| Number of culture positive/total number | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | Inoculum Dose | Lymph Node | Skin | Heart | Spleen | Bladder | Joint | All sites | No. of mice positive/total mice |
| ML23 pCADDY | 105 | 5/5 | 5/5 | 5/5 | 1/5 | 5/5 | 5/5 | 26/30 | 5/5 |
| 103 | 3/5 | 3/5 | 2/5 | 1/5 | 3/5 | 3/5 | 15/30 | 5/5 | |
| JH300 pCADDY | 105 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/30 | 0/5 |
| 104 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/18 | 0/3 | |
| 103 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/30 | 0/5 | |
DISCUSSION
With improved molecular genetic tools to evaluate various genes in Borrelia burgdorferi, we are beginning to get a better understanding of loci that are key for the pathogenesis of Lyme borreliosis. Although several important pathways have been identified, most notably the Rrp2-RpoN-RpoS regulatory cascade (Boardman et al., 2008; Burtnick et al., 2007; Caimano et al., 2004; Caimano et al., 2007; Fisher et al., 2005; Hubner et al., 2001; Yang et al., 2000b; Yang et al., 2003; Yang et al., 2005), it remains unclear how B. burgdorferi integrates the signals it perceives into a regulatory response. Several in vitro correlates have been established that simulate environmental conditions as the spirochete moves from an infected tick to a mammalian host following a blood meal, particularly increases in temperature, fluctuations in pH, dissolved oxygen, CO2 and other uncharacterized mammalian specific factors (Akins et al., 1995; Akins et al., 1998; Brooks et al., 2003; Carroll et al., 1999; Cassatt et al., 1998; Champion et al., 1994; Das et al., 1997; Fikrig et al., 1997; Hyde et al., 2007; Lybecker and Samuels, 2007; Ojaimi et al., 2003; Ojaimi et al., 2005; Revel et al., 2002; Schwan et al., 1995; Seshu et al., 2004a; Skare et al., 1999; Stevenson et al., 1995; Tokarz et al., 2004; Yang et al., 2000a). Despite these important advances in the molecular and genetic analyses of B. burgdorferi, much remains unknown regarding genes that contribute to the pathogenic potential of B. burgdorferi as well as the transcriptional regulatory proteins that affect the expression of these genes.
In this study, we demonstrate that the BosR regulatory protein is linked to the pathogenic potential of B. burgdorferi since it is needed for the synthesis of RpoS (Fig. 5B), which in turn is required for the expression of the known borrelial virulence determinants, most notably ospC and dbpBA (Blevins et al., 2008; Caimano et al., 2004; Gilbert et al., 2007; Grimm et al., 2004b; Pal et al., 2004; Shi et al., 2008; Tilly et al., 2007; Weening et al., 2008; Yang et al., 2005). We also show that BosR is linked to the optimal response to ROS and affects the production of NapA and Cdr, two proteins involved in combating oxidative stress and maintaining the proper redox balance in B. burgdorferi (Boylan et al., 2003; Boylan et al., 2006; Li et al., 2007). These data are consistent with the similarity that BosR shares with the oxidative stress regulator PerR from Bacillus spp. (Bsat et al., 1998a; Fuangthong et al., 2002; Herbig and Helmann, 2001b; Mongkolsuk and Helmann, 2002). As such, BosR is linked to both physiologically critical and virulence-associated pathways in a manner that we predict is RpoS-independent and RpoS-dependent, respectively. Our working hypothesis is that BosR responds to the redox environment of the cell and modulates gene expression accordingly in either a direct or indirect manner. Since the redox status is likely to change as B. burgdorferi traffics through the tick vector and mammalian host, regulation by BosR might represent an important adaptive response that this pathogen uses to colonize such disparate hosts.
The most direct approach to evaluate the importance of BosR is by genetically inactivating or deleting bosR; however, prior attempts to inactivate bosR in infectious B. burgdorferi were unsuccessful. All of the previous attempts to isolate bosR mutants used agarose plating and the ability to visualize colonies may have been impaired since the mutant exhibits a delayed growth rate. Because transformations involving B. burgdorferi are inefficient, one can dilute a transformation population in liquid media under appropriate selection and obtain clonal isolates (Yang et al., 2004). When this method was used to select bosR mutants, several ML23 bosR::kanR clones were isolated, characterized, and a representative strain designated as JH300 (Fig. 1).
The growth defect observed for JH300, especially when grown microaerobically, may have contributed to the difficulty in isolating a bosR mutant previously, given the inability of this strain to replicate to high densities (Fig. 2). One striking feature of JH300 is its increased ability to attain maximal densities when the cells are grown anaerobically. This is consistent with the idea that a critical detoxification function for BosR would not be required under anaerobiosis when the levels of dissolved oxygen, and thus levels of ROS, would be reduced.
The increased sensitivity of the bosR mutant to H2O2 is consistent with a role of BosR in regulating the oxidative stress response; however, we predicted that the differential would be more pronounced (Fig. 3). The high level resistance to H2O2 observed in the parental strain is consistent with recent results reported by Boylan et al. indicating that borrelial cells are more refractory to H2O2-mediated oxidation than other bacteria (Boylan et al., 2008), mostly due to their lack of iron and thus the concomitant absence of Fenton-based DNA damage (Posey and Gherardini, 2000). Boylan et al. also showed that B. burgdorferi are more sensitive to alkyl peroxides that can oxidize polyunsaturated lipids that these spirochetes incorporate into their cell envelope (Boylan et al., 2008). The difference in H2O2 sensitivity between the bosR and wild type strains observed here suggests that BosR does neutralize the oxidative stress imposed by H2O2, due instead to damage incurred by the oxidation of borrelial proteins and lipids, but not nucleic acids. Another protein whose levels we thought would be adversely affected by BosR levels was SodA since purified BosR was found to bind to a putative sodA operator domain (Seshu et al., 2006). However, the bosR mutant showed no change in the level of SodA (Fig. 4). Recently, Esteve-Gassent et al. demonstrated that sodA mutants show an increased sensitivity to superoxide anion generating compounds relative to their isogenic parent (Esteve-Gassent et al., 2009). While it is possible the bosR and sodA mutants may have similar sensitivity phenotypes to other oxidative stressors, including H2O2, it is equally as plausible that the response seen to H2O2 and other peroxides would be distinct given the fact the SodA provides a specific enzymatic function (i.e., conversion of superoxide anion to H2O2) whereas BosR presumably regulates many unlinked genes involved in the oxidative stress response (i.e., NapA and Cdr) and thus would likely have a more profound overall effect on the borrelial ROS detoxification. It is important to note that the analysis described herein only accounts for steady-state levels of SodA. It is possible that BosR levels affect sodA transcription and the lack of an observed effect may be due to the stability of the borrelial SodA protein.
The observation that the oxidative stress responsive proteins NapA and Cdr are induced when the wild-type (MSK5) and parental strain (ML23) are grown anaerobically (Fig. 4) is somewhat counterintuitive to the proposed functions of these proteins; that is, one might predict that the NapA and Cdr would be selectively produced under microaerobic growth and made at lower levels when borrelial cells are grown under hypoxic conditions. This observation may reflect differences in CO2 content in the growth of these spirochetes. Higher concentrations of CO2 (5%) simulate, in part, mammalian host conditions and serve as an environmental cue independent of oxygen (Hyde et al., 2007; Klengel et al., 2005; Mitchell, 2005). A CO2 effect was observed for NapA production such that when CO2 was limited, independent of the presence or absence of oxygen, less NapA was synthesized relative to conditions where CO2 levels were increased (Hyde et al., 2007). Thus, a CO2 regulatory effect is operative that limits NapA production when the media is purged of all gases, including CO2 (Hyde et al., 2007). Under these conditions one might expect that NapA and Cdr production would also be altered in the bosR mutant but, more importantly, would not be able to respond as effectively to make more of these proteins when either CO2 or O2 levels are increased.
Perhaps the most interesting characteristic of the bosR mutant is its link to RpoS, and thus OspC and DbpA, proteins that are important to establish infection (Blevins et al., 2008; Caimano et al., 2004; Grimm et al., 2004b; Pal et al., 2004; Shi et al., 2008; Tilly et al., 2007; Weening et al., 2008; Yang et al., 2005). The fact that OspC and DbpA levels are dependent on BosR in wild type B. burgdorferi suggests that BosR in some way interfaces with the Rrp2/RpoN/RpoS two-component regulatory system in a manner that results in the production of key virulence determinants in B. burgdorferi (Fig. 5B). Based on the BosR-dependent production of RpoS seen in ML23 and MSK5 relative to JH300 (Fig. 5B), BosR is required for rpoS expression, probably in an indirect but perhaps in a direct manner, or is involved (indirectly) in a post-transcriptional regulatory event that promotes the synthesis of RpoS. The connection of BosR to RpoS induction via BosR is not known but may involve the following: (i) alteration of the phosphorylation status of Hk2 (the cognate histidine kinase genetically linked to rrp2) or other phosphorylated intermediates that activate Rrp2 and subsequently rpoS (Boardman et al., 2008); (ii) expression of rpoN; and/or (iii) induction of DsrA, a small regulatory RNA required for efficient translation of rpoS transcripts into RpoS (Lybecker and Samuels, 2007).
In regard to the potential role of Hk2 in the BosR-specific induction of ospC and dbpBA, it is important to note that the absence of BosR in JH300 did not affect the production of Hk2 or Rrp2 (Fig. 5B for Hk2 and data not shown for Rrp2). This is consistent with the idea that the activation of these proteins does not require increased transcription since these two-component regulatory proteins are generally constitutively expressed and the resulting proteins activated post-translationally via a reversible phosphorylation event. Furthermore, the production of RpoS can occur independent of Hk2 given that an hk2 mutant can still transcribe rpoS via Rrp2 (Burtnick et al., 2007). The genes that may contribute to the phosphorylation status of Rrp2, in either an Hk2-dependent or independent manner, are not known.
While we were encouraged by the lack of infectivity seen for JH300 (Table 3), we were disappointed that we were unable to genetically complement this bosR mutant. Attempts to genetically complement JH300 with bosR expressed from its native promoter on a borrelial shuttle vector were unsuccessful, as were cis complementation approaches at a heterologous site in the chromosome. The inability to complement the bosR mutant with a shuttle vector construct may be due to a negative gene dosage effect due to the multicopy status of these plasmids. That is, if the balance of functional BosR is critical for cellular homeostasis, then increased levels of the regulator could result in anomalous expression of target genes with deleterious effects on B. burgdorferi growth. These concerns would presumably be allayed in a cis complementation strategy. However, since we designed our construct to recombine into a non-native sequence in the borrelial chromosome (Li et al., 2007), additional upstream sequences not found in Li et al. configuration, may be necessary for the native expression of bosR.
The lack of infectivity observed for JH300 was expected due to the absence of RpoS production and is consistent with the non-infectious phenotype of the B. burgdorferi rpoS mutant (Caimano et al., 2004). Based on the data obtained, we hypothesize that BosR regulated genes can be categorized as those that (i) maintain oxidative homeostasis in an RpoS-independent manner; (ii) affect the pathogenesis of B. burgdorferi due to RpoS-dependent regulatory events; and (iii) alter the pathogenic potential of the spirochete in an RpoS-independent context. Addition of inducible rpoS back into a bosR mutant, under conditions that synthesize native levels of RpoS, might provide a modality to determine how the BosR-dependent, RpoS-independent gene subset contributes to the infectivity potential of B. burgdorferi.
Another consideration for the bosR::kanR mutation in JH300 is the potential polar effect seen on the downstream gene bb0646, which encodes a putative lipase (Katona et al., 2004). To address the potential impact of the absence of bosR and bb0646 in JH300 relative to the loss of bb0646 alone in strain DS102, we compared some phenotypic traits of these two B. burgdorferi mutants. Strain DS102 exhibited no difference in growth rate relative to its parental strain ML23 (data not shown) and, in stark contrast to JH300, the absence of the putative lipase did not affect the production of RpoS (Fig. 6) or OspC (data not shown). These results suggest that the loss of RpoS and OspC in strain JH300 is due to the absence of BosR and not BB0646 and is consistent with the contention that a global regulator such as BosR would be associated with the regulatory response and/or production of several target proteins whereas a protein with a specific enzymatic function like BB0646 would not be involved in this type of regulation.
Recently an independently isolated strain was characterized whereby the native bosR promoter was replaced with a previously developed IPTG inducible promoter system (Pflac; Gilbert et al., 2007). Specifically, a Pflac-bosR construct was integrated into the borrelial chromosome (and linked with an antibiotic marker) replacing the native bosR locus (Hyde et al., 2010). When IPTG was selectively restricted, a conditional bosR mutant was obtained such that this strain exhibited reduced growth commensurate with that observed here for strain JH300 and did not make detectable levels of BosR. Conversely, if IPTG was added back to the culture, the growth of the cells mirrored that observed for strain ML23 and BosR was readily observed (Hyde et al., 2010). In addition, the induction of bosR was required for maximal resistance to H2O2 and resulted in the increased synthesis of NapA and SodA. Moreover, growth in the presence of IPTG and the concomitant production of BosR promoted the synthesis of OspC and DbpA, suggesting that BosR interfaced with rpoS (Hyde et al., 2010). B. burgdorferi cells grown in the absence of IPTG were non-infectious in immunodeficient mice (the parent strain lacks plasmid lp28-1 that is required for persistent infection; Labandeira-Rey et al., 2003) as observed for JH300 in immunocompetent mice described here (Table 3). Taken together with the data presented here, these results serve as an independent corroboration of the role of BosR in both oxidative stress and pathogenic properties of B. burgdorferi and further validate the importance of the BosR regulator in borrelial biology.
Herein we describe the isolation and characterization of a knockout mutant of bosR in a low passage isolate of B. burgdorferi. Initial characterization indicated that BosR is required for the normal replication of B. burgdorferi when the cells are cultured either microaerobically or anaerobically. Furthermore, B. burgdorferi cells to which BosR is absent are more sensitive to H2O2 (Fig. 3), suggesting that BosR regulatory events are required for the high level resistance to ROS seen for B. burgdorferi (Boylan et al., 2008). Moreover, bosR mutants are non-infectious presumably because the production of BosR is required for the optimal expression of genes involved in both the oxidative stress response (i.e., napA and cdr) and virulence (rpoS and rpoS-regulated genes). Array-based analyses will be helpful in identifying genes regulated by BosR to better define the loci needed for the borrelial oxidative detoxification response and to determine if and how BosR regulated RpoS-dependent and RpoS-independent genes factor into the pathogenesis of B. burgdorferi.
EXPERIMENTAL PROCEDURES
Bacterial Strains
B. burgdorferi strains, B31 derivatives, were grown in BSK-II medium supplemented with 6% normal rabbit serum (NRS) either microaerobically or anaerobically as described (Table 1) (Hyde et al., 2007). Borrelial strains were grown in the appropriate antibiotic for selective pressure as needed: kanamycin at 300 μg/ml; streptomycin at 50 μg/ml; and gentamicin at 50 μg/ml. The Institutional Biosafety Committee at Texas A&M University approved the use of infectious B. burgdorferi described in this study.
Table 1.
Strains and Plasmids used in this study
| B. burgdorferi strains used in this study: | ||
| Strain | Genotype | Reference |
| MSK5 | B31 derivative, all plasmids present | (Labandeira-Rey and Skare, 2001a) |
| ML23 | missing lp25 | (Labandeira-Rey and Skare, 2001a) |
| DS102 | ML23, bb0646::gentR | (Hyde et al., 2010) |
| JH300 | ML23, bosR::kanR | This study |
| E. coli strains used in this study: | ||
| Strain | Genotype | Source |
| Mach-1™-T1R | Φ80lacZΔM15 ΔlacX74 hsdR (rk−, mk+) ΔrecA1398 endA1 tonA |
Invitrogen |
| Plasmids used in this study: | ||
| Plasmid | Resistance | Comments/Source/Reference |
| pCR8/GW/TOPO | specR | Gateway PCR cloning/entry vector; Invitrogen |
| pJS167 | kanR | suicide vector containing bosR::kanR construct (Seshu et al., 2004b) |
| pBBE22 | kanR | borrelial shuttle vector pBSV2 containing pncA fragment to restore infectivity in ML23 (Purser et al., 2003) |
| pKFSS1 | specR/strepR | streptomycin resistant borrelial shuttle vector (Frank et al., 2003); confers specR in E. coli |
| pCADDY | specR/strepR | shuttle vector derived from pBBE22 (Purser et al., 2003) replacing the kanamycin resistance with streptomycin resistant determinant; confers specR in E. coli |
Escherichia coli Mach1™-T1R cells were used for all cloning steps and were transformed with appropriate PCR-amplified products cloned into pCR8/GW/TOPO (Invitrogen Corp., Carlsbad, CA). The E. coli cells were grown with aeration in LB media at 37 C. For experiments involving E. coli, antibiotics were used at the following concentrations: spectinomycin at 100 μg/ml and kanamycin at 50 μg/ml.
Plasmid constructs
A streptomycin-resistant shuttle vector carrying the bbe22 and bbe23 region of lp25 to restore infectivity was constructed and designated pCADDY (Table 1). To obtain pCADDY, pBBE22 and pKFSS1 were digested with NdeI and AatII to remove the aphI (confers resistance to kanamycin; kanR) and aadA (confers resistance to streptomycin; strR) genes, respectively. The pKFSS1 fragment containing aadA was then cloned into the pBBE22 backbone replacing the aphI gene. The construct was screened by restriction digest and candidates confirmed by sequence analysis.
Transformation of B. burgdorferi
B. burgdorferi strains ML23 and JH300 were made competent, electroporated, and transformants isolated with antibiotics (kanamycin or streptomycin as appropriate depending on the construct), as previously described (Samuels, 1995; Seshu et al., 2004b; Seshu et al., 2006; Weening et al., 2008). Electroporated cells were allowed to recover overnight in 15 ml of complete BSK media. Following an overnight incubation, the final volume was increased to 45 ml and 180 μl of 0.5% phenol red was added along with the appropriate antibiotic to allow for improved visibility due to a decrease in pH and to impose a selective pressure, respectively. Cultures were then distributed in 180 μl volumes across two 96 well plates and incubated for several weeks until a change in the pH indicator is detected, suggestive of growth (Yang et al., 2004). Putative transformants were then screened by PCR, Southern, and Western analysis.
PCR
PCR was done using Invitrogen Supermix High Fidelity as described previously with the oligonucleotide primers listed in Table 2 (Weening et al., 2008).
Table 2.
Oligonucleotides used in this study.
| Designation | Oligonucleotide sequence (5′-3′) | Description |
|---|---|---|
| bosRFBamHI | GGATCCTGCTCCAAATCCATGAATA | Amplifies bosR with 58 bp upstream sequence resulting in a 586 bp product |
| bosRRPstI | CTGCAGTTTAAATGTTGAAAAAGATA |
SDS-PAGE and Western Immunoblotting
B. burgdorferi cultures were grown to 5×107 cells/ml and samples were taken for Western analysis. Borrelial protein lysates were resolved by SDS-12.5% polyacrylamide gel electrophoresis (PAGE) as previously described (Hyde et al., 2007). Proteins were resolved by SDS-PAGE and transferred to PVDF membrane for Western analysis as indicated previously (Hyde et al., 2007; Seshu et al., 2004a; Seshu et al., 2004b; Seshu et al., 2006; Weening et al., 2008). Antibodies were used at the following dilutions: murine anti-flagellum (Affinity Bioreagent, Golden, CO) at 1:20,000; rabbit anti-NapA at 1:100,000 (generously provided by Frank Gherardini) (Boylan et al., 2003); rabbit anti-Cdr at 1:1000 (generously provided by Frank Gherardini) (Boylan et al., 2006); rabbit anti-OspC at 1:1,000 (generously provided by Richard Marconi); rabbit anti-DbpA at 1:10,000 (generously provided by Magnus Höök); and mouse anti-Rrp2 and anti-Hk2 each at 1:40, respectively (generously provided by Xiaofeng Yang). To detect Rrp2 or Hk2, an additional amplification step was required using 1:1000 goat anti-mouse Ig followed by a subsequent incubation with 1:1000 anti-goat conjugated horseradish peroxidase. Finally, secondary antibodies, e.g., anti-mouse or anti-rabbit with conjugated horseradish peroxidase, as appropriate, were used in conjunction with chemiluminescent substrates to detect the requisite antigen-antibody complexes.
Hydrogen peroxide assays and flow cytometry analysis
ML23 and JH300 cultures were inoculated at 105 cells/ml and incubated under anaerobic conditions until reaching a cell density of 5×107 cells per ml. For each assay conducted, 3 independent cultures were grown in triplicate. 5×107 cells were pelleted and resuspended in modified BSK-II lacking BSA and treated with 0, 1, 5, 10, or 50 mM hydrogen peroxide for 4 hours in a 1 ml final volume. Samples were centrifuged and washed with phosphate-buffered saline (PBS) containing 2% BSA and incubated with 100 ng/μl propidium iodide (PI) for 15 minutes in the dark. Following the incubation, the samples were fixed with 4% paraformaldehyde for 10 minutes, pelleted, and washed in PBS, 2% BSA. The resulting samples were resuspended in 1 ml PBS, 2% BSA and analyzed with a FACSCalibur flow cytometer in conjunction with CellQuest acquisition software (Becton Dickinson Immunocytometry Systems, San Jose, CA). The log amplification was used to collect forward scatter, side scatter, and PI fluorescence with the latter collected through a 650 nm longpass filter. The list mode data acquired for each strain, growth condition, and concentration of H2O2 tested was based on approximately 20,000 events (in duplicate) as indicated by light scatter gates. The subsequent data was analyzed with FlowJo (versions 8.7 and 8.8, Treestar, Inc., Ashland, OR), using forward and side light scatter to gate B. burgdorferi. Controls included heat-killed and untreated samples.
Statistical Analysis
The percentage of killed B. burgdorferi at various concentrations of H2O2 was analyzed using a logit regression model. The data analysis was done in R version 2.9.1 (http://CRAN.R-project.org) using the generalized linear models function in conjunction with a binomial error structure. The response data consisted of a two-column array whereby column 1 contained the number of cells killed and column 2 contained the number of viable cells. The explanatory variable used was log10[H2O2]. The various fractional cell kills following H2O2 exposure were predicted from the model for each strain analyzed, as were 95% confidence intervals around each concentration. When compared with log10[H2O2] by itself, the inclusion of the group factor significantly improved the fit of the model tested to P < 0.001, based on χ2 analyses of deviance.
Infectivity Studies
C3H mice at 8 weeks of age were infected intradermally with 103, 104, or 105 of B. burgdorferi strain ML23 pCADDY or JH300 pCADDY. Following 14 days of infection, the mice were sacrificed and skin, spleen, heart, bladder, lymph node, and tibiotarsal joint were aseptically removed and cultivated in BSK-II media supplemented with 6% NRS and appropriate antibiotics. The presence of B. burgdorferi was scored by darkfield microscopy (Gilbert et al., 2007; Labandeira-Rey and Skare, 2001b; Labandeira-Rey et al., 2003; Seshu et al., 2006; Weening et al., 2008). All animal work performed was reviewed and approved by the University Laboratory Animal Care Committee at Texas A&M University.
Acknowledgments
We thank Scott Samuels for generously providing the borrelial shuttle vector pKFSS1 and Patti Rosa and Philip Stewart for sharing the borrelial shuttle vectors pBSV2 and pBSV2G. We also gratefully acknowledge Magnus Höök for providing antibodies to DbpA, Frank Gherardini for generously providing antibodies to B. burgdorferi NapA and Cdr, Richard Marconi for antibodies to strain B31 OspC, and Xiaofeng Yang for providing antibodies specific for borrelial Rrp2 and Hk2. We also thank Rick Verbeek for purifying recombinant B. burgdorferi RpoS. This work was supported by the Public Health Service grant R01-AI042345 (to J.T.S) from the National Institute of Allergy and Infectious Diseases.
References
- Akins DR, Porcella SF, Popova TG, Shevchenko D, Baker SI, Li M, Norgard MV, Radolf JD. Evidence for in vivo but not in vitro expression of a Borrelia burgdorferi outer surface protein F (OspF) homologue. Mol Microbiol. 1995;18:507–520. doi: 10.1111/j.1365-2958.1995.mmi_18030507.x. [DOI] [PubMed] [Google Scholar]
- Akins DR, Bourell KW, Caimano MJ, Norgard MV, Radolf JD. A new animal model for studying Lyme disease spirochetes in a mammalian host-adapted state. J Clin Invest. 1998;101:2240–2250. doi: 10.1172/JCI2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins JS, Hagman KE, Norgard MV. Assessment of decorin-binding protein A to the infectivity of Borrelia burgdorferi in the murine models of needle and tick infection. BMC Microbiol. 2008;8:82. doi: 10.1186/1471-2180-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman BK, He M, Ouyang Z, Xu H, Pang X, Yang XF. Essential role of the response regulator Rrp2 in the infectious cycle of Borrelia burgdorferi. Infect Immun. 2008;76:3844–3853. doi: 10.1128/IAI.00467-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan JA, Posey JE, Gherardini FC. Borrelia oxidative stress response regulator, BosR: A distinctive Zn-dependent transcriptional activator. Proc Natl Acad Sci U S A. 2003;100:11684–11689. doi: 10.1073/pnas.2032956100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan JA, Hummel CS, Benoit S, Garcia-Lara J, Treglown-Downey J, Crane EJ, 3rd, Gherardini FC. Borrelia burgdorferi bb0728 encodes a coenzyme A disulphide reductase whose function suggests a role in intracellular redox and the oxidative stress response. Mol Microbiol. 2006;59:475–486. doi: 10.1111/j.1365-2958.2005.04963.x. [DOI] [PubMed] [Google Scholar]
- Boylan JA, Lawrence KA, Downey JS, Gherardini FC. Borrelia burgdorferi membranes are the primary targets of reactive oxygen species. Mol Microbiol. 2008;68:786–799. doi: 10.1111/j.1365-2958.2008.06204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CS, Hefty PS, Jolliff SE, Akins DR. Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals. Infect Immun. 2003;71:3371–3383. doi: 10.1128/IAI.71.6.3371-3383.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bsat N, Herbig A, Casillas-Martinez L, Setlow P, Helmann JD. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulon (PerR) repressors. Mol Microbiol. 1998a;29:189–198. doi: 10.1046/j.1365-2958.1998.00921.x. [DOI] [PubMed] [Google Scholar]
- Bsat N, Herbig A, Casillas-Martinez L, Setlow P, Helmann JD. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulon (PerR) repressors. Mol Microbiol. 1998b;29:189–198. doi: 10.1046/j.1365-2958.1998.00921.x. [DOI] [PubMed] [Google Scholar]
- Burtnick MN, Downey JS, Brett PJ, Boylan JA, Frye JG, Hoover TR, Gherardini FC. Insights into the complex regulation of rpoS in Borrelia burgdorferi. Mol Microbiol. 2007;65:277–293. doi: 10.1111/j.1365-2958.2007.05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caimano MJ, Eggers CH, Hazlett KR, Radolf JD. RpoS is not central to the general stress response in Borrelia burgdorferi but does control expression of one or more essential virulence determinants. Infect Immun. 2004;72:6433–6445. doi: 10.1128/IAI.72.11.6433-6445.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caimano MJ, Iyer R, Eggers CH, Gonzalez C, Morton EA, Gilbert MA, Schwartz I, Radolf JD. Analysis of the RpoS regulon in Borrelia burgdorferi in response to mammalian host signals provides insight into RpoS function during the enzootic cycle. Mol Microbiol. 2007;65:1193–1217. doi: 10.1111/j.1365-2958.2007.05860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, Garon CF, Schwan TG. Effects of environmental pH on membrane proteins in Borrelia burgdorferi. Infect Immun. 1999;67:3181–3187. doi: 10.1128/iai.67.7.3181-3187.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassatt DR, Patel NK, Ulbrandt ND, Hanson MS. DbpA, but not OspA, is expressed by Borrelia burgdorferi during spirochetemia and is a target for protective antibodies. Infect Immun. 1998;66:5379–5387. doi: 10.1128/iai.66.11.5379-5387.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion CI, Blanco DR, Skare JT, Haake DA, Giladi M, Foley D, Miller JN, Lovett MA. A 9.0-kilobase-pair circular plasmid of Borrelia burgdorferi encodes an exported protein: evidence for expression only during infection. Infect Immun. 1994;62:2653–2661. doi: 10.1128/iai.62.7.2653-2661.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Barthold SW, Giles SS, Montgomery RR, Telford SR, 3rd, Fikrig E. Temporal pattern of Borrelia burgdorferi p21 expression in ticks and the mammalian host. J Clin Invest. 1997;99:987–995. doi: 10.1172/JCI119264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve-Gassent MD, Elliott NL, Seshu J. sodA is essential for virulence of Borrelia burgdorferi in the murine model of Lyme disease. Mol Microbiol. 2009;71:594–612. doi: 10.1111/j.1365-2958.2008.06549.x. [DOI] [PubMed] [Google Scholar]
- Fikrig E, Barthold SW, Sun W, Feng W, Telford SR, 3rd, Flavell RA. Borrelia burgdorferi P35 and P37 proteins, expressed in vivo, elicit protective immunity. Immunity. 1997;6:531–539. doi: 10.1016/s1074-7613(00)80341-6. [DOI] [PubMed] [Google Scholar]
- Fisher MA, Grimm D, Henion AK, Elias AF, Stewart PE, Rosa PA, Gherardini FC. Borrelia burgdorferi sigma54 is required for mammalian infection and vector transmission but not for tick colonization. Proc Natl Acad Sci U S A. 2005;102:5162–5167. doi: 10.1073/pnas.0408536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. aadA confers streptomycin resistance in Borrelia burgdorferi. J Bacteriol. 2003;185:6723–6727. doi: 10.1128/JB.185.22.6723-6727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuangthong M, Herbig AF, Bsat N, Helmann JD. Regulation of the Bacillus subtilis fur and perR genes by PerR: not all members of the PerR regulon are peroxide inducible. J Bacteriol. 2002;184:3276–3286. doi: 10.1128/JB.184.12.3276-3286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou G. How to flip the (redox) switch. Cell. 2002;111:607–610. doi: 10.1016/s0092-8674(02)01165-0. [DOI] [PubMed] [Google Scholar]
- Gilbert MA, Morton EA, Bundle SF, Samuels DS. Artificial regulation of ospC expression in Borrelia burgdorferi. Mol Microbiol. 2007;63:1259–1273. doi: 10.1111/j.1365-2958.2007.05593.x. [DOI] [PubMed] [Google Scholar]
- Grimm D, Eggers CH, Caimano MJ, Tilly K, Stewart PE, Elias AF, Radolf JD, Rosa PA. Experimental assessment of the roles of linear plasmids lp25 and lp28-1 of Borrelia burgdorferi throughout the infectious cycle. Infect Immun. 2004a;72:5938–5946. doi: 10.1128/IAI.72.10.5938-5946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, Bueschel DM, Schwan TG, Policastro PF, Elias AF, Rosa PA. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci U S A. 2004b;101:3142–3147. doi: 10.1073/pnas.0306845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbig AF, Helmann JD. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol Microbiol. 2001a;41:849–859. doi: 10.1046/j.1365-2958.2001.02543.x. [DOI] [PubMed] [Google Scholar]
- Herbig AF, Helmann JD. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol Microbiol. 2001b;41:849–859. doi: 10.1046/j.1365-2958.2001.02543.x. [DOI] [PubMed] [Google Scholar]
- Hubner A, Yang X, Nolen DM, Popova TG, Cabello FC, Norgard MV. Expression of Borrelia burgdorferi OspC and DbpA is controlled by a RpoN-RpoS regulatory pathway. Proc Natl Acad Sci U S A. 2001;98:12724–12729. doi: 10.1073/pnas.231442498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JA, Seshu J, Skare JT. Transcriptional profiling of Borrelia burgdorferi containing a unique bosR allele identifies a putative oxidative stress regulon. Microbiology. 2006;152:2599–2609. doi: 10.1099/mic.0.28996-0. [DOI] [PubMed] [Google Scholar]
- Hyde JA, Trzeciakowski JP, Skare JT. Borrelia burgdorferi alters its gene expression and antigenic profile in response to CO2 levels. J Bacteriol. 2007;189:437–445. doi: 10.1128/JB.01109-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JA, Shaw DK, Smith R, III, Trzeciaowski JP, Skare JT. Characterization of a conditional bosR mutant in Borrelia burgdorferi. Infect Immun. 2010;78 doi: 10.1128/IAI.01018-09. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona LI, Tokarz R, Kuhlow CJ, Benach J, Benach JL. The fur homologue in Borrelia burgdorferi. J Bacteriol. 2004;186:6443–6456. doi: 10.1128/JB.186.19.6443-6456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, Liang WJ, Chaloupka J, Ruoff C, Schroppel K, Naglik JR, Eckert SE, Mogensen EG, Haynes K, Tuite MF, Levin LR, Buck J, Muhlschlegel FA. Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr Biol. 2005;15:2021–2026. doi: 10.1016/j.cub.2005.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun. 2001a;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun. 2001b;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira-Rey M, Seshu J, Skare JT. The absence of linear plasmid 25 or 28-1 of Borrelia burgdorferi dramatically alters the kinetics of experimental infection via distinct mechanisms. Infect Immun. 2003;71:4608–4613. doi: 10.1128/IAI.71.8.4608-4613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Pal U, Ramamoorthi N, Liu X, Desrosiers DC, Eggers CH, Anderson JF, Radolf JD, Fikrig E. The Lyme disease agent Borrelia burgdorferi requires BB0690, a Dps homologue, to persist within ticks. Mol Microbiol. 2007;63:694–710. doi: 10.1111/j.1365-2958.2006.05550.x. [DOI] [PubMed] [Google Scholar]
- Lybecker MC, Samuels DS. Temperature-induced regulation of RpoS by a small RNA in Borrelia burgdorferi. Mol Microbiol. 2007;64:1075–1089. doi: 10.1111/j.1365-2958.2007.05716.x. [DOI] [PubMed] [Google Scholar]
- Medrano MS, Ding Y, Wang XG, Lu P, Coburn J, Hu LT. Regulators of expression of the oligopeptide permease A proteins of Borrelia burgdorferi. J Bacteriol. 2007;189:2653–2659. doi: 10.1128/JB.01760-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AP. Fungal CO2 sensing: a breath of fresh air. Curr Biol. 2005;15:R934–936. doi: 10.1016/j.cub.2005.10.064. [DOI] [PubMed] [Google Scholar]
- Mongkolsuk S, Helmann JD. Regulation of inducible peroxide stress responses. Mol Microbiol. 2002;45:9–15. doi: 10.1046/j.1365-2958.2002.03015.x. [DOI] [PubMed] [Google Scholar]
- Nadelman RB, Wormser GP. Lyme borreliosis. Lancet. 1998;352:557–565. doi: 10.1016/S0140-6736(98)01146-5. [DOI] [PubMed] [Google Scholar]
- Ojaimi C, Brooks C, Casjens S, Rosa P, Elias A, Barbour A, Jasinskas A, Benach J, Katona L, Radolf J, Caimano M, Skare J, Swingle K, Akins D, Schwartz I. Profiling of temperature-induced changes in Borrelia burgdorferi gene expression by using whole genome arrays. Infect Immun. 2003;71:1689–1705. doi: 10.1128/IAI.71.4.1689-1705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojaimi C, Mulay V, Liveris D, Iyer R, Schwartz I. Comparative transcriptional profiling of Borrelia burgdorferi clinical isolates differing in capacities for hematogenous dissemination. Infect Immun. 2005;73:6791–6802. doi: 10.1128/IAI.73.10.6791-6802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal U, Yang X, Chen M, Bockenstedt LK, Anderson JF, Flavell RA, Norgard MV, Fikrig E. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J Clin Invest. 2004;113:220–230. doi: 10.1172/JCI19894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE, Gherardini FC. Lack of a role for iron in the Lyme disease pathogen. Science. 2000;288:1651–1653. doi: 10.1126/science.288.5471.1651. [DOI] [PubMed] [Google Scholar]
- Purser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, Norris SJ. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol Microbiol. 2003;48:753–764. doi: 10.1046/j.1365-2958.2003.03452.x. [DOI] [PubMed] [Google Scholar]
- Revel AT, Talaat AM, Norgard MV. DNA microarray analysis of differential gene expression in Borrelia burgdorferi, the Lyme disease spirochete. Proc Natl Acad Sci U S A. 2002;99:1562–1567. doi: 10.1073/pnas.032667699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa PA, Tilly K, Stewart PE. The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol. 2005;3:129–143. doi: 10.1038/nrmicro1086. [DOI] [PubMed] [Google Scholar]
- Samuels DS. Electrotransformation of the spirochete Borrelia burgdorferi. 1995 doi: 10.1385/0-89603-310-4:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan TG, Piesman J, Golde WT, Dolan MC, Rosa PA. Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proc Natl Acad Sci U S A. 1995;92:2909–2913. doi: 10.1073/pnas.92.7.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshu J, Boylan JA, Gherardini FC, Skare JT. Dissolved oxygen levels alter gene expression and antigen profiles in Borrelia burgdorferi. Infect Immun. 2004a;72:1580–1586. doi: 10.1128/IAI.72.3.1580-1586.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshu J, Boylan JA, Hyde JA, Swingle KL, Gherardini FC, Skare JT. A conservative amino acid change alters the function of BosR, the redox regulator of Borrelia burgdorferi. Mol Microbiol. 2004b;54:1352–1363. doi: 10.1111/j.1365-2958.2004.04352.x. [DOI] [PubMed] [Google Scholar]
- Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Hook M, Skare JT. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Mol Microbiol. 2006;59:1591–1601. doi: 10.1111/j.1365-2958.2005.05042.x. [DOI] [PubMed] [Google Scholar]
- Shi Y, Xu Q, McShan K, Liang FT. Both decorin-binding proteins A and B are critical for the overall virulence of Borrelia burgdorferi. Infect Immun. 2008;76:1239–1246. doi: 10.1128/IAI.00897-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skare JT, Foley DM, Hernandez SR, Moore DC, Blanco DR, Miller JN, Lovett MA. Cloning and molecular characterization of plasmid-encoded antigens of Borrelia burgdorferi. Infect Immun. 1999;67:4407–4417. doi: 10.1128/iai.67.9.4407-4417.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson B, Schwan TG, Rosa PA. Temperature-related differential expression of antigens in the Lyme disease spirochete, Borrelia burgdorferi. Infect Immun. 1995;63:4535–4539. doi: 10.1128/iai.63.11.4535-4539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Imlay JA. Oxidative stress. Curr Opin Microbiol. 1999;2:188–194. doi: 10.1016/s1369-5274(99)80033-2. [DOI] [PubMed] [Google Scholar]
- Tilly K, Bestor A, Jewett MW, Rosa P. Rapid clearance of Lyme disease spirochetes lacking OspC from skin. Infect Immun. 2007;75:1517–1519. doi: 10.1128/IAI.01725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarz R, Anderton JM, Katona LI, Benach JL. Combined effects of blood and temperature shift on Borrelia burgdorferi gene expression as determined by whole genome DNA array. Infect Immun. 2004;72:5419–5432. doi: 10.1128/IAI.72.9.5419-5432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weening EH, Parveen N, Trzeciakowski JP, Leong JM, Hook M, Skare JT. Borrelia burgdorferi lacking DbpBA exhibits an early survival defect during experimental infection. Infect Immun. 2008;76:5694–5705. doi: 10.1128/IAI.00690-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Goldberg MS, Popova TG, Schoeler GB, Wikel SK, Hagman KE, Norgard MV. Interdependence of environmental factors influencing reciprocal patterns of gene expression in virulent Borrelia burgdorferi. Mol Microbiol. 2000a;37:1470–1479. doi: 10.1046/j.1365-2958.2000.02104.x. [DOI] [PubMed] [Google Scholar]
- Yang X, Goldberg MS, Popova TG, Schoeler GB, Wikel SK, Hagman KE, Norgard MV. Interdependence of environmental factors influencing reciprocal patterns of gene expression in virulent Borrelia burgdorferi. Mol Microbiol. 2000b;37:1470–1479. doi: 10.1046/j.1365-2958.2000.02104.x. [DOI] [PubMed] [Google Scholar]
- Yang XF, Alani SM, Norgard MV. The response regulator Rrp2 is essential for the expression of major membrane lipoproteins in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2003;100:11001–11006. doi: 10.1073/pnas.1834315100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XF, Pal U, Alani SM, Fikrig E, Norgard MV. Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J Exp Med. 2004;199:641–648. doi: 10.1084/jem.20031960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XF, Lybecker MC, Pal U, Alani SM, Blevins J, Revel AT, Samuels DS, Norgard MV. Analysis of the ospC regulatory element controlled by the RpoN-RpoS regulatory pathway in Borrelia burgdorferi. J Bacteriol. 2005;187:4822–4829. doi: 10.1128/JB.187.14.4822-4829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]