

Abstract
Members of the AMP-activated protein kinase family, including the Snf1 kinase of Saccharomyces cerevisiae, are activated under conditions of nutrient stress. AMP-activated protein kinases are heterotrimeric complexes composed of a catalytic α subunit and regulatory β and γ subunits. In this study, the role of the β subunits in the regulation of Snf1 activity was examined. Yeasts express three isoforms of the AMP-activated protein kinase consisting of Snf1 (α), Snf4 (γ), and one of three alternative β subunits, either Sip1, Sip2, or Gal83. The Gal83 isoform of the Snf1 complex is the most abundant and was analyzed in the greatest detail. All three β subunits contain a conserved domain referred to as the glycogen-binding domain. The deletion of this domain from Gal83 results in a deregulation of the Snf1 kinase, as judged by a constitutive activity independent of glucose availability. In contrast, the deletion of this homologous domain from the Sip1 and Sip2 subunits had little effect on Snf1 kinase regulation. Therefore, the different Snf1 kinase isoforms are regulated through distinct mechanisms, which may contribute to their specialized roles in different stress response pathways. In addition, the β subunits are subjected to phosphorylation. The responsible kinases were identified as being Snf1 and casein kinase II. The significance of the phosphorylation is unclear since the deletion of the region containing the phosphorylation sites in Gal83 had little effect on the regulation of Snf1 in response to glucose limitation.
The Snf1 protein kinase of Saccharomyces cerevisiae is the yeast ortholog of the AMP-activated protein kinase (AMPK) found in mammals and other eukaryotes. AMPK acts as a nutrient and energy sensor, becoming activated under conditions of nutrient and energy depletion (6). In mammals, AMPK plays a key role in glucose homeostasis and is a target for drugs used to treat metabolic syndrome and type 2 diabetes (34). In yeast, the Snf1 kinase plays an essential role during aerobic growth and fermentative growth on alternative carbon sources. Cells lacking Snf1 kinase activity are viable but display numerous phenotypes including poor or no growth on alternative carbon sources, defects in meiosis and sporulation, defects in response to ion stress, and defects in pseudohyphal growth (7).
The Snf1 kinase and all members of the AMPK family function as heterotrimers composed of a catalytic α subunit complexed with regulatory β and γ subunits (2). The γ subunit in mammalian enzymes directly binds three molecules of AMP (26, 33), which stimulates enzyme activity by inhibiting the dephosphorylation of the conserved threonine residue in the kinase activation loop (23). In yeast, there is no evidence that the γ subunit binds AMP; however, similar to mammals, the key glucose-regulated step is the dephosphorylation of the kinase activation loop (22).
In this study, we examine the role of the β subunits in the regulation of the Snf1 kinase activity. Yeasts express three isoforms of the Snf1 kinase that differ depending on which of the three distinct β subunits, Sip1, Sip2, and Gal83, is incorporated into the enzyme. Previous studies have shown that the Snf1 isoforms have distinct substrate preferences (24), subcellular localizations (32), and stress response capacities (9). Only the Snf1 isoform containing Gal83 as the β subunit is able to localize to the cell nucleus in a process that requires Sak1, one of the three Snf1-activating protein kinases. Since all three of the Snf1-activating kinases (SAKs) are capable of phosphorylating Snf1 on its activation loop (3), it has remained a mystery as to why the Sak1 kinase is specifically required for Snf1 nuclear localization.
The β subunits of Snf1 as well as mammalian AMPK contain a domain that is referred to as either a carbohydrate-binding module (CBM) (11) or a glycogen-binding domain (GBD) (19). The structure of this domain has been solved (20), and it was previously shown that this domain binds most tightly to branched oligosaccharides like glycogen that contain α1→6 branches (12). The binding of glycogen to the β subunit causes an allosteric inhibition of AMPK activity and inhibits phosphorylation by the upstream activating kinase. The β subunits of yeast contain the GBDs, but the importance of binding glycogen is questionable since cells that lack the ability to make glycogen show a normal regulation of Snf1 kinase in response to glucose limitation (15). Nonetheless, the deletion of the GBD from the Gal83 protein caused an increased activity of the Snf1 enzyme and release from glucose repression. Therefore, the GBD acts as a negative regulator of kinase activity in both mammalian and fungal cells.
In this study we examine the role of the GBD present in the Sip2 and Sip1 proteins. We also extend the characterization of the Gal83 GBD by determining what connection this domain has with the regulated dephosphorylation of the Snf1 kinase. Finally, we have characterized other N-terminal domains in the β subunits that control accumulation and phosphorylation.
MATERIALS AND METHODS
Yeast strains and media.
The yeast strains used in this study (Table 1) were all derived from S288c. Cells were grown at 30°C in synthetic complete medium lacking the appropriate nutrient for plasmid selection. Glucose, sucrose, or raffinose was present as the carbon source at 2% (g/100 ml). Low-glucose medium contained glucose at 0.05% (g/100 ml). When indicated, 2-deoxyglucose was added to sucrose medium at a concentration of 0.02% (g/100 ml). Raffinose medium was supplemented with 1 μg/ml antimycin A to prevent aerobic growth. In a typical shift experiment, cells were grown in high-glucose medium to an optical density at 600 nm (OD600) value of between 0.5 and 0.8. Half of the cells were harvested by centrifugation and used as the high-glucose sample. The other half was washed, resuspended in low-glucose medium, and grown an additional 2 h prior to harvest.
TABLE 1.
S. cerevisiae strains
| Strain | Genotype |
|---|---|
| MSY557 | MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 sip1Δ::HIS3 sip2Δ::HIS3 gal83Δ::HIS3 |
| MSY920 | MATaura3-52 leu2Δ0 his3Δ200 sip1Δ::HIS3 sip2Δ::HIS3 gal83Δ::HIS3 snf1Δ10 |
| MSY1059 | MATaura3-52 leu2Δ1 trp1Δ63 sip1Δ::HIS3 sip2Δ::HIS3 gal83Δ::HIS3 SNF1-3HA |
| MSY1066 | MATaura3-52 leu2Δ1 trp1Δ63 sip1Δ::HIS3 sip2Δ::HIS3 gal83Δ::HIS3 SNF1-3HA reg1Δ::HIS3 |
Plasmids.
Plasmids used to express the Snf1 β subunits used the pRS315 vector (27) and contained the β subunit genes with a triple flag tag inserted near the N terminus of each protein. In each case, the β subunit gene is expressed from its own cognate promoter and terminator sequences. Plasmids used for tandem affinity purification (TAP) of the Snf1 heterotrimer and the Snf1-activating kinases were described previously (3). The entire REG1 gene (4.6-kb genomic fragment from EcoRI 954 nucleotides [nt] upstream of the REG1 open reading frame to the Sau3A site 602 nt downstream of the stop codon) was cloned into pRS316. The Reg1 protein was tagged with 5 copies of the V5 epitope (GKPIPNPLLGLDST), replacing C-terminal Reg1 residues 1005 to 1014 (29).
Protein purification.
Snf1 heterotrimers containing Snf1, Snf4, and either Sip1, Sip2, or Gal83 were coexpressed in BL21 Codon Plus bacteria using the pDUET coexpression system described previously by Sanders et al. (23). The kinase complexes were then purified by nickel-agarose chromatography and dialyzed against kinase buffer (20 mM HEPES [pH 7.0], 0.5 mM EDTA, 0.5 mM dithiothreitol, 5 mM magnesium acetate) and 5% (vol/vol) glycerol. Proteins were divided into aliquots and stored at −80°C. Sak1, Tos3, Elm1, and Snf1 complexes were TAP purified from yeast as described previously (3). The Mig1 protein (residues 207 to 413) with a His tag at its N terminus was expressed in Escherichia coli cells, purified by nickel agarose chromatography, and dialyzed against kinase buffer with 5% (vol/vol) glycerol.
Kinase assays.
Protein kinases were purified from yeast as TAP fusions (3) or from bacteria (17) as described previously. In vitro kinase reaction mixtures (20 μl) contained 0.2 mM [γ-32P]ATP (1,000 cpm/pmol), kinase buffer, and the indicated recombinant proteins. Reaction mixtures were incubated at 30°C for 1 h and then stopped by the addition of SDS sample buffer. Proteins were resolved on SDS-polyacrylamide gels. The gels were dried and subjected to autoradiography.
Western blotting and immunoprecipitation.
Snf1-hemagglutinin (HA) was detected with a 1:2,000 dilution of HA probe (Santa Cruz), Reg1-5V5 was detected with a 1:1,000 dilution of anti-V5 (Invitrogen), and β subunits tagged with the triple-flag epitope were detected with a 1:1,000 dilution of mouse monoclonal anti-FLAG (Sigma). Goat anti-mouse IgG DyLight 800 (Thermo) diluted 1:5,000 was used as the secondary antibody. Blots were processed by using the Snap i.d. system (Millipore) and scanned by using an Odyssey scanner (Li-Cor). Integrated intensity values of bands in triplicate were quantified by using Odyssey scanning software. To detect Snf1 activation loop (Thr210) phosphorylation, protein extracts were prepared using the NaOH cell lysis method (10, 13). Protein extracts (500 μg) were immunoprecipitated in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease and phosphatase inhibitors using 20 μl HA probe agarose conjugate (Santa Cruz). Bound proteins were eluted in SDS sample buffer and resolved on SDS gels. Blots were treated with Odyssey blocking buffer (Li-Cor). Rabbit polyclonal antibody (diluted 1:500) directed against phosphorylated Snf1 T210 (13) was used as the primary antibody to detect phosphorylated Snf1. Goat anti-rabbit IRDye 800CW (Li-Cor) (1:5,000 dilution) was used as a secondary antibody. Immunoprecipitation of Reg1-V5 and Snf1-HA was detected by preparing glass bead protein extracts in RIPA buffer supplemented with protease and phosphatase inhibitors. Extracts (500 μg) were incubated with 20 μl HA probe agarose conjugate (Santa Cruz) and washed extensively, and bound protein was eluted in SDS sample buffer.
Real-time qPCR.
Total RNA was isolated from cells grown in either high- or low-glucose medium using the RNeasy Plus minikit (Qiagen). cDNA was synthesized by using a mixture of random and oligo(dT) primers and the RT2 first-strand kit (SA Biosciences). Real-time quantitative PCR (qPCR) reaction mixtures (20 μl) contained 10 μl 2× SYBR green PCR master mix (Applied Biosystems) and 0.3 μl of cDNA. Primer efficiencies were determined experimentally (18), and only primer pairs with an efficiency of 2.0 ± 0.05 were used in this study (Table 2). Reactions were performed by using an Applied Biosystems 7300 real-time PCR machine. PCRs utilized a four-stage profile: stage 1, 50°C for 2 min; stage 2, 95°C for 10 min; stage 3, 95°C for 15 s and 60°C for 1 min (40 times); and stage 4 (dissociation), 95°C for 15 s, 60°C for 15 s, and 95°C for 15 s. Reactions were run in triplicate on a single 96-well plate. Mean ΔCT values were used for calculations of mRNA abundance by using the comparative threshold cycle (CT) method (25).
TABLE 2.
Oligonucleotides
| Primer | Sequence | Size of product (nt) | Efficiency |
|---|---|---|---|
| 18S-T401 | CGGCTACCACATCCAAGGAA | ||
| 18S-B587 | GCTGGAATTACCGCGGCT | 186 | 2.03 |
| HXT1-T4 | AATTCAACTCCCGATCTAATATCTCC | ||
| HXT1-B183 | GACACCTTTTCCGGTGTTTG | 179 | 2.05 |
| PCK1-T1245 | CTTCCTAGCCTTGCACCCTA | ||
| PCK1-B1432 | CATTGGCTAACGAACCATCA | 187 | 2.02 |
Invertase assays.
The invertase activity of mid-log-phase cells grown in high and low glucose was determined quantitatively by using a colorimetric assay coupled to glucose oxidase (5). Three independent cultures were assayed, and the mean values and standard errors were plotted. The units of invertase activity used were mU/OD, where 1 U equals one μmol glucose released per minute.
RESULTS
Expression of Snf1 kinase complexes with distinct β subunits.
Low-copy-number plasmids expressing a single Snf1 kinase β subunit were introduced into yeast cells with complete deletions of all three β subunit genes. For detection by Western blotting, the β subunits all contained three copies of the flag epitope inserted after residues 4, 4, and 5 of the Gal83, Sip2, and Sip1 proteins, respectively (Fig. 1A). The positions of the deletions of the conserved glycogen-binding domains (GBDs) are also shown. Extracts were prepared from cells transformed with these plasmids and examined by quantitative Western blotting (Fig. 1B and C). Proteins of the predicted molecular masses that were not present in cells transformed with empty plasmid vector were detected. Quantitation of the Western signal indicated that of all the β subunits, Gal83 accumulates to the highest level. These data indicate that Gal83 is expressed at a level almost 4 times higher than that of Sip2 and 16 times higher than that of Sip1. The higher abundance of the Gal83 protein was reported by studies using TAP and green fluorescent protein (GFP) tags at the C terminus (4, 32). Each of the tagged β subunits was functionally indistinguishable from the untagged β subunit genes (data not shown), and all could provide β subunit function for growth on sucrose medium (Fig. 1D). One measure of Snf1 kinase pathway deregulation is the ability to grow on sucrose medium in the presence of 2-deoxyglucose. The deletion of the GBD from the Gal83 subunit confers a limited ability to grow in the presence of 2-deoxyglucose (Fig. 1D). The deletion of the GBD from Sip1 and from Sip2 did not confer any growth advantage on this medium.
FIG. 1.

Glycogen-binding domain deletions. (A) The β subunits were tagged with three copies of the flag epitope (F3) at their N termini. The β subunits all expressed the C-terminal domain that mediates interactions with the α and γ subunits (AG). The β subunits were expressed as full-length proteins or as deletions lacking the glycogen-binding domain (GBD), as shown. (B) Western blotting of protein extracts from cells expressing the flag-tagged β subunits. The mobility of the β subunits is indicated (>), as is the position of a background band present in control cells (vec) lacking the flag epitope (*). M, molecular mass standard (in kilodaltons); WT, wild type. (C) Relative accumulation of the different β subunits. (D) Growth of cells on medium containing glucose, sucrose, or sucrose plus 2-deoxyglucose (2DG). Tenfold serial dilutions of cells expressing the indicated β subunits are shown.
Effect of GBD deletion on Snf1-mediated regulation of gene expression.
The Snf1 kinase signaling pathway regulates gene expression, stimulating the expression of genes needed for growth on alternative carbon sources and inhibiting genes that are not needed when glucose is scarce. We chose three genes to examine since their mechanisms of regulation by Snf1 are distinct. Snf1 induces the expression of invertase (SUC2) by phosphorylating the transcriptional repressor Mig1 (31). Snf1 induces PCK1 expression by phosphorylating Cat8, a transcriptional activator (1). Finally, we examined the Snf1-mediated repression of the low-affinity glucose transporter Hxt1, although the Snf1 substrate in this pathway is not known with certainty (30). To measure the regulation of the SUC2 gene, we measured the activity of invertase, the enzyme encoded by this gene (Fig. 2A). In the absence of a β subunit, the level of invertase expression is low and does not respond to glucose limitation. Providing the cell with any one of the β subunits restores the induction of invertase activity during glucose limitation, consistent with previously reported results using strains with different β subunit mutations (24). The deletion of the GBD from the Sip1 or Sip2 protein had little effect on invertase expression under conditions of either high or low glucose. In contrast, the deletion of the GBD from the Gal83 protein resulted in a large increase in the expression of invertase in the presence of high glucose, supporting the idea that Snf1 kinase containing the Gal83-ΔGBD as its β subunit is active even in the presence of high glucose.
FIG. 2.
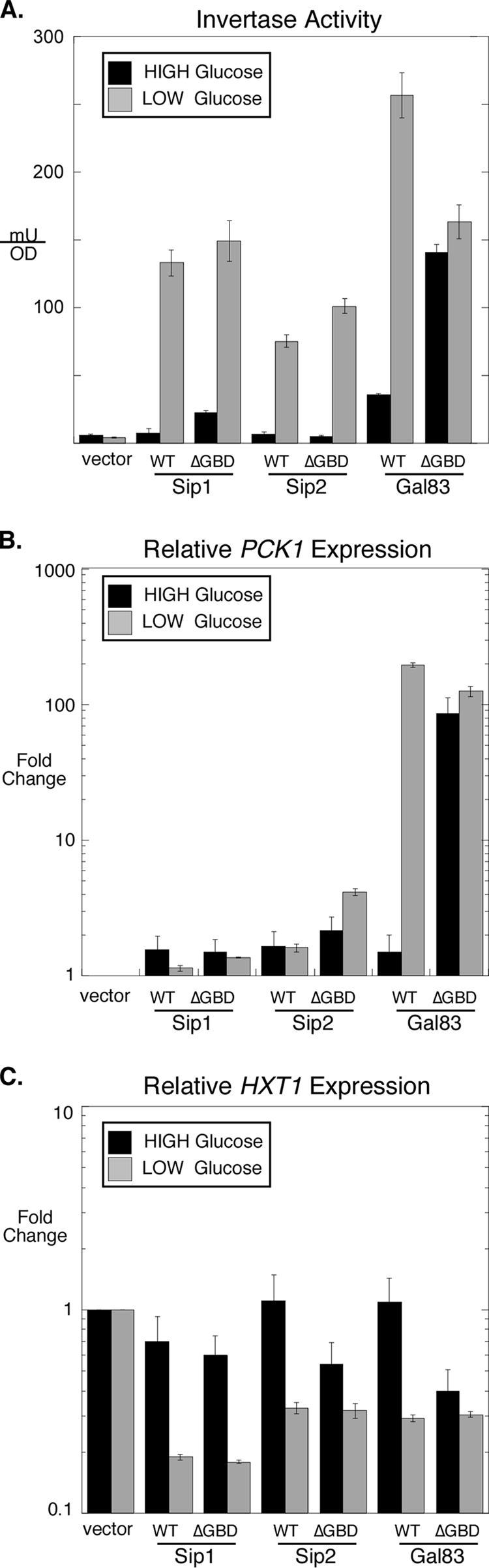
Effect of the GBD deletions on gene expression. (A) Invertase activity was measured for cells (MSY557) transformed with an empty plasmid vector or with a low-copy-number plasmid expressing a single β subunit as indicated. Cells were grown in either high or low glucose prior to harvesting. (B and C) PCK1 (B) and HXT1 (C) mRNA levels were measured by qRT-PCR. Cells were grown in either high or low glucose prior to harvesting. Mean values of fold changes in gene expression are plotted, with error bars representing 1 standard error.
The expression of the PCK1 gene was examined by quantitative reverse transcription-PCR (qRT-PCR) (Fig. 2B). PCK1 encodes phosphoenolpyruvate carboxykinase, a key enzyme required for gluconeogenesis whose expression is induced by Snf1 signaling (21). When Sip1 or Sip2 was expressed as the only β subunit, little or no induction of PCK1 mRNA was observed. With Gal83 as the lone β subunit, PCK1 expression was induced over 100-fold upon glucose limitation. The deletion of the GBD from Gal83 caused the induction of PCK1 even in the presence of high glucose.
The expression of HXT1 was also examined as a representative of genes whose levels of expression are inhibited by Snf1 signaling. In the absence of any β subunit, the HXT1 gene was not repressed in response to glucose limitation (Fig. 2C). When any one of the β subunits was provided, HXT1 expression was reduced by between 3- and 5-fold. When the GBD was deleted from the Sip1 protein, little change in HXT1 expression levels was observed relative to that of full-length Sip1. When the GBD was deleted from the Sip2 protein, HXT1 expression was reduced in the high-glucose sample by 2-fold compared to that of full-length Sip2. When the GBD was deleted from the Gal83 protein, HXT1 expression was reduced 3-fold. Thus, in the three genes examined, regardless of whether they are induced or repressed by Snf1 signaling, the deletion of the GBD from the Gal83 protein had the greatest effect on gene expression. Taken together, these data support the idea that the Snf1 kinase complex containing the Gal83 protein requires the GBD to be maintained in an inactive state.
The Gal83 GBD regulates Snf1 activation loop phosphorylation through Reg1 association.
The deletion of the GBD from the Gal83 protein had the greatest effect on Snf1-regulated gene expression and was chosen for further study. The activation of the Snf1 kinase closely parallels its phosphorylation on the activation loop threonine 210 (13). Cells expressing full-length Gal83 as the only β subunit showed glucose regulation of the T210 phosphorylation site (Fig. 3A). In contrast, the cells expressing Gal83-ΔGBD showed high levels of T210 phosphorylation even in the presence of high glucose. When this assay was repeated in triplicate and the Western signals were quantified, the high-glucose phosphorylation of T210 was increased over 7-fold when the GBD was deleted from Gal83 (Fig. 3B). The increased level of phosphorylation of Snf1 in cells expressing Gal83-ΔGBD was recently reported by Momcilovic et al. (15). Earlier studies from our laboratory have shown that the key glucose-regulated event controlling the phosphorylation status of T210 is the Glc7/Reg1-mediated dephosphorylation step (22). Therefore, we examined the association of the Reg1 protein with the Snf1 enzyme containing or lacking the Gal83 GBD. The Snf1 protein tagged with the HA epitope was collected from protein extracts, and bound Reg1 protein was detected by Western blotting (Fig. 3C). The Reg1 association with the Snf1 complex was detected in both high- and low-glucose media. When the GBD was deleted from Gal83, a significant reduction in the association of Reg1 with Snf1 was observed in the extracts prepared from cells in high glucose. These data suggest that in high-glucose medium, the Reg1 association with Snf1 correlates with T210 dephosphorylation. The immunoprecipitation was repeated in triplicate, and the signals were quantified (Fig. 3D). The deletion of the Gal83 GBD causes a 3-fold reduction in the Reg1 association in both high and low glucose.
FIG. 3.

Effect of the Gal83 glycogen-binding domain on Snf1 activation and Reg1 association. (A) Cells (MSY1059) were transformed with low-copy-number plasmids expressing either Gal83 or Gal83-ΔGBD as indicated. Extracts were prepared and probed with antibodies that detect either total Snf1 (HA) or phosphorylated Snf1 (PT210). (B) The relative phosphorylation of Snf1 T210 was determined in triplicate as described above (A) by using the Li-Cor Odyssey scanner. The mean value of phosphorylated Snf1 normalized with total Snf1 is plotted, with 1 standard error shown. (C) Cells (MSY1066) transformed with the plasmids shown were grown in high (H) or low (L) glucose as indicated. Extracts were prepared, and the Snf1 protein was recovered from 500 μg protein using anti-HA-conjugated agarose beads. Associated proteins were eluted in SDS sample buffer, and Reg1 was detected by Western blotting with anti-V5 antibodies. Control Western blots of 20 μg protein with antibodies against the HA, V5, and flag epitopes are shown. IP, immunoprecipitation. (D) The relative association of Reg1 with Snf1 was determined in triplicate as described above (C) by using the Li-Cor Odyssey scanner. The mean values for the amount of bound Reg1 divided by the total amount of Reg1 are plotted, and 1 standard error is indicated.
Snf1 β subunits are phosphorylated in vitro.
Previous studies with Snf1 heterotrimers TAP purified from yeast showed that the β subunits Sip2 and Gal83 were phosphorylated in vitro (16). Here we further investigated the phosphorylation of the Snf1 subunits using the Snf1-Snf4-Gal83 isoform purified from cells lacking any Snf1-activating kinases and containing either wild-type Snf1 or Snf1 with the activation loop mutation T210A that prevents phosphorylation by the Snf1-activating kinases (Fig. 4A). Complexes were incubated with either Sak1, Tos3, or Elm1 kinases that were TAP purified from snf1Δ cells. Mass spectrometry of the proteins associated with Sak1 identified the casein kinase II (CKII) proteins (3). Therefore, the Sak1 preparation is referred to as Sak1+CKII. When the Snf1 heterotrimers were incubated without any Snf1-activating kinase (Fig. 4A, lanes 1 and 2), we were unable to detect any autophosphorylation of Snf1 subunits. The addition of the Snf1-activating kinases leads to the incorporation of 32P into the Snf1 but not the Snf1-T210A protein. We also detected an incorporation of 32P into the Gal83 protein in reaction mixtures that contained wild-type Snf1 (Fig. 4A, lanes 4, 7, and 10). The phosphorylation of Gal83 was prevented by the T210A mutation when the Snf1 complex was activated by Tos3 or Elm1 (Fig. 4A, lanes 8 and 11). However, when the Sak1+CKII kinase preparation was used, the Gal83 protein in the Snf1 and Snf1-T210A complexes was phosphorylated (lanes 4 and 5). We interpret these data to mean that the Gal83 protein is subjected to phosphorylation by more than one kinase. The activation of Snf1 leads to Gal83 phosphorylation, as was seen when Tos3 and Elm1 were used as SAKs. However, when the Snf1-T210A complex was used with the Sak1+CKII preparation, phosphorylation of Gal83 was still observed. Therefore, a kinase other than Snf1 (Sak1 or CKII) must be responsible for the phosphorylation of Gal83.
FIG. 4.

In vitro phosphorylation of the β subunits. (A) In vitro kinase assay using the TAP-purified Snf1-Snf4-Gal83 complex with wild-type Snf1 or with the T210A mutation. Snf1 complexes were incubated with TAP-purified Sak1, Tos3, or Elm1. Mobilities of the Snf1, Gal83, Sak1, Tos3, and Elm1 proteins are indicated. (B) Coomassie-stained protein gel of the Snf1 heterotrimer complexes purified from bacteria. Mobilities of the Snf1 subunits are indicated. (C) In vitro kinase assay with Snf1 heterotrimers purified from bacteria with and without incubation with Sak1 TAP purified from yeast. A shorter exposure of this autoradiogram showing incorporation into the recombinant Mig1 protein (bottom) was included to assess Snf1 activation.
With the availability of highly purified recombinant Snf1 heterotrimers purified from bacteria, we decided to reexamine the phosphorylation events within the Snf1 kinase complex. Snf1 heterotrimers containing Snf1, Snf4, and either Gal83, Sip2, or Sip1 were purified from bacteria, and their purity was analyzed with a Coomassie-stained SDS gel (Fig. 4B). The Gal83 isoform produced the greatest yield and had the lowest levels of contaminants and degradation products; the Sip2 isoform was intermediate, and the Sip1 isoform yielded small quantities of the Snf1 complex with substoichiometric quantities of Snf4 and full-length Sip1. All three Snf1 enzymes could be activated by Sak1, as shown in kinase assays with [γ-32P]ATP and the recombinant Mig1 protein as a substrate (Fig. 4C). None of the enzymes purified from bacteria showed any autophosphorylation activity when incubated by themselves (Fig. 4C, lanes 1, 3, and 5). When the Snf1-activating kinase Sak1 was added, phosphorylation was readily detected in the α and β subunits of all three Snf1 heterotrimers (lanes 2, 4, and 6). Note that the level of activity of the Sip1 isoform was lower than that of the Gal83 or Sip2 isoform, and a 4-fold-longer exposure of the autoradiogram in Fig. 4C, lanes 5 and 6, is shown. This finding may reflect the inherent differences in the activities of these isoforms. However, it seems more likely that the Sip1 form is less active due to the substoichiometric presence of its β and γ subunits. These data indicate that all three β subunits of the Snf1 kinase complex are subject to phosphorylation in vitro.
Mapping of Gal83 phosphorylation sites.
Since the Gal83 isoform is the most abundant in vivo and most easily purified from bacteria, it was chosen for further studies. The position of the Gal83 phosphorylation sites were mapped by creating glutathione S-transferase (GST) fusion proteins that were purified and assayed as Snf1 kinase substrates. Residues 1 to 63, 1 to 110, and 111 to 417 were expressed as GST fusions and affinity purified from bacteria (Fig. 5B). When used as substrates in in vitro kinase assays, phosphorylation was detected with the fusion of residues 1 to 110 but not of residues 1 to 63 or 111 to 417. Therefore, the phosphorylation sites of Gal83 were present in the region between residues 63 and 110. This mapping result was true for the Snf1-dependent sites (Fig. 5B, lane 8) as well as the Sak1+CKII-dependent sites (lane 2). Examination of the primary sequence of Gal83 in this region showed the presence of serine residues that fit the consensus Snf1 recognition sequence at positions 64 and 65 as well as three potential CKII sites at positions 87, 90, and 93 (Fig. 5C). Mutagenesis of these residues from serine and threonine into alanine greatly reduced the level of incorporation of 32P, indicating that they are indeed the major sites of phosphorylation. The alignment of this region of Gal83 with the other β subunits from yeast showed that the Snf1 site is strongly conserved with Sip2 and Sip1. Neither Sip1 nor Sip2 showed primary sequence conservation of the CKII sites.
FIG. 5.

Mapping of the Gal83 phosphorylation sites. (A) In vitro kinase assay mixtures contained [γ-32P]ATP and the TAP-purified proteins indicated above the lanes. GST-GAL83 substrates contained residues 1 to 63, 1 to 110, or 111 to 417. (B) Western blotting of purified GST-Gal83 fusion proteins. The predicted mobilities of the full-length constructs are indicated on the left. (C) Multiple-sequence alignment of residues 55 to 110 in Gal83, residues 67 to 115 in Sip2, and residues 172 to 223 in Sip1. The positions of Gal83 residues 63 and 110 and the positions of the Snf1 and CKII sites are indicated.
Gal83 is phosphorylated by Snf1 and casein kinase II.
The finding that purified Sak1 kinase (with associated CKII) could directly phosphorylate residues in Gal83 was intriguing. Studies of the localization of the Gal83 isoform of Snf1 showed that of the three Snf1-activating kinases, only Sak1 could promote nuclear localization (8). Therefore, we sought to determine whether the phosphorylation was catalyzed by Sak1 or the associated CKII. Wild-type Sak1 and a kinase-dead mutant of Sak1 containing the D277A mutation were TAP purified from yeast. Wild-type Sak1 but not the D277A form is able to activate Snf1, as judged by the phosphorylation of the Mig1 protein (Fig. 6A, lanes 1 to 3). However, the D277A mutant enzyme demonstrates a comparable phosphorylation of the Gal83 protein at residues 1 to 110 (lanes 5 and 7). Therefore, the Sak1 kinase is not responsible for the phosphorylation of Gal83.
FIG. 6.
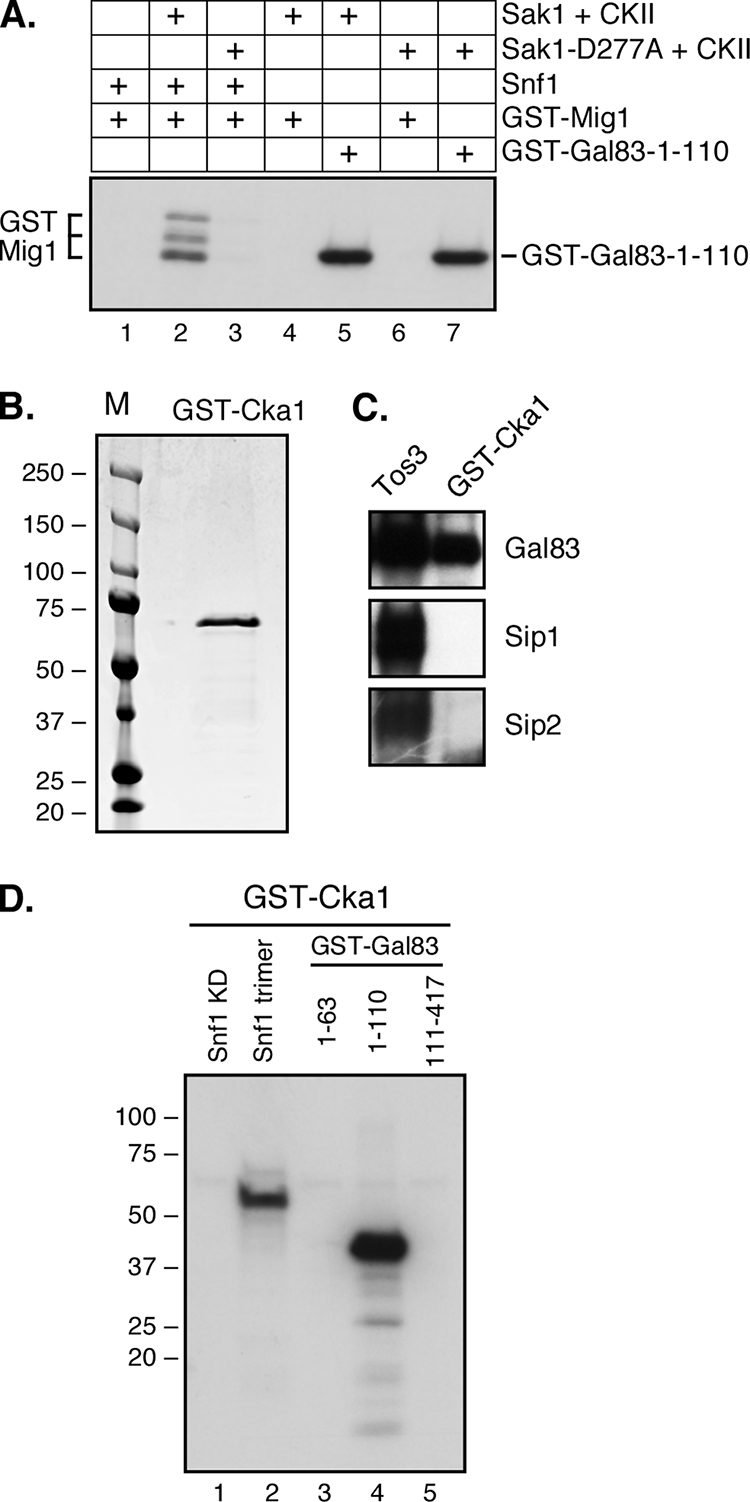
Purified casein kinase II phosphorylates Gal83. (A) In vitro kinase assays with wild-type and kinase-dead Sak1-D277A preparations. Substrate proteins were included as indicated. (B) GST-Cka1 was purified from yeast and analyzed for purity on a Coomassie-stained SDS gel. (C) In vitro kinase assays with [γ-32P]ATP and purified GST-Cka1 or TAP-purified Tos3 incubated with Snf1 heterotrimers purified from bacteria. The incorporation of 32P into the β subunit is shown. (D) In vitro kinase assays with purified GST-Cka1, [γ-32P]ATP, and the indicated substrate proteins. Snf1 kinase was included as the kinase domain (KD) purified from E. coli cells or as a heterotrimer with Snf4 and Gal83 purified from yeast.
In order to confirm that the CKII present in the Sak1 preparation is directly responsible for the phosphorylation of Gal83, we purified GST-tagged Cka1 from yeast (Fig. 6B). CKA1 encodes one of the two catalytic subunits of CKII in yeast. The GST-purified Cka1 protein showed no evidence of an association of the CKII regulatory subunits Ckb1 or Ckb2 (30 and 32 kDa, respectively). Purified GST-Cka1 kinase was able to phosphorylate the Gal83 protein in the context of the Snf1 heterotrimer (Fig. 6C, lane 2). Furthermore, the purified GST-Cka1 kinase showed the same specificity for the GST-Gal83 fusion proteins, with phosphorylation of the fusion at residues 1 to 110 but not the fusion at residues 1 to 63 or 111 to 417. Taken together, our data indicate that the kinases responsible for the in vitro phosphorylation of Gal83 are Snf1 and CKII.
Effect of Gal83 phosphorylation in vivo.
To analyze the effect of the Gal83 phosphorylation in vivo, we generated a series of low-copy-number plasmids expressing Gal83 proteins from its cognate promoter and with three copies of the V5 epitope at the N terminus. These plasmids expressed either full-length Gal83 or Gal83 with specific deletions that removed the region containing the GBD, the phosphorylation sites, or both (Fig. 7A). All proteins were expressed in vivo and detectable by Western blotting (Fig. 7B). The N1 and GBD deletions appeared to accumulate at roughly the same level as the full-length protein. The N2 deletion, which removes residues 12 to 243, was expressed although at a much lower level than the other constructs. A slower-migrating band clearly visible in the GBD deletion was determined to be due to phosphorylation since phosphatase treatment eliminated the band (Fig. 7G). The function of these Gal83 constructs was measured by examining gene expression (Fig. 7D, E, and F) and cell growth on different carbon sources (Fig. 7C). All of the Gal83 deletion constructs were functional, as judged by their ability to regulate gene expression and to grow by the fermentation of raffinose. Both of these abilities require a functional Snf1 kinase. Only the deletion of the GBD, either with or without the N-terminal region containing the phosphorylation sites, caused a dysregulation of Snf1 signaling. Cells expressing the GBD deletion of Gal83 showed an aberrant regulation of invertase, PCK1, and HXT1 in high glucose and the ability to grow in the presence of 2-deoxyglucose, both indicative of constitutive Snf1 activity. We also sought to determine whether the Gal83 protein was phosphorylated by both Snf1 and CKII in vivo. We noticed that the Gal83-ΔGBD protein showed a reduced level of SDS gel migration when cells were grown in low glucose. This slower-mobility band is the result of in vivo phosphorylation since it can be converted to the higher-mobility species by treatment with phosphatase (Fig. 7G). The phosphorylation status of the Gal83-ΔGBD protein in cells expressing either no Snf1 protein, wild-type Snf1, or the Snf1-T210A mutant was examined. In the presence of wild-type Snf1, phosphorylated Gal83-ΔGBD is apparent (Fig. 7G, lanes 5 and 6). When the activation-impaired allele of SNF1 is present, the level of phosphorylation of Gal83-ΔGBD is reduced but detectable (Fig. 7G, lanes 7 and 8). Finally, in the absence of any Snf1 protein, the phosphorylation of the Gal83-ΔGBD protein was not observed (lanes 1 and 2). Therefore, the efficient phosphorylation of Gal83-ΔGBD in vivo most likely requires the action of both Snf1 kinase and a second kinase.
FIG. 7.

Effect of Gal83 deletions in vivo. (A) Schematic representation of Gal83 constructs used. All constructs contained 3 copies of the V5 epitope (3V5) at the N terminus and the α-γ interaction domain (AG) at the C terminus. Amino acids deleted in the GBD, N1, and N2 mutants are shown. (B) Anti-V5 Western blotting of cells (MSY557) transformed with plasmid vector (vec) or plasmid expressing wild-type (WT) Gal83 or a Gal83 deletion mutant as shown. (C) Growth assay of cells spotted onto medium containing glucose, raffinose, or sucrose with 2-deoxyglucose. (D) Invertase assay of cells grown in high and low glucose as shown. Mean values from three independent cultures are plotted with 1 standard error. (E and F) PCK1 (E) and HXT1 (F) mRNA levels were measured by qRT-PCR. Cells were grown in either high or low glucose prior to harvest. Mean values of fold changes in gene expression are plotted, with error bars representing 1 standard error. (G) Anti-V5 Western blotting of cells (MSY920) transformed with plasmids expressing Gal83-ΔGBD and either vector, Snf1, or Snf1-T210A as indicated. Extracts were prepared from cells grown in high (H) or low (L) glucose as shown. Extracts were treated with or without lambda phosphatase (lanes 1 and 2).
The N terminus of Sip1 inhibits accumulation and activity.
During the course of our investigations, we screened a high-copy-number genomic library for genes that could compensate for defects in the Snf1 kinase pathway. In two independent genetic screens, we isolated plasmids that contained genomic DNA that encoded the C-terminal fragment of the Sip1 protein. The fact that we never recovered the full-length SIP1 gene in these screens led us to suspect that a truncated Sip1 protein either was expressed at a higher level or was more active than full-length Sip1. To test this hypothesis, we generated recombinant Sip1 proteins tagged with the flag epitope at the N terminus between residues 5 and 6 (Fig. 8A). The truncated Sip1ΔN lacks residues 13 to 436. These two SIP1 genes were introduced into yeast cells on either low-copy-number (CEN) or high-copy-number (2μm) plasmids. Western blotting with anti-flag antibodies readily detected the Sip1 and Sip1ΔN proteins migrating somewhat slower than their predicted molecular masses (95 and 48 kDa, respectively) (Fig. 8B). The quantitation of the Sip1 signals indicated that the deletion of the N terminus results in an increase in protein accumulation in both the low- and high-copy-number plasmids. The Sip1ΔN protein expressed from the low-copy-number plasmid and the full-length Sip1 protein expressed from the high-copy-number plasmid are expressed at an equivalent level that is about 15-fold higher than that of the full-length protein expressed from a low-copy-number plasmid (Fig. 8B). The increased level of accumulation of the Sip1ΔN construct is also reflected by an increase in Snf1 kinase signaling, as judged by increased invertase induction (Fig. 8C) and increased aerobic growth on glycerol-ethanol medium (Fig. 8D). The deletion of the Sip1 N terminus appears to increase levels of activity as well as accumulation since the Sip1ΔN (CEN) protein shows a higher level of Snf1 signaling than Sip1 (2μm), even though both proteins are expressed at the same level.
FIG. 8.

Inhibitory effect of the Sip1 N terminus. (A) Schematic representation of the Sip1 constructs used showing three copies of the flag epitope (F3) at the N terminus and the α-γ interaction domain (AG) at the C terminus. Amino acids deleted in the ΔN construct are indicated. (B) Anti-flag Western blotting of cells (MSY557) transformed with either low-copy-number (CEN) or high-copy-number (2μm) plasmids expressing full-length Sip1 or the ΔN form of Sip1. The migration of the Sip1 protein is indicated (>), as is a band present in cells lacking the flag epitope (*). The relative abundance of the Sip1 proteins as determined by quantitative Western blotting is indicated. (C) Invertase expression in cells transformed with empty vector (vec) or with either high- or low-copy-number plasmids expressing Sip1. (D) Growth assays of cells expressing Sip1 from high- or low-copy-number plasmids. Tenfold serial dilutions of cells were spotted onto media containing glucose (Glu), raffinose (Raf), or a mixture of glycerol and ethanol (GE) as the carbon sources.
DISCUSSION
The discovery of carbohydrate-binding motifs in the β subunits of the AMPK enzymes (11, 20) suggests that these domains may bind a ligand and participate in the regulation of the AMPK pathway. Indeed, studies described previously by McBride et al. have shown that α1→6-branched carbohydrates bind the GBD and act as allosteric inhibitors of mammalian AMPK activity (12). In yeast, the involvement of glycogen in the regulation of Snf1 is uncertain, since mutations that block glycogen synthesis do not appear to impact Snf1 regulation (15). However, the deletion of the GBD from the Gal83 subunit resulted in the constitutive activation of Snf1, demonstrating that this domain is required for limiting Snf1 activity under conditions of glucose abundance (15). In this study, we sought to uncover the underlying mechanism of Snf1 regulation by the GBD and to extend the study to the other β subunits, Sip1 and Sip2.
The deletion of the GBD from the three β subunits resulted in very different outcomes. We found that the deletion of the GBD from Gal83 causes constitutive activation, consistent with data from studies described previously by Momcilovic et al. (15). When Gal83-ΔGBD was the sole β subunit, the expression of SUC2, HXT1, and PCK1 was deregulated, consistent with the constitutive activation of Snf1. These three genes were chosen since they are mechanistically distinct, with two genes (SUC1 and PCK1) being induced by Snf1 and one (HXT1) being repressed by Snf1. In contrast with the case of Gal83, very little change in Snf1-mediated gene regulation was observed when the GBD was deleted from Sip1 or Sip2. Little induction of invertase was observed, and little change in HXT1 or PCK1 mRNA abundance was detected. Thus, the three different isoforms of Snf1 appear to be regulated through distinct means. We performed quantitative Western blots to examine the phosphorylation state of the Snf1 activation loop. As was reported for experiments described above, the deletion of the GBD from Gal83 caused an increase in the level of phosphorylation of the Snf1 activation loop threonine. Our data showed a 7-fold increase in Snf1 phosphorylation when cells were grown in high glucose. Finally, we showed that this increase in activation loop phosphorylation may be caused by defects in the association of Reg1. Coimmunoprecipitation of Reg1 and Snf1 in cells expressing either full-length Gal83 or the Gal83-ΔGBD construct was examined. We detected a 3-fold reduction in the level of association of Reg1 with the Snf1 complex when the GBD was deleted. These data suggest that there is a direct correlation between the Reg1 association with Snf1 and the phosphorylation status of the Snf1 activation loop. We propose that the mechanism by which the Gal83 GBD maintains Snf1 in an inactive state is the recruitment of the Reg1-Glc7 complex (Fig. 9A).
FIG. 9.

Model for β subunit regulation of Snf1 kinase. (A) The Gal83 glycogen-binding domain downregulates the activity of the Snf1 kinase by helping to recruit the Glc7/Reg1 phosphatase complex. (B) The phosphorylation of the Snf1 kinase subunits is mediated by the three Snf1-activating kinases, each of which is capable of phosphorylating the activation loop threonine 210. Active Snf1 can then phosphorylate a conserved site present in Gal83 as well as in Sip1 and Sip2. Casein kinase II (CKII), recruited to the Snf1 complex through its association with the Sak1 protein, recognizes phosphorylation sites present in Gal83 but not in Sip1 or Sip2.
During the course of our studies with purified Snf1 kinase enzymes, we noticed that the Snf1 and Gal83 subunits could be phosphorylated in vitro. Incubation of the Snf1 heterotrimer with [γ-32P]ATP and the Snf1-activating kinase Sak1 showed an incorporation of radioactivity into Snf1 and Gal83 but not Snf4. Here we show that the other β subunits, Sip1 and Sip2, are also subjected to phosphorylation in vitro (Fig. 4C). The Gal83 protein is phosphorylated by both Snf1 and a kinase that is present in our TAP-purified preparations of Sak1. Mass spectrometry analysis indicated that the Sak1 protein was purified in a complex with CKII. Experiments with kinase-dead Sak1 and with recombinant purified CKII showed that the kinases responsible for the phosphorylation of Gal83 in vitro were Snf1 and CKII but not Sak1 (Fig. 6). We mapped the predominant sites of phosphorylation in Gal83 to between residues 63 and 110 (Fig. 5). A sequence that fits with the consensus Snf1 recognition sequence (28) is found in this region and is conserved with the Sip1 and Sip2 subunits (Fig. 5C). Also present were sites that fit the consensus recognition sequence for CKII (14); however, the CKII sites were present only in Gal83 and were not conserved in Sip1 or Sip2. This observation is consistent with data from in vitro kinase assays in which purified CKII could phosphorylate Gal83 but not Sip1 or Sip2.
Identifying the responsible kinase and mapping the sites of phosphorylation have proven to be easier tasks than uncovering the biological significance of these sites. First, the deletion of the region encompassing the phosphorylation sites of Gal83 did not appear to alter the function or regulation of the Snf1 kinase (Fig. 7). Nonetheless, it seems unlikely that the phosphorylation of Gal83 is a mere in vitro artifact. The phosphorylation of Gal83 was observed in vivo, as judged by a gel mobility shift of the Gal83-ΔGBD protein that was reversed by phosphatase treatment (Fig. 7G). The fact that the mobility shift was reduced but still observed when Gal83-ΔGBD was incorporated into a heterotrimer with activation-impaired Snf1 suggests that the phosphorylation of Gal83 in vivo is catalyzed by both Snf1 and a second kinase. Circumstantial evidence suggests that CKII may be that second kinase in vivo. First, CKII phosphorylates Gal83 in vitro. Second, Gal83-ΔGBD shows no evidence of phosphorylation when Snf1 is absent. Previous studies have shown that Snf1 and Sak1 associate in vivo and that CKII is a component of the Sak1 complex. Thus, we propose a model in which the phosphorylation of Gal83 in vivo is catalyzed by both Snf1 and CKII, with CKII being directed to Gal83 through its association with Sak1 (Fig. 9B).
Yeasts express three β subunits. Two of the β subunits, Gal83 and Sip2, are very similar in size (415 and 417 residues) and sequence (55% identity). We show here that they differ in abundance and their mechanisms of regulation. The third β subunit, Sip1, is almost twice as large as the other two (815 residues) and shows much less sequence similarity. We noted in previous studies that the expression of Sip1 as the only β subunit resulted in Snf1 kinase that had a reduced level of activity, as judged by the reduced ability to grow aerobically. The reduced level of activity of the Sip1 isoform is due in part to its low abundance. Using β subunits containing a triple-flag tag at their N termini and quantitative Western blotting, we show here that the Gal83 subunit is most abundant, being in excess over Sip2 and Sip1 by 4- and 16-fold, respectively (Fig. 1C). In this study we show that the nonconserved N terminus of Sip1 is in part responsible for its low abundance and for the reduced function of Snf1. The deletion of the Sip1 N terminus resulted in an 18-fold increase in its abundance along with a concomitant increase in Snf1 function (Fig. 8). This finding solved a curious genetic puzzle in that we had isolated clones of Sip1 in genetic screens selecting for increased Snf1 function. In two independent screens, we isolated fragments of the SIP1 gene that lacked the promoter region and over half of the protein-coding region. In spite of this large deletion, the SIP1 gene fragment provided more β function than the full-length SIP1 gene. Here we show that the deletion of the N terminus of Sip1 results in an increased accumulation of the Sip1 protein. This further suggests that the abundance of Sip1 may be a key regulator of its function.
Acknowledgments
We thank Tony Meyn and Diane Lenhart for help with qRT-PCR.
This work was supported by Public Health Service grant GM46443 from the National Institutes of Health.
Footnotes
Published ahead of print on 6 November 2009.
REFERENCES
- 1.Charbon, G., et al. 2004. Key role of Ser562/661 in Snf1-dependent regulation of Cat8p in Saccharomyces cerevisiae and Kluyveromyces lactis. Mol. Cell. Biol. 24:4083-4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dyck, J. R., et al. 1996. Regulation of 5′-AMP-activated protein kinase activity by the noncatalytic beta and gamma subunits. J. Biol. Chem. 271:17798-17803. [DOI] [PubMed] [Google Scholar]
- 3.Elbing, K., et al. 2006. Purification and characterization of the three Snf1-activating kinases of Saccharomyces cerevisiae. Biochem. J. 393:797-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghaemmaghami, S., et al. 2003. Global analysis of protein expression in yeast. Nature 425:737-741. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein, A., and J. O. Lampen. 1975. β-D-Fructofuranoside fructohydrolase from yeast. Methods Enzymol. 42C:504-511. [DOI] [PubMed] [Google Scholar]
- 6.Hardie, D. G. 2007. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8:774-785. [DOI] [PubMed] [Google Scholar]
- 7.Hedbacker, K., and M. Carlson. 2008. SNF1/AMPK pathways in yeast. Front. Biosci. 13:2408-2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedbacker, K., et al. 2004. Pak1 protein kinase regulates activation and nuclear localization of Snf1-Gal83 protein kinase. Mol. Cell. Biol. 24:8255-8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong, S. P., and M. Carlson. 2007. Regulation of snf1 protein kinase in response to environmental stress. J. Biol. Chem. 282:16838-16845. [DOI] [PubMed] [Google Scholar]
- 10.Kushnirov, V. V. 2000. Rapid and reliable protein extraction from yeast. Yeast 16:857-860. [DOI] [PubMed] [Google Scholar]
- 11.Machovic, M., and S. Janecek. 2006. The evolution of putative starch-binding domains. FEBS Lett. 580:6349-6356. [DOI] [PubMed] [Google Scholar]
- 12.McBride, A., et al. 2009. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 9:23-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCartney, R. R., and M. C. Schmidt. 2001. Regulation of Snf1 kinase. Activation requires phosphorylation of threonine 210 by an upstream kinase as well as a distinct step mediated by the Snf4 subunit. J. Biol. Chem. 276:36460-36466. [DOI] [PubMed] [Google Scholar]
- 14.Meggio, F., et al. 1994. Substrate specificity of protein kinase CK2. Cell. Mol. Biol. Res. 40:401-409. [PubMed] [Google Scholar]
- 15.Momcilovic, M., et al. 2008. Roles of the glycogen-binding domain and Snf4 in glucose inhibition of SNF1 protein kinase. J. Biol. Chem. 283:19521-19529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nath, N., et al. 2002. Purification and characterization of Snf1 kinase complexes containing a defined beta subunit composition. J. Biol. Chem. 277:50403-50408. [DOI] [PubMed] [Google Scholar]
- 17.Neumann, D., et al. 2003. Mammalian AMP-activated protein kinase: functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr. Purif. 30:230-237. [DOI] [PubMed] [Google Scholar]
- 18.Nolan, T., et al. 2006. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1:1559-1582. [DOI] [PubMed] [Google Scholar]
- 19.Polekhina, G., et al. 2003. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr. Biol. 13:867-871. [DOI] [PubMed] [Google Scholar]
- 20.Polekhina, G., et al. 2005. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure 13:1453-1462. [DOI] [PubMed] [Google Scholar]
- 21.Proft, M., et al. 1995. CAT5, a new gene necessary for derepression of gluconeogenic enzymes in Saccharomyces cerevisiae. EMBO J. 14:6116-6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubenstein, E. M., et al. 2008. Access denied: Snf1 activation loop phosphorylation is controlled by availability of the phosphorylated threonine 210 to the PP1 phosphatase. J. Biol. Chem. 283:222-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanders, M. J., et al. 2007. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 403:139-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt, M. C., and R. R. McCartney. 2000. Beta-subunits of Snf1 kinase are required for kinase function and substrate definition. EMBO J. 19:4936-4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmittgen, T. D., and K. J. Livak. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101-1108. [DOI] [PubMed] [Google Scholar]
- 26.Scott, J. W., et al. 2004. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 113:274-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith, F. C., et al. 1999. The SNF1 kinase complex from Saccharomyces cerevisiae phosphorylates the transcriptional repressor protein Mig1p in vitro at four sites within or near regulatory domain 1. FEBS Lett. 453:219-223. [DOI] [PubMed] [Google Scholar]
- 29.Southern, J. A., et al. 1991. Identification of an epitope on the P and V proteins of simian virus 5 that distinguishes between two isolates with different biological characteristics. J. Gen. Virol. 72:1551-1557. [DOI] [PubMed] [Google Scholar]
- 30.Tomas-Cobos, L., and P. Sanz. 2002. Active Snf1 protein kinase inhibits expression of the Saccharomyces cerevisiae HXT1 glucose transporter gene. Biochem. J. 368:657-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treitel, M. A., et al. 1998. Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol. Cell. Biol. 18:6273-6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vincent, O., et al. 2001. Subcellular localization of the Snf1 kinase is regulated by specific beta subunits and a novel glucose signaling mechanism. Genes Dev. 15:1104-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao, B., et al. 2007. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449:496-500. [DOI] [PubMed] [Google Scholar]
- 34.Zhang, B. B., et al. 2009. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 9:407-416. [DOI] [PubMed] [Google Scholar]