

Abstract
Two major issues are presented. First, a challenge is made by us that a misunderstanding of physiology has led to incomplete or wrong functional designations of genes in some cases. Normal physiological processes are dynamic, integrated and periodic, and, therefore, it is difficult to define normal physiological function by looking at a single time point or single process in a non-stressed subject. The ability of the organism to successfully respond to homeostatic disruptions defines normal physiology. Genes were selected for survival and to appropriately respond to stresses, such as physical activity. Omitting gene functions by restricting them to non-stressful conditions could lead to less than optimal primary preventions, treatments and cures for diseases. Physical exercise, as a stressor, should be used to better demonstrate the complete functional classifications of some genes. Second, the challenge from others of an ‘exercise pill’ as a mimetic of natural physical activity will be shown to be lacking a scientific basis. The concept of an ‘exercise pill’/‘exercise mimetic’ demonstrates an inadequate appreciation of the complexities in integrating cell, tissue, organ and systems during both acute disruptions in homeostasis by a single bout of exercise, and longer-term chronic adaptations to different types of exercise such as resistance and endurance. It is our opinion that those promoting drugs targeting a single or few molecules should not redefine the term ‘exercise’ and exercise concepts in an attempt to sensationalize findings. Additionally, the scientific criteria that the authors demand to be met to legitimately use the terms ‘exercise pill’ and ‘exercise mimetic’ are presented.
Is normal physiological function being obtained from abnormal pathalogical models?
What is normal function?
A fundamental question for medicine is What is normal physiology? Physiology is defined in the Oxford English Dictionary (2009, online) as ‘the branch of science that deals with the normal functioning of living organisms and their systems and organs’. Pathology is defined as ‘the branch of science that deals with the causes and nature of diseases and abnormal anatomical and physiological conditions’. The logical construct is then set where any cause resulting in a switch from normal to abnormal function is pathological. One such cause that fits this construct is a reduction in daily physical activity. A reduction in physical activity levels cause abnormal function, initiating both increases in risk factors for chronic disease in the short term and pathology associated with overt clinical disease in the longer term. In support of an increase in risk factors for chronic diseases in response to even short-term reductions in physical activity, we have recently reported that removal of 8500 steps (dropping from ∼10 000 to ∼1500) in the absence of a structured exercise programme for 2 weeks results in abnormal physiological changes in healthy young men. Such changes included decreases in maximal 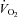 and in whole body insulin sensitivity and increases in intra-abdominal fat. Epidemiological data show that if reductions in physical activity are extended to the longer term, prevalence of many chronic diseases increases. For example, physically inactive humans have increased risks of breast cancer, colon cancer, coronary heart disease, hypertension, osteoporosis, stroke and type 2 diabetes that are ∼30%, ∼40%, ∼40%, ∼30%, ∼60%, ∼60% and ∼50%, respectively (Katzmarzyk & Janssen, 2004). Thus, studying physiological conditions in a population of sedentary or less active humans is more a study of pathology than normal physiology. Medicine must know what biologically normal physiology is in order to know how to prescribe the most optimal treatments to maintain optimal health of all organ systems. Obviously, according to this paragraph, low levels of physical activity do not produce optimal health.
and in whole body insulin sensitivity and increases in intra-abdominal fat. Epidemiological data show that if reductions in physical activity are extended to the longer term, prevalence of many chronic diseases increases. For example, physically inactive humans have increased risks of breast cancer, colon cancer, coronary heart disease, hypertension, osteoporosis, stroke and type 2 diabetes that are ∼30%, ∼40%, ∼40%, ∼30%, ∼60%, ∼60% and ∼50%, respectively (Katzmarzyk & Janssen, 2004). Thus, studying physiological conditions in a population of sedentary or less active humans is more a study of pathology than normal physiology. Medicine must know what biologically normal physiology is in order to know how to prescribe the most optimal treatments to maintain optimal health of all organ systems. Obviously, according to this paragraph, low levels of physical activity do not produce optimal health.
A fundamental question for physiology is ‘What is normal function?’. Physiological processes are dynamic, integrated and periodic, and therefore it is difficult to define normal physiological function by looking at a single time point or single process. First, normal physiology contains circadian/diurnal rhythms modifying hormone levels and behaviour though intrinsic mechanisms. Conversely, the extrinsic decision to have meals or participate in physical activity alters fluxes of metabolic and signalling pathways. However, whether responding to intrinsic or extrinsic, acute or chronic perturbations, physiological processes react by minimizing disruptions to homeostasis. While acute disruptions to homeostasis are normalized by rapid changes in existing protein function, chronic disruptions to homeostasis are often minimized by a more permanent alteration of basal gene expression. Consequently, normal physiology can be defined by the capacity of systems to respond to acute as well as successfully adapt to chronic disruptions in homeostasis. When responding to disruptions in homeostasis, the greater the functional capacity an organ system has, the greater functional reserve is present, and thus the greater the likelihood of survival if faced with an external challenge to homeostasis. An example is a decline in organ systems’ capacity with ageing, which leads to increased likelihood of death due to the inability to counter a homeostatic disruption. Biologists must recognize that mechanisms minimizing disruptions to homeostasis by exercise are normal functionand that they provide the proper context from which to understand gene–environment interaction.
Important differences exist between the normal physiological periodicity of daily exercise and the abnormal pathology caused when the exercise signal is continuously stimulated. Kramer & Goodyear (2007) discuss the paradox of healthy episodic vs. chronic diseased cellular stress; and Carey & Kingwell (2009) provide examples of how sustained effects of some genetic/pharmacological manipulations ‘do not accurately model the episodic nature of exercise’. Knocking out of c-Jun amino-terminus kinases (JNKs) (Hirosumi et al. 2002) or using salicylates to inactivate IκB kinase-β (Kim et al. 2001) diminishes adiposity, improves insulin sensitivity and enhances insulin receptor signalling capacity. On the other hand, exercise increases the activation of NFκB (Kramer & Goodyear, 2007) and JNK (Goodyear et al. 1996), but repeated daily exercise also diminishes adiposity and improves insulin sensitivity. Another example is the episodic release of IL-6 from contracting skeletal muscle, and its consequent increase in blood and physiological effect on target organs vs. its pathological effects during its chronic increase in low-grade chronic inflammatory diseases (Mathur & Pedersen, 2008).
What is the optimal way to study normal physiology? Normal physiology is the dynamic interaction between resting and stressed systems. Therefore understanding a physiological process requires explanation of gene or protein function at both rest and in response to homeostatic disruptions. For example, studying people at rest would not elucidate any of the commonly known beneficial adaptations to chronic exercise training that include (1) lowering cardiac work at a given absolute workload (the adaptation to endurance exercise training that switches to a higher metabolic cardiac work efficiency by increasing stroke volume more and increasing exercise tachycardia less, thus lowering myocardial O2 uptake at any given cardiac output); and (2) oxidizing more fatty acids at a similar workload (the endurance exercise training adaptation by switching away from glucose as a fuel to using greater fatty acids as a fuel). Conversely, while remaining sedentary certainly disrupts homeostasis, it is a cause of pathological maladaptations (see above) and thus not appropriate for understanding normal physiology.
Normal physiology includes the organism's response to daily or repeated high levels of physical stress. The response allows for the physiological study of the massive integration of cells, organs and organ systems attempting to minimize homeostatic disruptions during and following exercise stress. For this integration to occur, thousands of changes in protein post-translational modifications, protein abundance, and gene expression happen simultaneously. Repeated moderate–high levels of exercise stress are normal physiology because in their absence, the sedentary organism loses the maximal capacity of organ systems, sliding toward pathology. Thus, individuals with high levels of exercise capacity (high fitness) have become targets for physiological study because they have lower prevalence of pathological chronic diseases and of mortality.
Taken together, we contend gene functions differ at low and high levels of daily physical activity. Clearly then the definition of normal physiology is not limited to rest, but in fact must extend to the capacity of organ systems to maintain homeostasis when disrupted by stresses such as physical activity. Categorization of genes by normal function only at rest or in subjects who have little daily physical activity leads to an incomplete understanding of physiology and may eventually hurt the health of patients. We contend protein functions determined only at rest in sedentary subjects are being listed in protein function databases as absolutes without proper interpretation, and thus the functional databases are not completely defined. The resulting problem is that no one today knows how much physiology is incomplete or wrong because of the near-exclusive usage of low activity subjects at rest that omits gene functions in response to acute and chronic environmental stresses such as exercise or exercise training. A common way that these improperly defined gene functions are being designated is through the expanded use of transgenic animals in determining gene functions. The next section will consider examples of wrong functions being attributed to genes by the using transgenic/knockout mice with restricted physical activity.
Transgenic/knockout animals
We will contend here that the physical activity level of genetically modified animals, in some cases, is critical to the interpretation of a gene's complete function. This is important because the gene often operates with fundamentally different functions depending on the physiological context of high or low physical activity (as described above). If the function of a gene is characterized only during low physical activity, but the function of this same gene differs in response to high physical activity, then the incomplete function of the gene is obtained and placed into the database. Our contention is demonstrated by two scenarios (Fig. 1).
Figure 1. Presence or absence of phenotype when comparing forced inactivity to physically active in Scenarios A and B and resultant conventional and correct interpretations.

See the text for detailed explanations.
Scenario A of Fig. 1 is demonstrates how exercise can reveal phenotypes not present in resting transgenic mice. For example mice with a mutated myosin binding protein C appeared overtly healthy at rest, but they exhibited a phenotype of decreased exercise capacity and death in four out of eight of the mice within 24 h of ending treadmill running exercise in one study (Yang et al. 2001). A review wrote, ‘There are numerous examples of transgenic models in which the baseline cardiovascular phenotype is unchanged or minimally changed from the wild type, only to become manifest during the stress of exercise testing (Bernstein, 2003). Another example is hormone sensitive lipase (HSL) knockout mice that at rest have no differences in circulating fatty acids or liver glycogen content, but exercise reveals an inability to increase circulating fatty acids and a accelerated reduction in liver glycogen, thereby appropriately defining a physiological role for HSL (Fernandez et al. 2008). By exercising, disruption of physiological homeostasis necessitates attempts to restore homeostasis through the activation of the specifically targeted genes. Thus, physiological function of the genes can better be elucidated.
To ensure proper characterization of gene function, we believe that one must test the function of a gene in response to physiological stresses (i.e. high physical activity) that occurred during the evolution of our present genome. Throughout evolution, high levels of physical activity were obligatory for survival of animals and humans (Booth et al. 2000; Booth et al. 2002; Chakravarthy & Booth, 2004; Booth & Lees, 2007). Bennett and Ruben elegantly wrote, ‘The selective advantages of increased activity capacity are not subtle but rather are central to survival and reproduction. An animal with greater stamina has an advantage that is readily comprehensible in selective terms. It can sustain greater levels of pursuit or flight in gathering food or avoiding becoming food. It will be superior in territorial defense or invasion. It will be more successful in courtship and mating’ (Bennett & Ruben, 1979). Until only recently, physical activity was obligatory for survival. Today, mice and rats housed in cages unable to mimic evolution's physical activity levels may not completely elucidate the evolved gene's function. Thus, the failure to disturb homeostasis in a sedentary animal (without access to voluntary wheel running) will often fail to uncover the true evolutionary function of the gene. Humans also inherited evolutionary gene functions during a physically active lifestyle. For instance in scenario A, the appearance of a new phenotype following voluntary physical activity reveals the biological purpose for which the gene was selected.
In scenario B of Fig. 1 we discuss the situation where normal physiology rescues the disease phenotype that occurs at rest. As an example, we will use melanocortin-4 receptor knockout (MC4R−/−) mice that develop a maturity-onset obesity syndrome associated with hyperphagia, hyperinsulinaemia, and hyperglycaemia (Huszar et al. 1997). In response to this phenotype, the Mouse Genome Informatics Website of Jackson Laboratories states, ‘Mutations in this gene (MC4R) result in hyperglycemia and weight gain’. However, this phenotypic information was obtained from laboratory mice with a low level of physical activity. When MC4R−/− mice are allowed to increase physical activity levels with voluntary wheel running, this led Haskell-Luevano et al. (2004) to write, ‘Herein, we present the effects of voluntary exercise on the MC4R knockout mice in terms of bypassing the morbid obesity and hyperphagia phenotypes associated with this genetic obesity model’ (italics added); and also wrote (Haskell-Luevano et al. 2009), ‘voluntary exercise can prevent the genetic predisposition of MC4R-associated obesity and diabetes.’ Allowing mice to have normal physiology by placing wheels for running into a cage prevented obesity, making the interpretation of Jackson Laboratories only valid with sedentary mice. The opposite conclusion is reached when the mutation interacts with increased levels of physical activity, suggesting that MC4R plays no role in metabolic dysfunction. Studied in the context of a sedentary environment, gene interaction is more akin to a gene's role in pathology than in normal physiology. In this way the interaction between sedentary behavior and gene function may provide information about current sociological levels of physical activity in a pathological context, but not a physiological context. Indeed in the context of normal physical activity, the effect of MC4R on the lean phenotype is minimal. A revised entry into the functional protein database should read, ‘The response to the physiological stress of increased physical activity results in no phenotype in the MC4R mouse. However, in response to a sedentary pathological stress, the MC4R mouse develops obesity, hyperinsulaemia, and hyperleptaemia associated with type 2 diabetes.’ An amazing discovery is thus made for an interaction between the MC4R gene and low physical activity compared to evolutionary levels.
OLETF rats, have a spontaneous mutation to inactivate the cholecystokinin-1 receptor (CCKR1), causing hyperphagia. Similar to MC4R−/− rats, above, no disease phenotype exists when OLETF rats are permitted voluntary running (Morris et al. 2008). However, a disease phenotype of obesity, type 2 diabetes and non-alcoholic fatty liver disease is revealed when voluntary running is abolished with CCKR1 mutation (Rector et al. 2008b), indicating that CCKR1, like MC4R−/−, should be labelled as having no phenotype in physically active animals. If protein function databases wish to designate a disease phenotype, they need to indicate that the interaction of low physical activity is necessary to show disease. These examples provide evidence to study knockout transgenic models both during an acute exercise bout and at rest following chronic repeated exercise training better insight into ‘normal’ physiology is gained.
Summary
A challenge has been presented. What is normal physiology? Are correct models of chronic diseases being tested to obtain correct biological functions of genes as orchestrated for survival in the last thousands of years? These questions challenge whether biologists are studying and interpreting gene functions correctly.
Liabilities of the terms ‘exercise pill’ and ‘exercise mimetics’
The term ‘exercise mimetic’ has recently been used in numerous publications. In the first publication, to the best we can determine gene expression for PGC-1α and PKC-ζ along with 10 other genes were said to suit their ‘goal of identifying a tractable number of intervention points for the development of “exercise mimetic” pharmaceuticals that improve integrated area under the curve for glucose (Ort et al. 2007). The second publication was professor Evans paper in Cell entitled ‘AMPK and PPARδ agonists are exercise mimetics’ (Narkar et al. 2008). The 2008 Scientific Report of the Salk Institute page for the accomplishments of Professor Evans, states, ‘By tinkering with a metabolic program in muscle, we stumbled upon “exercise in a pill”.’ This finding begs the question: what is an exercise mimetic? The term ‘mimetic’ is defined by the Oxford English Dictionary (1989, online) under the subheading “Pharmacology and Biochemistry” as ‘relating to or practicing mimesis or mimicry synthetic compound that produces the same (or a very similar) effect as another (esp. a naturally occurring) compound’. The term ‘exercise’ is defined by the Oxford English Dictionary (1989, online) as ‘exertion undertaken with a view to the maintenance or improvement of health’ and by the Merriam–Webster Medical Dictionary (2002) as ‘Active bodily exertion performed to develop or maintain fitness’. It is our opinion that drugs targeting a single or few molecules should not be used to redefine the term ‘exercise’ to fit the need to sensationalize their findings. Exercise scientists have defined hundreds of biochemical, molecular, and physiological adaptations to ‘exercise’ for two score years. The cumulative body of exercise publications shows adaptations to exercise in almost every cell type, tissue, organ and system (Fig. 2). The term exercise should be reserved to describe all complex regulatory and adaptive responses to the physiological perturbations of physical exertion rather than being used to describe the limited adaptive responses to a drug. A third publication (Carey & Kingwell, 2009) concludes, ‘the term ‘exercise mimetic’ is probably not appropriate; rather a term that better reflects the specific aspect of exercise being targeted may achieve better acceptance’.
Figure 2. Changes induced upon decreasing physical activity from high to low levels.

A proposed exercise mimetic would have to prevent the same changes as natural exercise or the exercise pill would not be an authentic mimetic. Original drawing was generously made by Gheorghe Constantinescu.
The second part of this review will consider our opinions of the concept of drugs mimicking exercise. The consideration is divided into two sections – science, and economics.
Science
The question of whether, in our opinion, science supports the possibility of a true ‘exercise mimetic’ will be split into three sections – exercise sciences, pharmacological sciences and health sciences.
Exercise sciences
The first reservation is that human physiological adaptations to disruptions in homeostasis caused by exercise are so complex that no mono- or poly-pill will ever be able to mimic every exercise adaptation for six reasons.
Exercise adaptations are multi-organ. Adaptations to exercise and lack of exercise occur in almost every organ system, such as circulatory, neural, endocrine, skeletal muscle, connective tissue (bones, ligaments and tendons), gastrointestinal, immune and kidney (Fig. 2). Systems where exercise adaptations don't occur or are not well documented to occur are lungs and senses. Pharmaceuticals, on the other hand, must limit their effects to single organs. This is recognized in the Concluding Remarks and Future Perspectives in a review article by Puigserver and Spiegelman, writing, ‘Because of the role of PGC-1α in many important metabolic processes, it is worth asking whether and how the activities of PGC-1α might be modulated for therapeutic purposes …[of course, this wide range] wide range of physiological actions [by PGC-1α in multiple tissues] also points out a problem: activation or inhibition is going to have to be tissue selective or tissue specific to be useful. … This suggests that the usefulness of PGC-1α as a therapeutic target will all come down to an ability to achieve biological specificity’ (Puigserver & Spiegelman, 2003). Since exercise produces adaptations in multiple organs, exercise has a natural ‘built-in’ specificity that is unmatched by the therapeutic limitations of drugs.
Molecular biology has led to the idea that exercise is polygenetic. Already in 1996, 50 mRNAs had been reported to be altered by exercise/exercise training in skeletal muscle alone (Booth & Baldwin, 1996). More recently, 900 mRNAs in skeletal muscle had greater than 2-fold changes during the 24 h after either 3 or 12 h of voluntary running (Choi et al. 2005). Interestingly, lack of physical activity by hindlimb immobilization of rat (involving the soleus muscle) required an exhaustive search for an mRNA in skeletal muscle that did not change (Pattison et al. 2003). Both of these studies indicate that many more than just one or two mRNAs change after alterations in contractile activity. The cumulative data demonstrate that the complexity of tissue physiological responses to alterations in exercise levels extends to changes in thousands of mRNAs in skeletal muscle. In addition to skeletal muscle, data exist to support several hundred mRNAs changing in peripheral blood mononuclear cells (Radom-Aizik et al. 2009) and neurophils (Radom-Aizik et al. 2008) of humans, and in the liver (Colombo et al. 2005) and cardiac muscle of rats (Strom et al. 2005). Carey & Kingwell (2009) conclude with regard to health benefits of regular exercise. ‘Obviously no single pharmaceutical agent could mimic this response.’ Altogether, the combination of a number of different organs having individualized changes of hundreds of genes in response to exercise presents science that makes it very unlikely that one or two molecules could prove to be therapeutic targets for an ‘exercise pill’ in the near future.
Different exercise modalities result in different mRNA expressions, protein expressions, and physiological phenotypes. At the mRNA levels, differential gene expression occurs in the same tissue in response to resistance and endurance exercises (Holloszy & Booth, 1976; Stepto et al. 2009). For example, at the protein level, contractile protein content (per whole muscle) increases per whole skeletal muscle with resistance training, but remains unchanged per unit of mass. On the other hand in response to endurance training, contractile protein content and concentration remain largely unchanged. Conversely, both mitochondrial protein concentration and content (per whole muscle) increase in endurance-trained individuals, while for strength training only total content and not concentration increases. Lastly, an example of differing phenotypes in the same organ in response to two types of endurance exercise is provided by changes in bone mass. While bone mass increases in endurance running, it decreases in endurance cycling (Rector et al. 2008a). Thus even if an ‘exercise pill’ produced the same (or a very similar) effect as one type of exercise, it would have difficulty replicating other types of exercise that have their own unique health benefits.
Knockout animals show us that one key exercise gene is unlikely to exist. The observation that most mitochondrial genes were reduced in peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1α) knockout mice led to the conclusion that PGC-1α was the ‘master regulator gene of mitochondrial biogenesis’ (Wu et al. 2002). However, these results were obtained with sedentary mice. When PGC-1α knockout mice were endurance-exercise trained, they still had some or all exercise adaptations in two different studies. Piltgaard and colleagues (Leick et al. 2008) found that endurance exercise training increased aminolevulinate synthase, cytochrome oxidase I, cytochrome c, and hexokinase II mRNA and protein levels in skeletal muscle equally in wild-type and PGC-1α whole-body knockout (KO) mice. They concluded that ‘PGC-1α is not mandatory for exercise training-induced adaptations in skeletal muscle mitochondrial proteins.’ (This is another example of an error in gene classification presented earlier in the first portion of this review.) Another putative master regulator of exercise induced phenotypes is AMPK, which is the target of the proposed ‘exercise mimetic’ AICAR. However, when Pilegaard and colleagues tested the response of whole-body α2- and α1-AMPK knockout mice to an endurance-exercise training protocol, a different conclusion was reached (Jorgensen et al. 2005). They stated, ‘In conclusion, KO of the α2- but not the α1-AMPK isoform markedly diminished AMPK activation during running. Nevertheless, exercise-induced activation of the investigated genes in mouse skeletal muscle was not impaired in α1- or α2-AMPK KO muscles.’ Pilegaard and colleagues also concluded, ‘other factors can take over for training-induced responses in muscles when either AMPK or PGC-1α is lacking’ (Leick et al. 2008).
Furthermore, AICAR does not mimic many biochemical adaptations to either an acute exercise bout or endurance exercise training. For example, (a) after a single injection of AICAR, plasma fatty acids fell with no change in plasma glycerol while an exercise bout increases both of them – in the same rats, AICAR had no effect on skeletal muscle glucose-6-phosphate or glycogen concentration, but both decreased after a single bout of exercise (Rantzau et al. 2008); (b) AICAR is less effective in attenuating ACCβ Ser218 phosphorylation (inferring less AMPK activation) than exercise training (both 10 days of treatment) (McConell et al. 2008); (c) AICAR reduces IL-6 mRNA expression and protein release (Glund et al. 2009), while exercise increases both of them (Ostrowski et al. 1998); and (d) AICAR does not increase sarcolemmal fatty acid binding protein concentration, but muscle contraction does (Jeppesen et al. 2009). Therefore, AICAR does mimic biochemical adaptations to endurance exercise as claimed in a recent publication (Narkar et al. 2008).
These examples clearly show that no one putative master regulator of exercise phenotype can completely prevent exercise-induced adaptations from occurring. Indeed, from an evolutionary viewpoint, the ability to adapt to increased levels of physical activity was necessary for survival, likely containing multiple overlapping pathways responsible for single adaptations. Thus, both the science and evolutionary logic predict that a single molecule will be unable to ‘mimic’ exercise-induced adaptations.
(5) Multiple signalling transduction. It is beyond the permitted word length here to review the tremendous number of signalling pathways activated by exercise, but others have just commented on the complexity of metabolic homeostasis during exercise and the inappropriateness of the term ‘exercise mimetic’ (Carey & Kingwell, 2009). In the above examples, two such pathways are examined. What will be mentioned is that some of these pathways are likely to be differentially modified by resistance- and endurance-type exercise. It is unlikely that a single molecular target, like AMPK, would activate the same, or similar, pathways for increasing mitochondrial concentration and skeletal muscle mass to meet the definition of mimetic as claimed (Narkar et al. 2008). It would be dangerous to claim, as is being done on Nova's Marathon Mouse web site (http://www.pbs.org/wgbh/nova/sciencenow/0403/03.html), that AICAR is an exercise pill for the elderly because AICAR does not counter loss of muscle strength, a major cause of mortality in elderly (Metter et al. 2002; Newman et al. 2006; Cesari et al. 2009), but decreases signalling by inhibiting signalling proteins that regulate the initiation of protein synthesis in skeletal muscle (Deshmukh et al. 2008).
(6) Claims of ‘exercise pill’ are flawed. Methodological concerns for the paper claiming that AMP kinase is an exercise mimetic have been presented in our recent Viewpoint article (Booth et al. 2009).
Pharmacological sciences
ED50 and LD50. The dose of drug required to produce a specified effect in 50% of the population is the median effective dose (ED), abbreviated as ED50. In preclinical studies of drugs, the median lethal dose (LD), as determined in experimental animals, is abbreviated LD50. The ratio of LD50 to ED50 is an indication of the therapeutic index. An exercise pill would be required by the US Food and Drug Administration to obtain a therapeutic index in pre-clinical testing. Comparisons of therapeutic indices would be demanded by us in order to prove that an ‘exercise pill’ was as effective as natural physical activity in healthy populations.
No known LD50 exists for exercise. It is unlikely that an exercise LD50 exists. In order to determine whether LD50 exists for exercise or not, an American Heart Association Statement on Exercise and Acute Cardiovascular Events – Placing the Risks into Perspective was used as a reference source by Thompson et al. (2007) and some of their cited studies were used to obtain the following death rates. The absolute rate of exercise-related death among US high-school and college athletes was 1 per 133 000 men and 1 per 769 000 women (numbers include all sports-related non-traumatic deaths, so other causes than cardiovascular events were included) (Van Camp et al. 1995). An incidence of 1 sudden death per 33 000 was reported in Italy in older individuals participating in more intense sports. In less intense sports, death rates in adults have been reported in two articles. The first indicated that 1 death occurred per 396 000 person-hours of jogging, or at a rate of 1 death per year for every 7620 joggers; these numbers include known or readily diagnosed coronary artery disease (Thompson et al. 1982). However, when only previously healthy individuals were examined in the same study, death rates were about half – 1 death per 792 000 hours or 15 260 subjects. The second article reported 1 death per 82 000 club members at a large commercial fitness chain, a rate of 1 death per 2.57 million workouts (Thompson et al. 2007). Club members who exercised infrequently or less than once a week composed ∼50% of the exercise-related deaths. In agreement, individuals who exercise regularly have both a lower base-line risk of myocardial infarction and lower relative risk during heavy physical exertion (Mittleman et al. 1993).
Therapeutic index (LD50/ED50). Drugs with wide therapeutic indices are better than ones with narrow indices. No level of exercise is known to be so toxic as to produce 50% mortality. Therefore, no LD50 exists for exercise. Theoretically then the dosage (duration × intensity) of exercise to cause a LD50 must be very high. Therefore, the LD50/ED50 ratio approaches infinity. Any ‘exercise mimetic’ drug must also have an extremely high LD50 and a therapeutic index of near infinity to perform as well as natural exercise. Without an exercise mimetic as therapeutically effective as exercise, it is questionable whether it would be ethical to give an exercise pill to someone capable of being physically active.
Health sciences
In addition to meeting scientific criteria to qualify as an exercise mimetic, any such pharmaceutical should ultimately improve health with a similar efficacy as exercise. Simply put, an exercise mimetic must have, at least, similar ED50 levels to physical activity guidelines to justify cost effectiveness. While the health benefits of exercise are undisputed based on numerous epidemiological and clinical studies, it will be difficult to obtain a single ED50. For each patient, the ED50 of exercise for a specific chronic disease will vary depending on exercise type, age, sex and exercise frequency, intensity and duration (Church & Blair, 2009). Nevertheless in attempting to determine the level of physical activity that an exercise mimetic must meet for substantial health benefits to be achieved, we defer to the Physical Activity Guidelines Advisory Committee Report (Physical Activity Guidelines Advisory Committee, 2008). While the report astutely states that there are no simple guidelines for daily physical activity, they make the following statement as a general overall recommendation for aerobic exercise: ‘However, as a starting place for overall public health benefit, data from a large number of studies evaluating a wide variety of benefits in diverse populations generally support 30 to 60 minutes per day of moderate- to vigorous-intensity physical activity on 5 or more days of the week. For a number of benefits, such as lower risk for all-cause mortality, coronary heart disease, stroke, hypertension, and type 2 diabetes in adults and older adults, lower risk is consistently observed at 2.5 hours per week (equivalent to 30 minutes per day, 5 days per week) of moderate- to vigorous intensity activity’ (Physical Activity Guidelines Advisory Committee, 2008). The Physical Activity Guidelines Advisory Committee Report presents data showing that this amount of aerobic physical activity reduces mortality risk by 30%, cardiovascular diseases by 20–35%, and metabolic diseases by 30–40% in healthy populations. In addition the Committee recommends that progressive muscle strengthening exercises that target all major muscle groups be performed on two or more days per week consisting of 8–12 repetitions of each exercise performed to volitional fatigue in order to improve muscle strength.
Known negative health consequences of drugs that enhance physical performance
Although there are many current drugs that mimic some adaptations to exercise, none have been considered exercise pills. For example, while never being called exercise pills, anabolic steroids and recombinant erythropoietin both mimic and enhance a minimal number of exercise adaptations. However, these drugs contain major side effects. For example, Sjoqvist et al. (2008) list some side effects of anabolic steroids as endocrinological, blood lipid dysfunction, cardiac arrest, premature mortality and neuropsychiatrical. Side effects of erythropoietin are myocardial infarction, cerebrovascular disease and serious thromoembolic events. A true exercise mimetic would not have any side effects that do not occur with exercise (normal side effects of exercise can be muscle soreness, fatigue, etc.), and certainly no major side effects. Carey & Kingwell (2009) contend that an exercise polypill would ‘likely be associated with unwanted effects.’
In summary, an exercise mimetic would not be able to have a side effect that exercise does not have. It is simple logic, with reference to the definition of ‘mimetic’, that an exercise pill cannot increase the risk of any unhealthy condition that exercise itself doesn't cause. Again the ethics of harming the patient must be considered.
Potential negative health consequences from altered behaviour by the public using ‘exercise mimetics’
If individuals believe exercise is a treatment that can be replaced by drugs, harm to the individual will occur in two ways. First, the patient will fail to achieve optimal health. As outlined above, the beneficial effects of exercise are so widespread and diverse (Fig. 2) that current drug approaches directed at mimicking a limited number of exercise pathways only primarily prevent a limited number of chronic diseases. Due to the limited effectiveness such a lifestyle supplement, a false sense of pharmacological health would be created, leading many people to stop exercising altogether. The fact that such a drug would be called an ‘exercise mimetic’ rather than a more appropriate title such as an anti-diabetic drug or anti-obesity drug is purely sensationalizing the particular drug, again leading to a false sense of health security in the general public. Obviously, the goal of biomedical research is to make people healthier; however, continued use of the term ‘exercise pill’ or ‘exercise mimetic’ for drugs incapable of truly mimicking exercise according to the definition of mimetic in the Oxford English Dictionary online may actually lead to a reduction in health of our general population. If this were to occur, we would consider it as unethical.
When drug targets of exercise pathways make sense
We agree that it is a worthwhile goal to find drug targets to a small number of the total exercise adaptations that will keep patients from losing functional capacity in a small number of the total organ systems that decondition when they are unable to exercise. Such pharmaceuticals could be clinically advantageous during recovery from illness or surgery or in patients too frail to engage in exercise. Additionally, pharmaceuticals that attenuate losses of organ or exercise capacities for those incapable of sufficient of amounts of exercise would be beneficial. The current concept of an ‘exercise pill’ illustrates a lack of knowledge about the totality of adaptations to exercise. In summary, implying that a single ‘exercise pill’ will provide all adaptations to exercise when the ‘exercise pill’ only provides a clinically significant gain in physiological capacity for one specific exercise target (out of the multitude of adaptations from actual exercise) is deemed by us as medical malpractice, because the single target pill would not mimic the overwhelming majority of all healthy adaptations to exercise. The ethically correct terminology is ‘partial exercise pill’ to reflect it will improve the clinical condition of patients unable to exercise but the ‘term also reflects that the partial exercise pill’ will not replace all the health benefits obtained with natural exercise.
Summary
Those using the term ‘exercise pill’ to mimic all adaptations and health benefits to exercise reveal their lack of appreciation for complex integrative physiological responses throughout the body that occur in response exercise, often preventing appropriate design and interpretation in scientific discovery. Our critique of the liabilities of an ‘exercise pill’ adds to a growing number of reviews (Goodyear, 2008; Richter et al. 2008; Warden & Fuchs, 2008; Carey & Kingwell, 2009; Church & Blair, 2009; Hawley & Holloszy, 2009) about the unreality of an ‘exercise pill’ from investigators whose long careers studying adaptations to exercise have given them an adequate appreciation of physical exercise's complexities. Selected quotations from these publications are –‘It is unlikely that a single ‘exercise pill’ will ever supply most of the benefits of regular exercise’ (Goodyear, 2008); ‘Given the complexity of exercise as a stimulus that affects multiple (all?) organs, it would seem somewhat premature to conclude that AMPK and PPARδ agonists are exercise mimetics’ (Richter et al. 2008); ‘It is somewhat audacious to suggest that we can mimic the health benefits of exercise with a pill’ (Church & Blair, 2009); ‘if finding orally active exercise mimetics is really a longstanding medical goal, we believe it will continue to be elusive’ (Hawley & Holloszy, 2009); and ‘no single agent will ever mimic the broad range of exercise-related health benefits’ (Carey & Kingwell, 2009).
Economics
Development costs of a new drug in the USA
The true cost of drug development is unknown. Estimates range from only $110 million per new drug for the average cost of drug development in the 1990s according to Public Citizen (Rx R&D Myths: The Case Against The Drug Industry's R&D ‘Scare Card’ July 2001; http://www.citizen.org/documents/ACFDC.PDF) to $800 million in 2000 (DiMasi et al. 2003) to $1.2 billion in 2006 as the average cost of developing a new biotechnology product according to The Tufts Center for the Study of Drug Development (http://csdd.tufts.edu/NewsEvents/NewsArticle.asp?newsid=69). A very contentious debate as to the true cost of developing one drug is ongoing between various groups (Light, 2007; Dimasi et al. 2008); this debate is beyond the scope of the current review.
Net cost savings
What do pills cost? Will the pharmaceutical industry give free ‘exercise pills’ to everyone? An answer of ‘Yes’ would seem doubtful. It seems likely that everyone will directly or indirectly pay for ‘exercise pills’. Therefore, we propose a direct comparison between cost-effectiveness ratios between natural exercise and ‘exercise pills’. The cost effectiveness ratio is determined by the cost of gain in health from a candidate intervention in the denominator to the cost of obtaining health gain in the numerator. Depending on the originator, what is cost-effective may well be viewed differently by pharmaceutical companies and society-at-large (the reader is referred to Gold et al. 1996 for greater detail). To better understand the potential cost and cost-effectiveness of an ‘exercise pill’, statins will be considered because they are one of the most prescribed drugs in the world, and an extensive literature exists on their cost effectiveness.
Statin cost-effectiveness. We have selected only two of the most recent reports. One recent review has evaluated all literature relating to the clinical and cost effectiveness of statins in the primary prevention of coronary heart disease and cardiovascular disease in the United Kingdom (Franco et al. 2007). One of the review's conclusions was that statin therapy is the most expensive option in primary prevention for those individuals who have a 10-year coronary heart disease risk below 30%; the review indicates that statins should not constitute the first choice of treatment in these populations. A major unanswered concern was the uncertainty that asymptomatic individuals would be compliant for lifelong medication. This concern is particularly poignant for younger patients where the putative long-term health saving through primary prevention would be the greatest. With such a concern, the review calls for significant efforts to be made to ensure that other interventions to reduce coronary heart disease risk (including smoking cessation, exercise and a healthy diet, as well a range of drug treatments, such as antihypertensives, β-blockers and aspirin) which are of equivalent proven efficacy are optimized to lessen the cost to the United Kingdom health care system. Thus, statins are not recommended for primary prevention and rather exercise is recommended as an alternative.
Our purpose of presenting statin pill costs is to demonstrate the type of comparison that would be required to decide the cost–benefit ratio of replacing exercise with a pill. An analysis by Fletcher (2009) uses a cost-effectiveness ratio, as defined under the paragraph What do pills cost? For the next example, the denominator is one-quality-adjusted-life year gained and the numerator is gained at $50 000 cost. If health care costs were to be rationed to $50 000/quality-adjusted life year, then the cost effectiveness ratio of each daily statin pill (numerator) progressively declines as the category of health risk improves (denominator). Examples given in (Pletcher et al. 2009) follow. The cost effectiveness of a stain pill could not exceed $2.22–2.83 per statin pill for those individuals having LDL-cholesterol levels >130 mg dl−1 with a 15% 10-year coronary heart risk to gain one-quality-adjusted-life year; no more than $0.70–1.53 for >130 mg LDL dl−1 with a 5% 10-year coronary heart risk; and no more than $0.68 per pill for individuals >45 years of age having LDL-cholesterol levels >130 mg dl−1 (Pletcher et al. 2009). If cost was only $0.10 or less per statin pill, then everyone with low-density lipoprotein cholesterol levels >130 mg dl−1 could take the pill at a net cost saving to the health system (Pletcher et al. 2009). The analysis of the cost-effectiveness ratio of low-intensity statin pills indicates that ‘exercise pill’ costs would have to be very cheap to be more cost-effective to the health care system than natural physical activity itself. On the other hand, we expect any ‘exercise pill’ that mimicked all responses to exercise would have to be a poly pill and would be quite costly based upon the unhealthy conditions it would have to prevent in Fig. 2. Exercise itself is better primary medicine than any current drug for low-risk groups.
What are the costs of physical inactivity? An extremely wide range of estimations has been made; a US government study will be cited here. Total direct and indirect costs of physical inactivity were estimated by US Center for Disease to be $150 billion dollars per year based upon 1986 disease statistics in the USA and presented in year-2000 dollars (Pratt et al. 2000). Direct costs of physical inactivity were $77 billion in this study. Conservative extrapolation of these costs to 2010 would at least double the total and direct costs of physical inactivity to $300 and $154 billion, respectively. For comparison to disease, the American Heart Association puts the economic costs of cardiovascular disease at 403.1 billion in 2004.
Summary
Our analysis of projected economic costs, in concert with the diseases whose prevalence is increased by the lack of exercise (Fig. 2), if correct, does not justify the cost-effectiveness of replacing natural exercise in those physically capable of exercising for primary prevention of all chronic diseases. If pills only mimicking a small percentage of all health benefits of a physically active lifestyle, but labelled incorrectly as ‘exercise pills’, were to cause individuals to believe they could get all the benefits of exercise without daily exercise, then the following would likely ensue. The prevalence of chronic health disorders shown in Fig. 2 that the ‘exercise pill’ does not prevent would increase, potentially decreasing quality of life, increasing sick days, increasing disabilities, resulting in more chronic illness, and more mental health issues in those who give up a physically active lifestyle. If all US individuals were physically active to the level recommended to the Secretary of Health and Humans Services (240 minutes of moderate to vigorous exercise per week), then a significant portion of the annual $150 billion in direct health care costs as a result of inactivity could be saved.
Criteria for an ‘exercise pill’ or ‘exercise mimetic’ must:
Usage of “mimetic” must be consonant with its definition which is ‘Same or similar’ adaptive effects of exercise training in preventing chronic disorders (Fig. 2) (Oxford English Dictionary, 1989, online).
Not produce unhealthy side effects that are not produced by moderate exercise training itself.
Be cost effective, thus lowering US health care costs in populations unable to exercise.
Decrease mortality rate and have a therapeutic index more favourable than exercise training.
Be ethical.
The entirety of molecular, physiological and epidemiological data on the benefits exercise strongly supports the criteria listed above.
Conclusion
Thomas Kuhn's book The Structure of Scientific Revolutions (Kuhn, 1962) proposes that a crisis exists when an anomaly becomes apparent in an existing paradigm. An anomaly in an existing paradigm for exercise has been perceived by us. The existing paradigm is that the biology of sedentary animals represents normal biology, capable of explaining the evolved functions of proteins. Implicit in this current paradigm is the assumption that differences between low physical activity levels by caged animals and their counterparts in the wild are of minimal importance in both wild-type and transgenic/knockout animals. Examples in both rodents and humans definitively show differences in biology between wild/highly active subjects and those confined to cage-activity/sedentary behaviour. (Cordain et al. 1998; Hayes et al. 2005; Kump & Booth, 2005; & Olsen et al. 2008). Furthermore, as discussed earlier in this review, phenotypes in some transgenic animals with low levels of physical activity do not match functions of the same proteins at higher physical activity levels, Thus, because substantial differences exist in animals undergoing low versus high amounts of physical activity, the current paradigm of what is normal biology and protein function (gene expression) is challenged by us. According to Bennett & Ruben (1979), some genes had to be selected to support high level physical activity to provide a survival advantage. In our proposed new paradigm, habitually states of high physical activity are considered as the appropriate biological control condition reflecting the evolutionarily developed genetic and biological norm. Acceptance of our new paradigm would resolve the crisis by recognizing models with high levels of physical activity are needed to better appreciate and understand normal biology and normal protein function (gene expression).
A second crisis in exercise physiology has occurred. The current paradigm is that no drug can closely mimic all health benefits of habitual natural exercise that combines both aerobic and resistance types. However, a new competing paradigm is presented by Narkar et al. (2008) as ‘exercise in a pill’ (Salk Institute, 2008). We contend their ‘exercise in a pill’ does not have all and health benefits of natural exercise and therefore the proposed new paradigm by Narkar et al. to replace natural physical activity with an ‘exercise in a pill’ is an anomaly without merit. Indeed the complexity of exercise adaptation is highlighted by numerous studies whereby removal of putative master regulators of exercise adaptation did not prevent exercise adaptation. This has led other experts (Goodyear, 2008; Richter et al., 2008; Warden & Fuchs, 2008; Carey & Kingwell, 2009; Church & Blair, 2009; Hawley & Holloszy, 2009) and us studying the adaptations to exercise to contend that all health benefits of exercise are too complex to be replaced by a single pill.
Kuhn indicates that all crises close in one of three ways – (1) normal science handles the crisis-provoking problem; (2) the problem resists even apparently radical new approaches; and (3) the crisis may end with a new candidate for paradigm and with the ensuing battle over its acceptance. Only biologists with an adequate appreciation of physical exercise's complexities and its role in shaping our current genome can resolve the two anomalies discussed in this review, as only they can provide appropriate design and interpretation in scientific discovery involving physical activity/inactivity research.
Acknowledgments
We thank Espen Spangenburg, Eva Chin and Grant Simmons for reading parts or all of manuscript and offering important suggestions; Ronald Terjung for offering the title. Grant was written while authors were supported by University of Missouri Research Board and NIH AG18780 (F.W.B.) and Danish National Research Foundation (M.J.L.). We thank Donald Connor and Howard Wilson for assistance in labelling Fig. 2.
Author contributions
Both authors contributed equally to this review and approved the final version.
References
- Bennett AF, Ruben JA. Endothermy and activity in vertebrates. Science. 1979;206:649–654. doi: 10.1126/science.493968. [DOI] [PubMed] [Google Scholar]
- Bernstein D. Exercise assessment of transgenic models of human cardiovascular disease. Physiol Genomics. 2003;13:217–226. doi: 10.1152/physiolgenomics.00188.2002. [DOI] [PubMed] [Google Scholar]
- Booth FW, Baldwin KM. In: Handbook of Physiology: section 12, Exercise: Regulation and Integration of Multiple Systems. Rowell LB, Shepherd JT, editors. New York: Oxford University Press; 1996. pp. 1075–1123. Muscle plasticity; energy demand and supply. In. [Google Scholar]
- Booth FW, Chakravarthy MV, Gordon SE, Spangenburg EE. Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. J Appl Physiol. 2002;93:3–30. doi: 10.1152/japplphysiol.00073.2002. [DOI] [PubMed] [Google Scholar]
- Booth FW, Gordon SE, Carlson CJ, Hamilton MT. Waging war on modern chronic diseases: primary prevention through exercise biology. J Appl Physiol. 2000;88:774–787. doi: 10.1152/jappl.2000.88.2.774. [DOI] [PubMed] [Google Scholar]
- Booth FW, Laye MJ, Spangenburg EE. Gold standards for scientists who are conducting animal-based exercise studies. J Appl Physiol. 2009 doi: 10.1152/japplphysiol.00125.2009. in press. [DOI] [PubMed] [Google Scholar]
- Booth FW, Lees SJ. Fundamental questions about genes, inactivity, and chronic diseases. Physiol Genomics. 2007;28:146–157. doi: 10.1152/physiolgenomics.00174.2006. [DOI] [PubMed] [Google Scholar]
- Carey AL, Kingwell BA. Novel pharmacological approaches to combat obesity and insulin resistance: targeting skeletal muscle with ‘exercise mimetics’. Diabetologia. 2009;52:2015–2026. doi: 10.1007/s00125-009-1420-x. [DOI] [PubMed] [Google Scholar]
- Cesari M, Pahor M, Lauretani F, Zamboni V, Bandinelli S, Bernabei R, Guralnik JM, Ferrucci L. Skeletal muscle and mortality results from the InCHIANTI Study. J Gerontol A Biol Sci Med Sci. 2009;64:377–384. doi: 10.1093/gerona/gln031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy MV, Booth FW. Eating, exercise, and ‘thrifty’ genotypes: connecting the dots toward an evolutionary understanding of modern chronic diseases. J Appl Physiol. 2004;96:3–10. doi: 10.1152/japplphysiol.00757.2003. [DOI] [PubMed] [Google Scholar]
- Choi S, Liu X, Li P, Akimoto T, Lee SY, Zhang M, Yan Z. Transcriptional profiling in mouse skeletal muscle following a single bout of voluntary running: evidence of increased cell proliferation. J Appl Physiol. 2005;99:2406–2415. doi: 10.1152/japplphysiol.00545.2005. [DOI] [PubMed] [Google Scholar]
- Church TS, Blair SN. When will we treat physical activity as a legitimate medical therapy … even though it does not come in a pill? Br J Sports Med. 2009;43:80–81. doi: 10.1136/bjsm.2008.053850. [DOI] [PubMed] [Google Scholar]
- Colombo M, Gregersen S, Kruhoeffer M, Agger A, Xiao J, Jeppesen PB, Orntoft T, Ploug T, Galbo H, Hermansen K. Prevention of hyperglycemia in Zucker diabetic fatty rats by exercise training: effects on gene expression in insulin-sensitive tissues determined by high-density oligonucleotide microarray analysis. Metabolism. 2005;54:1571–1581. doi: 10.1016/j.metabol.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Cordain L, Gotshall RW, Eaton SB, Eaton SB., 3rd Physical activity, energy expenditure and fitness: an evolutionary perspective. Int J Sports Med. 1998;19:328–335. doi: 10.1055/s-2007-971926. [DOI] [PubMed] [Google Scholar]
- Deshmukh AS, Treebak JT, Long YC, Viollet B, Wojtaszewski JF, Zierath JR. Role of adenosine 5′-monophosphate-activated protein kinase subunits in skeletal muscle mammalian target of rapamycin signalling. Mol Endocrinol. 2008;22:1105–1112. doi: 10.1210/me.2007-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22:151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- Dimasi JA, Hansen RW, Grabowski HG. Misleading congress about drug development: Reply. J Health Polit Policy Law. 2008;33:319–324. doi: 10.1215/03616878-2007-063. discussion 325–317. [DOI] [PubMed] [Google Scholar]
- Fernandez C, Hansson O, Nevsten P, Holm C, Klint C. Hormone-sensitive lipase is necessary for normal mobilization of lipids during submaximal exercise. Am J Physiol Endocrinol Metab. 2008;295:E179–186. doi: 10.1152/ajpendo.00282.2007. [DOI] [PubMed] [Google Scholar]
- Franco OH, der Kinderen AJ, De Laet C, Peeters A, Bonneux L. Primary prevention of cardiovascular disease: cost-effectiveness comparison. Int J Technol Assess Health Care. 2007;23:71–79. doi: 10.1017/S0266462307051598. [DOI] [PubMed] [Google Scholar]
- Glund S, Treebak JT, Long YC, Barres R, Viollet B, Wojtaszewski JF, Zierath JR. Role of adenosine 5′-monophosphate-activated protein kinase in interleukin-6 release from isolated mouse skeletal muscle. Endocrinology. 2009;150:600–606. doi: 10.1210/en.2008-1204. [DOI] [PubMed] [Google Scholar]
- Gold MR, Siegel JE, Russell LB, Weinstein MC, editors. Cost-effectiveness in Health and Medicine. New York: Oxford University Press; 1996. [Google Scholar]
- Goodyear LJ. The exercise pill – too good to be true? N Engl J Med. 2008;359:1842–1844. doi: 10.1056/NEJMcibr0806723. [DOI] [PubMed] [Google Scholar]
- Goodyear LJ, Chang PY, Sherwood DJ, Dufresne SD, Moller DE. Effects of exercise and insulin on mitogen-activated protein kinase signalling pathways in rat skeletal muscle. Am J Physiol Endocrinol Metab. 1996;271:E403–408. doi: 10.1152/ajpendo.1996.271.2.E403. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C, Schaub JW, Andreasen A, Haskell KR, Moore MC, Koerper LM, Rouzaud F, Baker HV, Millard WJ, Walter G, Litherland SA, Xiang Z. Voluntary exercise prevents the obese and diabetic metabolic syndrome of the melanocortin-4 receptor knockout mouse. FASEB J. 2009;23:642–655. doi: 10.1096/fj.08-109686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskell-Luevano C, Todorovic A, Gridley K, Sorenson N, Irani B, Xiang Z. The melanocortin pathway: effects of voluntary exercise on the melanocortin-4 receptor knockout mice and ACTH(1–24) ligand structure activity relationships at the melanocortin-2 receptor. Endocr Res. 2004;30:591–597. doi: 10.1081/erc-200043759. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Holloszy JO. Exercise: it's the real thing! Nutr Rev. 2009;67:172–178. doi: 10.1111/j.1753-4887.2009.00185.x. [DOI] [PubMed] [Google Scholar]
- Hayes M, Chustek M, Heshka S, Wang Z, Pietrobelli A, Heymsfield SB. Low physical activity levels of modern Homo sapiens among free-ranging mammals. Int J Obes. 2005;29:151–156. doi: 10.1038/sj.ijo.0802842. [DOI] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol. 1976;38:273–291. doi: 10.1146/annurev.ph.38.030176.001421. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Jeppesen J, Albers P, Luiken JJ, Glatz JF, Kiens B. Contractions but not AICAR increase FABPpm content in rat muscle sarcolemma. Mol Cell Biochem. 2009;326:45–53. doi: 10.1007/s11010-008-0006-0. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, Pilegaard H. Effects of α-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005;19:1146–1148. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- Katzmarzyk PT, Janssen I. The economic costs associated with physical inactivity and obesity in Canada: an update. Can J Appl Physiol. 2004;29:90–115. doi: 10.1139/h04-008. [DOI] [PubMed] [Google Scholar]
- Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, Shoelson SE, Shulman GI. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108:437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer HF, Goodyear LJ. Exercise, MAPK, and NF-κB signalling in skeletal muscle. J Appl Physiol. 2007;103:388–395. doi: 10.1152/japplphysiol.00085.2007. [DOI] [PubMed] [Google Scholar]
- Kuhn TS. The Structure of Scientific Revolutions. Chicago: University of Chicago Press; 1962. [Google Scholar]
- Kump DS, Booth FW. Alterations in insulin receptor signalling in the rat epitrochlearis muscle upon cessation of voluntary exercise. J Physiol. 2005;562:829–838. doi: 10.1113/jphysiol.2004.073593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leick L, Wojtaszewski JF, Johansen ST, Kiilerich K, Comes G, Hellsten Y, Hidalgo J, Pilegaard H. PGC-1α is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E463–474. doi: 10.1152/ajpendo.00666.2007. [DOI] [PubMed] [Google Scholar]
- Light DW. Misleading congress about drug development. J Health Politics Policy Law. 2007;32:895–913. doi: 10.1215/03616878-2007-063. [DOI] [PubMed] [Google Scholar]
- Mathur N, Pedersen BK. Exercise as a mean to control low-grade systemic inflammation. Mediators Inflamm. 2008;2008:109502. doi: 10.1155/2008/109502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConell GK, Manimmanakorn A, Lee-Young RS, Kemp BE, Linden KC, Wadley GD. Differential attenuation of AMPK activation during acute exercise following exercise training or AICAR treatment. J Appl Physiol. 2008;105:1422–1427. doi: 10.1152/japplphysiol.01371.2007. [DOI] [PubMed] [Google Scholar]
- Metter EJ, Talbot LA, Schrager M, Conwit R. Skeletal muscle strength as a predictor of all-cause mortality in healthy men. J Gerontol A Biol Sci Med Sci. 2002;57:B359–365. doi: 10.1093/gerona/57.10.b359. [DOI] [PubMed] [Google Scholar]
- Mittleman MA, Maclure M, Tofler GH, Sherwood JB, Goldberg RJ, Muller JE. Triggering of acute myocardial infarction by heavy physical exertion. Protection against triggering by regular exertion. Determinants of Myocardial Infarction Onset Study Investigators. N Engl J Med. 1993;329:1677–1683. doi: 10.1056/NEJM199312023292301. [DOI] [PubMed] [Google Scholar]
- Morris RT, Laye MJ, Lees SJ, Rector RS, Thyfault JP, Booth FW. Exercise-induced attenuation of obesity, hyperinsulinemia, and skeletal muscle lipid peroxidation in the OLETF rat. J Appl Physiol. 2008;104:708–715. doi: 10.1152/japplphysiol.01034.2007. [DOI] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM. AMPK and PPARδ agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AB, Kupelian V, Visser M, Simonsick EM, Goodpaster BH, Kritchevsky SB, Tylavsky FA, Rubin SM, Harris TB. Strength, but not muscle mass, is associated with mortality in the health, aging and body composition study cohort. J Gerontol A Biol Sci Med Sci. 2006;61:72–77. doi: 10.1093/gerona/61.1.72. [DOI] [PubMed] [Google Scholar]
- Olsen RH, Krogh-Madsen R, Thomsen C, Booth FW, Pedersen BK. Metabolic responses to reduced daily steps in healthy nonexercising men. JAMA. 2008;299:1261–1263. doi: 10.1001/jama.299.11.1259. [DOI] [PubMed] [Google Scholar]
- Ort T, Gerwien R, Lindborg KA, Diehl CJ, Lemieux AM, Eisen A, Henriksen EJ. Alterations in soleus muscle gene expression associated with a metabolic endpoint following exercise training by lean and obese Zucker rats. Physiol Genomics. 2007;29:302–311. doi: 10.1152/physiolgenomics.00257.2006. [DOI] [PubMed] [Google Scholar]
- Ostrowski K, Rohde T, Zacho M, Asp S, Pedersen BK. Evidence that interleukin-6 is produced in human skeletal muscle during prolonged running. J Physiol. 1998;508:949–953. doi: 10.1111/j.1469-7793.1998.949bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison JS, Folk LC, Madsen RW, Childs TE, Spangenburg EE, Booth FW. Expression profiling identifies dysregulation of myosin heavy chains IIb and IIx during limb immobilization in the soleus muscles of old rats. J Physiol. 2003;553:357–368. doi: 10.1113/jphysiol.2003.047233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Physical Activity Guidelines Advisory Committee. Physical Activity Guidelines Advisory Committee Report. Washington: US Department of Health and Human Services; 2008. [DOI] [PubMed] [Google Scholar]
- Pletcher MJ, Lazar L, Bibbins-Domingo K, Moran A, Rodondi N, Coxson P, Lightwood J, Williams L, Goldman L. Comparing impact and cost-effectiveness of primary prevention strategies for lipid-lowering. Ann Intern Med. 2009;150:243–254. doi: 10.7326/0003-4819-150-4-200902170-00005. [DOI] [PubMed] [Google Scholar]
- Pratt M, Macera CA, Wang G. Higher direct medical costs associated with physical inactivity. Phys Sports Med. 2000;28:63–70. doi: 10.3810/psm.2000.10.1237. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Radom-Aizik S, Zaldivar F, Jr, Leu SY, Cooper DM. A brief bout of exercise alters gene expression and distinct gene pathways in peripheral blood mononuclear cells of early- and late-pubertal females. J Appl Physiol. 2009;107:168–175. doi: 10.1152/japplphysiol.00121.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radom-Aizik S, Zaldivar F, Jr, Leu SY, Galassetti P, Cooper DM. Effects of 30 min of aerobic exercise on gene expression in human neutrophils. J Appl Physiol. 2008;104:236–243. doi: 10.1152/japplphysiol.00872.2007. [DOI] [PubMed] [Google Scholar]
- Rantzau C, Christopher M, Alford FP. Contrasting effects of exercise, AICAR, and increased fatty acid supply on in vivo and skeletal muscle glucose metabolism. J Appl Physiol. 2008;104:363–370. doi: 10.1152/japplphysiol.00500.2007. [DOI] [PubMed] [Google Scholar]
- Rector RS, Rogers R, Ruebel M, Hinton PS. Participation in road cycling vs. running is associated with lower bone mineral density in men. Metabolism. 2008a;57:226–232. doi: 10.1016/j.metabol.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Rector RS, Thyfault JP, Morris RT, Laye MJ, Borengasser SJ, Booth FW, Ibdah JA. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol. 2008b;294:G619–626. doi: 10.1152/ajpgi.00428.2007. [DOI] [PubMed] [Google Scholar]
- Richter EA, Kiens B, Wojtaszewski JF. Can exercise mimetics substitute for exercise? Cell Metab. 2008;8:96–98. doi: 10.1016/j.cmet.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Salk Institute for Biological Studies. pp. 26–27. Scientific Report http://www.salk.edu/pdf/scientific_report/scientific_report08.pdf.
- Sjoqvist F, Garle M, Rane A. Use of doping agents, particularly anabolic steroids, in sports and society. Lancet. 2008;371:1872–1882. doi: 10.1016/S0140-6736(08)60801-6. [DOI] [PubMed] [Google Scholar]
- Stepto NK, Coffey VG, Carey AL, Ponnampalam AP, Canny BJ, Powell D, Hawley JA. Global gene expression in skeletal muscle from well-trained strength and endurance athletes. Med Sci Sports Exerc. 2009;41:546–565. doi: 10.1249/MSS.0b013e31818c6be9. [DOI] [PubMed] [Google Scholar]
- Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, Galbo H, Haunso S, Sheikh SP. Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. FEBS J. 2005;272:2684–2695. doi: 10.1111/j.1742-4658.2005.04684.x. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Franklin BA, Balady GJ, Blair SN, Corrado D, Estes NA, 3rd, Fulton JE, Gordon NF, Haskell WL, Link MS, Maron BJ, Mittleman MA, Pelliccia A, Wenger NK, Willich SN, Costa F. Exercise and acute cardiovascular events placing the risks into perspective: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism and the Council on Clinical Cardiology. Circulation. 2007;115:2358–2368. doi: 10.1161/CIRCULATIONAHA.107.181485. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Funk EJ, Carleton RA, Sturner WQ. Incidence of death during jogging in Rhode Island from 1975 through 1980. JAMA. 1982;247:2535–2538. [PubMed] [Google Scholar]
- Van Camp SP, Bloor CM, Mueller FO, Cantu RC, Olson HG. Nontraumatic sports death in high school and college athletes. Med Sci Sports Exerc. 1995;27:641–647. [PubMed] [Google Scholar]
- Warden SJ, Fuchs RK. Are ‘exercise pills’ the answer to the growing problem of physical inactivity? Br J Sports Med. 2008;42:562–563. doi: 10.1136/bjsm.2008.053512. [DOI] [PubMed] [Google Scholar]
- Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams RS. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- Yang Q, Osinska H, Klevitsky R, Robbins J. Phenotypic deficits in mice expressing a myosin binding protein C lacking the titin and myosin binding domains. J Mol Cell Cardiol. 2001;33:1649–1658. doi: 10.1006/jmcc.2001.1417. [DOI] [PubMed] [Google Scholar]