

Abstract
Skeletal muscles produce transient reactive oxygen species (ROS) in response to intense stimulation, disuse atrophy, heat stress, hypoxia, osmotic stress, stretch and cell receptor activation. The physiological significance is not well understood. Protein phosphatases (PPases) are known to be highly sensitive to oxidants and could contribute to many different signalling responses in muscle. We tested whether broad categories of PPases are inhibited by levels of acute oxidant exposure that do not result in loss of contractile function or gross oxidative stress. We also tested if this exposure results in elevated levels of global protein phosphorylation. Rat diaphragm muscles were treated with either 2,3-dimethoxy-1-naphthoquinone (DMNQ; 1, 10, 100 μm; a mitochondrial O2•−/H2O2 generator) or exogenous H2O2 (5, 50, 500 μm) for 30 min. Supernatants were assayed for serine/threonine PPase (Ser/Thr-PPase) or protein tyrosine PPase (PTP) activities. With the exception of 500 μm H2O2, no other oxidant exposures significantly elevated protein carbonyl formation, nor did they alter the magnitude of twitch force. DMNQ significantly decreased all categories of PPase activity at 10 and 100 μm and reduced PTP at 1 μm. Similar reductions in Ser/Thr-PPase activity were seen in response to 50 and 500 μm H2O2 and PTP at 500 μm H2O2. ROS treatments resulted a dose-dependent increase in the phosphorylation states of many proteins. The data are consistent with the concept that PPases, within intact skeletal muscles, are highly sensitive to acute changes in ROS activity and that localized ROS play a critical role in lowering the barriers for effective phosphorylation events to occur in muscle cells, thus increasing the probability for cell signalling responses to proceed.
Introduction
Increased oxidant production in skeletal muscle and other tissues has been demonstrated in response to a variety of widely divergent stress stimuli, including, intense contractile activity (Reid et al. 1992; Diaz et al. 1993), heat stress (Zuo et al. 2000), short term disuse atrophy (Falk et al. 2006), acute hypoxia (Zuo & Clanton, 2005), acute osmotic stress (Martins et al. 2008) and stretch (Cheng et al. 1995). In addition, skeletal muscle and other non-inflammatory cells are known to produce waves of local reactive oxygen in response to cell surface receptor activation via cytokines, hormones or growth factors (Goosens et al. 1995; Thannickal & Fanburg, 2000; Espinosa et al. 2009) and some non-inflammatory cells produce transient ROS in response to nuclear receptor activation (Felty et al. 2005; Zhang et al. 2007). Collectively, these observations lead to the question, why are oxidants produced so ubiquitously during such widely divergent stimuli and do they have a functional role? Obviously oxidants can influence numerous cell signalling pathways (Thannickal & Fanburg, 2000), but their lack of chemical specificity makes it difficult to conceive of how they might contribute to the responses from such divergent environmental signals. In this study we explored the possibility that oxidants can function to regulate in vivo global ‘phosphatase tone’ and thus influence the kinetics and amplification of many kinase signalling pathways. This general idea has been discussed by others, particularly in reference to receptor–ligand signalling and tyrosine phosphatase activity (Xu et al. 2002; Meng et al. 2004; Tonks, 2005; Salmeen & Barford, 2005; Chiarugi, 2005; Maher, 2006) but it has not been tested or framed in the context of normal physiological responses, particularly in skeletal muscle.
Protein phosphatases (PPase) are one of the most abundant proteins encoded by the mammalian genome (Almo et al. 2007). There are two broad families, the protein tyrosine PPases (PTPs) comprising about 112 human proteins, and the serine/threonine PPases (Ser/Thr PPase), making up about 31 proteins. Within the PTP family there are several classes; most belong to a class that is specific to tyrosine targets but there is also a dual-specific class of phosphatases (33 proteins) that interact with both serine/threonine and tyrosine phosphorylation sites. The second broad family, the Ser/Thr PPases, are also divided into two classes, the PPP family which use a Zn2+/Fe2+ complex at the catalytic site (15 proteins) and the PPM family, which are Mn2+/Mg2+ dependent (16 proteins). There is essentially no sequence homology between these two Ser/Thr PPase subclasses.
The redox sensitivity of the PTPs, has been well described in vitro and in cell culture systems since the early 1990s (Heffetz et al. 1990; Hecht & Zick, 1992). Subsequent work with a variety of PTP species has clearly established that this sensitivity has much potential biological importance (e.g. Lee et al. 1998; Takakura et al. 1999; Meng et al. 2002; Pyner et al. 2003; Salsman et al. 2005). In contrast, the redox sensitivity of Ser/Thr PPases is not well described, with the exception of calcineurin (Namgaladze et al. 2002; Namgaladze et al. 2005; Agbas et al. 2007; Lee et al. 2007). However, understanding whether Ser/Thr PPases are redox sensitive in a physiological context is important, since it has been estimated that 98% of all protein phosphorylation events occur on Ser/Thr residues in eukaryotic cells (Honkanen & Golden, 2002).
Other previous investigators have evaluated the effects of exogenous or cell generated reactive oxygen species (ROS) on PPase activity, using in vitro protein and/or cell culture preparations (e.g. Caselli et al. 1998; Denu & Tanner, 1998; Lee et al. 1998; Takakura et al. 1999; Meng et al. 2002; Namgaladze et al. 2002; Pyner et al. 2003; Pieri et al. 2003; Salmeen et al. 2003; Meng et al. 2004; Salmeen & Barford, 2005; Salsman et al. 2005; Namgaladze et al. 2005; Agbas et al. 2007; Chiarugi & Buricchi, 2007; Lee et al. 2007). However, to our knowledge, the influences of oxidants on PPases in intact tissues or animals and particularly in skeletal muscles have not been studied. Therefore, the relevance of this potential redox sensitivity to living, intact muscle is not known. It is difficult to extrapolate experiments on in vitro protein biochemistry or in cell culture to understand living muscle. First, it is reasonable to predict that fresh tissue, taken quickly from an animal, has a nearly intact antioxidant and SH-reducing network that could differ from cell culture. Second, the relative proportion and expression of specific phosphatases and kinases in fresh skeletal muscle could differ markedly from cultured cells, which show little similarity to adult muscle cells. Third, for phosphatase inhibition to be physiologically relevant in skeletal muscle, it should be sensitive to oxidants in a low enough range of concentrations that that there is no significant cell damage and no significant diminution in contractile function. Fourth, although we can predict, based on biochemical experiments, that some specific PPases are undoubtedly inhibited by oxidants in skeletal muscle, it is not clear the extent to which this causes an overall change in PPase activities. If only a few of the multitude of phosphatases are actually affected by physiologically relevant oxidants in intact tissue, it would impact our understanding of how generalized the influence of oxidants might be within a variety of signalling systems. Fifth, we do not know if redox modulation of ‘phosphatase tone’ translates into a measurable effect on overall protein phosphorylation.
This study was designed to test the hypothesis that short duration exposure to oxidants, at levels that appear to be physiologically relevant and below the threshold of clearly evident oxidative stress can have an overall global impact on the activity of phosphatases in living muscle. The functional role of oxidants could then be, in part, assigned to influencing the kinetics of broad categories of intracellular signalling events, thus consisting of a pathway for cross-talk between cell redox state and phosphorylation events. Secondly, we tested the hypothesis that oxidant exposure potentially has similar effects on Ser/Thr PPase activity when compared to the well known effects on PTP activity. Third, we tested whether exogenously applied H2O2 would have substantially different effects on PPase signalling compared to an internal redox cycling ROS generating system, 2,3 dimethoxy-1,3-naphoquinone (DMNQ), known to be a O2•− and H2O2 generator in the mitochondria (Tchivilev et al. 2008). Fourth, we tested whether oxidant exposure would result in a dose-dependent increase in overall protein phosphorylation, a finding that would be consistent with a role of oxidants in reducing overall ‘phosphatase tone’.
Methods
Ethical approval
All animal housing and procedures were performed at Ohio State University and were approved by the Ohio State Institutional Animal Care and Use Committee. All ethical guidelines established by The Journal of Physiology were adhered to (Drummond, 2009).
Tissue preparation
A total of 35 adult male Sprague–Dawley rats (294–470 g) were anaesthetized with intraperitoneal ketamine (100 mg kg−1) and xylazine (20 mg kg−1), tracheotomized and mechanically ventilated. After absence of corneal and withdrawal reflexes, the chest was opened and the intact diaphragm was excised and immediately placed in oxygenated (95% O2–5% CO2) Ringer solution (in mm: 21 NaHCO3, 1.0 MgCl2, 0.6 Na2HPO4, 0.45 Na2SO4, 2.0 CaCl2, 5.9 KCl, 121 NaCl; 11.5 glucose, and 10 μm d-tubocurarine). The animals were then killed by removing the heart, under anaesthesia. Four ∼5–8 mm wide diaphragm strips were cut from the costal diaphragm with ribs and tendon attached. The strips were mounted vertically in temperature-controlled tissue baths with oxygenated Ringer solution at 37°C. Muscle length was set to produce a maximum active peak twitch force (∼1–3 g preload) and field stimulation current was adjusted to exceed by >20% the current required for maximal twitch force. The tissues were allowed to equilibrate for 30 min. The buffer was then changed to fresh, preheated and pre-oxygenated Ringer solution. Throughout all protocols, the tissues were stimulated at 1 twitch min−1, a frequency that does not cause any run-down of the muscle and allows continuous monitoring of twitch contractile force. Tetanic stimulation was not used during the experiment to avoid any effects of intense stimulation on phosphorylation signalling.
In one group of experiments, sufficient H2O2 was added directly to the bath to produce initial concentrations of 5 μm, 50 μm or 500 μm. In a second set of experiments, the same procedures were employed except that the tissues were exposed to 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), prepared as a stock solution dissolved in DMSO, then diluted to 1, 10 or 100 μm in the tissue baths, based on the methods of Liu et al. (1998). The control bath contained 3 mm DMSO vehicle, a concentration that does not greatly affect contractile function (Reid & Moody, 1994). DMNQ is reduced to hydroquinone and semiquinone radicals by cellular oxidoreductases, which then undergo autoxidation to form intracellular O2•− and H2O2 (Liu et al. 1998). DMNQ is believed to primarily create ROS in the mitochondria (Tchivilev et al. 2008).
Analysis of phosphatase activities
All tissues were incubated in their respective oxidant-generating solutions for 30 min. They were then rapidly removed, carefully trimmed of non-diaphragm tissue and placed in ice cold Tris buffer (50 mm, pH 7.0) with an antiprotease cocktail containing 1 μm pepstatin, 2 μm leupeptin and 100 μm phenylmethanesulphonylfluoride. The tissues were never frozen and all homogenization and enzymatic assays were completed within 4 h of the in vitro exposure of the muscle. During all times (except during the assay measurement) the tissue or its supernatant were kept on ice. Assays for both Ser/Thr PPase and PTP at each concentration of a given category of ROS exposure were always run on the same tissues from the same animals, i.e. one animal provided the control and the three additional exposures to a given oxidant. Tissue homogenates were prepared by placing the ice-cold tissue in an ice-cold mortar with Tris/anti-protease cocktail (1: 1 w/v) and ground to a fine paste that included small pieces of connective tissue. Homogenates were then transferred to a 2.0 ml microcentrifuge tube, allowed to sit on ice for >30 min, and then centrifuged at 12 000 g for 15 min at 4°C. The supernatant was decanted into a new tube and the pellet discarded. Total protein content of each sample was determined using a modified Lowry method, as described by Peterson (1983) with bovine serum albumin as the standard.
Assays for PTP and Ser/Thr PPase activities were performed using the Invitrogen EnzChek Phosphatase Assay Kits (R-22067 Tyrosine phosphatases and R-33700 Ser/Thr phosphatases, respectively). These assays use the probe, 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) as a substrate to mimic phosphorylated proteins (Pastula et al. 2003; Welte et al. 2005; Montalibet et al. 2005). DiFMUP does not fluoresce in its phosphorylated state, but upon dephosphorylation, becomes highly fluorescent with excitation/emission maxima of ∼358/455 nm. Quantification is possible because 1 pmol of DiFMUP is equivalent to release of 1 pmol of phosphate. Linearity of each assay as a function of PPase protein concentration was tested using standards of commercially available potato phosphatases. Calibration curves of fluorescence, as a function of [DifMUP], were run for each assay type to calculate absolute levels of dephosphorylation rates. The PTP and Ser/Thr PPase assay kits contained specific inhibitors and buffer components that block the non-targeted categories of phosphatase in a specific manner. Although considered proprietary by the manufacturer (by response to direct communication), the specific blockers used and the specificity of the assays were tested and previously reported over a spectrum of widely different phosphatase species (Pastula et al. 2003). Sample protein supernatants were diluted with reaction buffer in two ways. One set was diluted in Tris/anti-protease cocktail alone and another matched sample was diluted in the same buffer but with 500 μm freshly dissolved dithiothreitol (DTT), to yield a final assay concentration of 500 μg protein ml−1. Matched samples in the presence or absence of DTT were always tested concurrently to identify the reversibility of sulfhydryl (−SH) oxidations on the proteins. Duplicate 50 μl aliquots of the diluted samples were added to microplate wells and the fluorescence measured using a fluorescence microplate reader (Spectramax Gemini XS, Molecular Devices or CytoFluor II, PerSeptive Biosystems) at 25°C, over 10 min, which is a highly linear portion of the kinetics curve for all samples. Readings were taken every 15 s with the slope of the fluorescence curve obtained by linear regression and used to determine absolute activity of each PPase category.
Measurements of protein carbonyl formation
Protein oxidation (i.e. carbonyl formation) was determined in muscle samples which were rapidly frozen after 30 min of exposure to oxidants, using 2,4-dinitrophenylhydrazine (OxyBlot Protein Oxidation Detection Kit, Chemicon International/Millipore). Carbonyl groups were derivitized to 2,4-dinitrophenylhydrazone after reaction with 2,4-dinitrophenylhydrazine. Detection and analysis was accomplished by SDS-PAGE, followed by Western blotting with the primary antibody specific to the dinitrophenyl. Stored protein samples were thawed on ice and two aliquots of each supernatant were diluted to 10 μg in 5 μl with water and denatured with 6% SDS. Derivatization solution was added to one sample, while derivatization-free solution was added to the other to provide a negative control. Both were incubated at room temperature for 15–30 min. Neutralization solution was added to both the treated sample and the negative control. Samples were then used immediately or stored for up to 3 days at 4°C, run on 10% Tris-HCl (Bio-Rad) gels and transferred to polyvinylidene fluoride transfer membranes. After incubating with primary (rabbit anti-DNP) and secondary (goat anti-rabbit IgG, horseradish peroxidase-conjugated) antibodies, the oxidized protein bands were visualized with enhanced chemiluminesence Western blotting detection reagents (Amersham) and the film was exposed. All scanned films were analysed using ImageJ software. The lanes in a given gel were scanned with identical areas between molecular weight markers of 20–150 kDa. A total of five distinct bands could be quantified in all gels and were used for further analysis. The densitometry area under each peak for each band was recorded for statistical analysis.
Measurements of H2O2 concentration
Concentrations of H2O2 in the baths, with and without tissues, were determined using 10-acetyl-3,7-dihydroxyphenoxazine (Amplex Red; Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit, Invitrogen). Amplex Red in the presence of horseradish peroxidase (HRP) reacts with H2O2 stoichiometrically 1: 1 to produce resorufin, which has a fluorescence maximum of ∼585 nm (Zhou et al. 1997). Tissue baths were maintained at 37°C, equilibrated with 95% O2–5% CO2, and loaded with H2O2 (initial concentration: 0, 5, 50, 500 μm). Buffer samples were withdrawn from tissue baths, with or without tissue, at various time points (0, 10, 20, 30 min) and placed in tightly caped vials. In separate experiments with tissue, the tissues were equilibrated and twitch stimulated 1 min−1 during exposure to H2O2 to simulate the same protocol used for PPase assays. H2O2 standards were prepared from a known stock provided with the assay kit and samples were diluted to fall within this range. Standards, blank and samples were added to microplate wells in duplicate. Amplex Red/HRP was added to each well at room temperature, the plate incubated for 30 minutes and end-point fluorescence was measured (ex 544/em 590). A standard curve was generated and sample concentrations were calculated based on linear regression of the standards.
Measurements of protein phosphorylation
Polyacrylamide gels were selectively stained for phosphoproteins using Pro-Q Diamond Phosphoprotein Gel Stain (Invitrogen) and then stained for total protein with SYPRO Ruby Protein Gel Stain (Invitrogen) to determine the total amount of phosphorylation caused by exposure to oxidants. Staining was done according to manufacturer's instructions and once the Pro-Q Diamond stain was added, everything was performed at room temperature and in reduced light. Samples were diluted to 10 μg with sample buffer containing urea and run on 12% acrylamide gels that were then fixed overnight in 50% methanol–10% acetic acid at room temperature. After three washes with water, gels were incubated with Pro-Q Diamond gel stain for 60 min, then destained using gel destaining solution (Invitrogen) for a total of 90 min and washed for an additional 10 min. Gels were imaged using excitation/emission 532/605 with a Molecular Imager PharosFX system (Bio-Rad) and Quantity One software. After imaging, gels were stained with SYPRO Ruby gel stain overnight, washed in 10% methanol + 7% acetic acid for 30 min, then rinsed with water for 10 min, and imaged again with the PharosFX system, which was set up for SYPRO Ruby (excitation/emission 532/605). The H2O2 and DMNQ exposed tissues were run on separate gels, each composed of 20 lanes of five experiments each. Therefore, all tissues from the H2O2 experiments were run together on the same gel, as were the DMNQ experiments.
Each lane of the ProQ-treated gels was scanned for phosphorylated proteins using a fixed width for all bands that spanned the entire range of proteins detected on the gel. Density profiles were then generated representing the average density across each lane using Scion Image. The raw data were transferred to a spreadsheet and twenty dominant peaks were identified that could be detected across all lanes. Changes in the profile during oxidant exposure were then determined for each sample against its matched control from the same experiment. For Sypro Ruby, total protein detection of the same gels, a fixed length and width scanning region was also used for each lane to detect the relative protein density of that particular lane. This was used to normalize the corresponding ProQ density bands to the relative amounts of total protein identified in the specific lane.
Statistical analysis
All data are expressed as means ± standard error of the mean. For the effects of oxidant exposure, multiway repeated ANOVAs were performed with dose of oxidant and DTT treatment being the factors of interest. Post-analysis was performed by comparing individual means using least squares contrast comparisons (SAS JMP® software). For comparisons of control PPase activities, a multiway ANOVA was done with PPase group, DTT treatment and animal (random) being the factors of interest. For analysis of carbonyl formation and protein phosphorylation events, multiway ANOVA was performed with oxidant treatment, position of the band on the gel, and animal (random variable) being the factors of interest. Crossed effects between treatment and specific bands were included in the model. As before, least squares mean contrasts were performed to distinguish individual effects between treatment means or, for example, to detect differences in individual bands of the phosphorylation gels from the matched control. All other comparisons (e.g. muscle twitch force values) used one-way ANOVA with post hoc comparisons against controls using Dunnett's test.
Results
Comparison of relative baseline PPase activities in control tissues
The relative rates of absolute Ser/Thr PPase and PTP activities were compared between control tissues that were placed in the oxygenated tissue baths but were not exposed to oxidants (Fig. 1). Absolute PTP activity was significantly higher than Ser/Thr PPase activity in all samples. The supernatants from DMNQ-control experiments for PTP were observed to have much higher activities than were supernatants from H2O2 controls. The only experimental difference between these groups was that controls for DMNQ were exposed to the sham vehicle (3 mm DMSO) for 30 min during incubation (Fig. 1). This concentration of DMSO has been previously shown to augment phosphatase activity (Misch & Misch, 1967; Fjeld et al. 2000; Santos et al. 2002). DTT treatment of the supernatants generally resulted in significant elevations in PPase activity. However, for PTP in DMSO-treated tissues, DTT slightly reduced PTP activity of matched samples.
Figure 1. Phosphatase activities in matched, untreated control tissues.

Hatched bars: data from controls of DMNQ experiments in which the tissues were exposed to the 3 mm DMSO solvent. Non-hatched bars: data from controls of H2O2 experiments in which tissues were not exposed to any solvent. Light grey bars: supernatants without DTT treatment. Dark grey bars: duplicate, matched supernatants treated with DTT prior to the phosphatase assays. Statistical differences from repeated measures multi-way ANOVA and contrasts between DTT and non-DTT treated supernatants (n= 5–6 in each group; **P < 0.01, *P < 0.05).
Effects of DMNQ and H2O2 treatment on PPase activities
Because of the different effects of baseline conditions on control PPase activity (Fig. 1), the dose responses to low level ROS exposures were analysed by comparing activity as a fraction of each matched control sample. Increasing doses of the intracellular O2•−/H2O2 generator, DMNQ, systematically reduced global Ser/Thr PPase (Fig. 2A) and PTP (Fig. 2B) activities at doses from 10 to 100 μm. PTP was also significantly reduced at 1 μm DMNQ. Post-experimental treatment of the supernatant proteins with 0.5 mm DTT reversed a portion of the inhibition of all treatments (dashed lines). The fact that in some cases DTT treatment did not return PPase activities to baseline is not surprising, since Hecht & Zick (1992) showed that it requires up to 10 mm to completely reverse some forms of oxidized PPase activity.
Figure 2. Effects of DMNQ treatment on relative phosphatase activity.

All data expressed as a fraction of matched baseline control tissues. A: Ser/Thr PPase (n= 5); B: PTP (n= 5). Points represent percentage differences from matched controls from the same animal. Statistical tests were post-ANOVA contrasts between sample means. Dashed lines, DTT treated supernatants, continuous lines, no DTT treatment. **P < 0.01, *P < 0.05 from matched control, ++P < 0.01, from matched DTT treatment at the same dose of DMNQ.
In general, increasing doses of H2O2 also decreased Ser/Thr PPase and PTP activities (Fig. 3). The effects were similar to those observed in the DMNQ experiments, i.e. increasing levels of H2O2 reduced activity, and treatment of the supernatant proteins with DTT partially reversed these effects (Fig. 3B). PTP activity was not greatly affected by H2O2 exposure, with slight stimulation at 5 μm H2O2, and modest inhibition at 500 μm, the latter only statistically significant in the DTT treated supernatants compared to the matched DTT-treated controls.
Figure 3. Effects of H2O2 treatment on relative phosphatase activity.

A, Ser/Thr PPase (n= 5); B, PTP (n= 5). Statistical tests as in Fig. 2. Dashed lines, DTT treated supernatants; continuous lines, no DTT treatment. **P < 0.01, *P < 0.05 from matched control, ++P < 0.01, from matched DTT treatment at same dose of H2O2.
Measurements of H2O2 in the tissue baths
H2O2 is a volatile, reactive molecule and our tissue baths were open, bubbled chambers. Furthermore, H2O2 is lipid soluble and penetrates cell membranes easily, where it can be scavenged by endogenous antioxidant systems. To understand the magnitude of these effects on the dosage of H2O2 exposure over the course of the experiment, we determined the changes in [H2O2] over 30 min in the tissue baths (Fig. 4). The same assay was used to test the stock H2O2 solutions added to baths. Three different rates of change can be observed. First, there is an immediate drop in [H2O2] to a value of ∼50–75% of the target concentration. These same reductions occurred whether or not the tissue was in the bath. Second, there was a slow reduction of [H2O2] in the bath when there was no tissue present (∼15% over 30 min). Third, when muscle strips were included in the bath, the decrease in [H2O2] was greatly accelerated (20–80% reduction over 30 min). With 5 μm exposure, there was essentially no measurable H2O2 after 20 min (Fig. 4, inset). With 50 μm exposure, the value was reduced to near zero within 30 min. In the first 10 min of the 500 μm H2O2 exposure, the average diaphragm consumed ∼3.75 μmol H2O2 g−1 min−1. Therefore, the actual H2O2 exposure was substantially lower over the course of the exposure period compared to the targeted value of [H2O2].
Figure 4. Decay of [H2O2] over time in the tissue baths.

Inset: expanded scale to demonstrate measurements in control tissue (no H2O2) and with 5 μm H2O2. Dashed lines are baths containing muscle tissue; continuous lines are baths without tissue. All measurements were compared against standards using Amplex Red fluorescence as the detection system (n= 4).
Effects of oxidant exposure on contractile function and oxidative stress
The intention of this study was to provide oxidant stimuli that might influence PPase activity and perhaps the kinetics of intracellular signalling pathways but not significantly diminish contractile function or cause oxidative stress. To this end, neither H2O2 nor DMNQ, at any concentrations tested, had any significant effects on maximum active twitch force at 30 min, when compared to baseline. Figure 5A shows the twitch amplitude before and throughout the DMNQ exposure. There were no changes in twitch amplitude at any dose and there were no changes in passive force (not shown). Similar results were seen at 5 and 50 μm exposure to H2O2 (Fig. 5B). However, in response to 500 μm H2O2, there was a significant elevation in passive force (P < 0.01) that began within 6 min of treatment. A similar proportional increase in total force occurred, resulting in no significant change in peak active twitch force.
Figure 5. Dose effects of oxidant exposure on twitch force.

A, response of peak twitch force (% of initial force prior to exposure) to DMNQ at all doses (1 min−1). There were no changes or trends in baseline force (not shown). B, effects of H2O2 on the percentage change in peak twitch force; lower curve represents the changes in passive force during 500 μm exposure (P < 0.001). No other doses affected passive force. Statistical differences measured using one-way ANOVA Dunnett's test from control for the last data point. Results are means ±s.e.m. for each point during the exposure (n= 6 in each; **P < 0.01).
To determine if the levels of oxidants given resulted in marked oxidative stress, we tested carbonyl formation of supernatant protein samples from tissues exposed to the same experimental protocol. Five strongly evident bands were evaluated in each gel (Fig. 6A and C) and the mean values for the summed densitometry areas are expressed in Fig. 6B and D. There were no significant effects or trends in carbonyl formation in any of the DMNQ exposures but at the highest dose of H2O2 given, there was a modest but highly significant elevation in protein carbonyls. With the exception of this group, however, and when coupled with the lack of effect on twitch force, we conclude that the overall oxidative stress imposed by these oxidant exposures must have been relatively minimal and below that seen in other physiological and pathophysiological conditions where carbonyl formation is often evident.
Figure 6. Dose effects of oxidant treatment on protein carbonyl formation.

Left panels, A and C, are representative gels showing effects of increasing oxidant exposure on carbonyl formation for DMNQ (A) and H2O2 (B). Intermittent lanes between doses are matched, non-derivatized negative control samples. B and D are the corresponding summed densitometry of the gels expressed as a sum of the areas under five distinct carbonyl bands for each lane. Only 500 μm H2O2 resulted in a significant elevation in carbonyl formation (n= 6 in each group).
Effects of oxidant exposure on net protein phosphorylation
Overall protein phosphorylation was evaluated using the ProQ/Sypro Ruby detection system. Typical gels are illustrated for DMNQ in Fig. 7A and for H2O2 in Fig. 7C. Relatively subtle changes in peak density were observed in superimposed density scans, as shown in the corresponding example traces in Fig. 7B (DMNQ) and 7D (H2O2). In some sets of matched scans, more dominant elevations in phosphorylation were observed in specific locations of the gel (Fig. 7B for DMNQ), whereas in other sets of scans, particularly for H2O2 exposure, many individual bands were more dense after treatment (Fig. 7D). The grouped data are shown in Fig. 8. The effect of DMNQ on protein phosphorylation is statistically significant at each dosage, with more predominant and significant increases as the dosage of DMNQ increased, particularly within specific molecular weight regions. The effects of increasing H2O2 exposure differed in that there was little effect of 5 μm exposure but with a much wider range of phosphorylated proteins, particularly at the 50 μm dose. To determine if the increases in phosphorylation across all of the bands in all gels were a common phenomenon, the mean relative densities of each band for each dose were compared directly against their respective control bands and plotted on a line of identity (Fig. 9). Eighty-six per cent of the measured phosphorylation bands showed increased phosphorylation after treatment with oxidants compared to the matched controls; the binomial probability of this being a random distribution is < 0.001. Many of these increases were small, but in total they suggest higher levels of protein phosphorylation across much of the phosphoproteome following oxidant exposure.
Figure 7. Examples of protein phosphorylation measurements in response to ROS.

A and C, ProQ Diamond gels (left) and Sypro Ruby gels (right) showing phosphorylated and total proteins, respectively. A, one matched set of samples showing all doses of DMNQ exposure; C, a matched set of samples for all doses of H2O2 exposure. Protein names on the gels are estimates from known locations of these proteins on standard gels. B and D, typical density scans from corresponding gels in A and C. B, scan from one set of control and 3 superimposed DMNQ dose exposures in which only some isolated bands demonstrated increased phosphorylation compared to matched controls. D, scan from one set of H2O2 exposures in which many bands at some concentrations of H2O2 exposure showed increased phosphorylation. Shaded areas highlight the control responses in both B and D. Stars represent areas on the gel that were consistently more phosphorylated for each type of oxidant exposure.
Figure 8. Grouped data for protein phosphorylation in response to ROS exposure.

Data expressed as differences from matched controls in densitometry of phosphorylated proteins. Left: DMNQ exposures; right: H2O2 exposures. *P < 0.05, **P < 0.01 using post-ANOVA least squares contrasts. The individual points were chosen from 20 of the most quantifiable peaks selected for each ROS exposure. P for individual ANOVAs are listed under each graph title.
Figure 9. Overall comparisons of 20 dominant phosphorylation bands with controls.

An identity plot (line) with individual values for the averaged peak heights in response to all levels of oxidant exposure in all 6 oxidant exposures (i.e. 120 average values in the plot) vs. the peak amplitude of the corresponding matched control peaks. Results show >85% of the values lie above the identity line (slope of 1).
Discussion
The results of this study are consistent with the hypothesis that low levels of oxidants, at concentrations that have little effect on twitch force generation over a 30 min exposure, have relatively consistent inhibitory influences on global PPase activities within intact, isolated skeletal muscle. This effect was particularly clear for the mitochondrial ROS producer, DMNQ. Importantly, Ser/Thr PPase proteins appeared as sensitive and, in some cases, more sensitive to the inhibitory effects of oxidants, compared to PTPs. Although this study does not distinguish which phosphatases undergo oxidation, results of oxidant exposure on the phosphorylation state of a wide spectrum of divergent proteins suggests that low levels of oxidants are broadly affecting phosphorylation events throughout the cell, in part through PPase inhibition. Therefore, these results are in general agreement with the concept that localized production of ROS functions as a coordinated response to promote regional phosphorylation signalling, an effect that is likely to apply to responses to wide ranges of physiological stimuli.
Mechanisms of action of oxidants on PPases
Our results for PTPs are consistent with the well recognized oxidative vulnerability of a ubiquitous and conserved cysteine-based active site on PTPs (Caselli et al. 1998; Denu & Tanner, 1998; Lee et al. 1998; Salmeen et al. 2003; Meng et al. 2004; Salmeen & Barford, 2005; Chiarugi & Buricchi, 2007). This signature cysteine, unlike free cysteine, is deprotonated at physiological pH, making it very vulnerable to oxidation by ROS (Salmeen & Barford, 2005) and resulting in loss of its capacity for catalytic dephosphorylation. The Cys-S− is easily oxidized to the suflonic acid form (–cys-SOH) and then, under continued oxidant exposure, to more stable –cys-SO2 (sulfinic) and –cys-SO3 (sulfonic) acids, which are not easily reversible, as reviewed in (Salmeen & Barford, 2005). Some PTPs, such as PTP1B have also been shown to form a sulfonamide bond, following oxidation, between the cysteine and a nearby nitrogen on serine (Salmeen et al. 2003). Other oxidation states, such as disulfide bonds and glutathiolation are common occurrences with other specific PTPs (Salmeen & Barford, 2005).
More surprising was the extent to which Ser/Thr PPases were sensitive to oxidative inhibition, which is less well described and is in contrast to a considerable body of literature on the topic. For example, Denu & Tanner (1998) found no effects of H2O2 on three members of the Ser/Thr PPase family, calcineurin, PP2Cα and γPPase, when exposed in vitro, concluding that Ser/Thr PPases are ‘relatively immune to H2O2 oxidation.’ Likewise, Sommer et al. (2002) studied PP1, PP2A and calcineurin and found a ‘very low sensitivity’ of cell lysates to H2O2, with some small effect on calcineurin. The intracellular redox sensitivity of calcineurin has subsequently been shown to be dependent on an intact intracellular environment via multiple mechanisms of superoxide dismutase interaction (Agbas et al. 2007), oxidation of the metal centre (Namgaladze et al. 2002), or redox-dependent proteolytic pathways (Lee et al. 2007). Only one previous study, on intact melanoma cells which were exposed to 10 μm H2O2, has examined global Ser/Thr PPase activity (Pieri et al. 2003). A ∼25% reduction in activity was observed that was partially reversible with DTT.
One of the unexpected findings was that H2O2 seemed to have only modest influences on PTP activity (Fig. 3B), with a small stimulation of activity at 5 μm H2O2, whereas the responses to DMNQ were robust and inhibitory over the entire range of exposures (Fig. 2B). Pieri et al. (2003) also noted a similar stimulation of PTP in intact melanoma cells exposed to 5 μm H2O2. They saw no effect of H2O2 at 10 μm and inhibition at higher exposures from an enzymatic H2O2 generating system. Although we cannot explain this paradoxical response that both labs have observed, it could reflect contrasting and varying sensitivities of different pools of PTPs to different levels or types of oxidants. However, there are other compelling possibilities that might explain this. For example, a low level of baseline control PTP activity was seen in the H2O2 exposure experiments (Fig. 1). Low baseline PTP activity could be due to the influence of the elevated 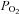 used in tissue bath experiments (
used in tissue bath experiments (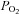 ∼650 mmHg). In contrast, normal muscle intracellular
∼650 mmHg). In contrast, normal muscle intracellular 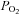 at rest is believed to be no more than 30 mmHg (Richardson et al. 2006). Such levels of hyperoxic exposure have been shown by previous investigators, in other model systems, to result in strong inhibition of PPase activities and this has been proposed as a possible contributing mechanism to O2 toxicity and O2 sensing (Kerepesi et al. 1984; Sheth et al. 1997; Nyunoya et al. 2005). Therefore, if baseline PTP activity was depressed to begin with by the high O2 environment in the baths, it would dampen or distort the response to exogenous H2O2. The lack of response of this group to DTT that is shown in Fig. 2B is misleading and is partly a reflection of the normalization to controls, also treated with DTT. Absolute values of activity were significantly elevated in all groups following DTT exposure.
at rest is believed to be no more than 30 mmHg (Richardson et al. 2006). Such levels of hyperoxic exposure have been shown by previous investigators, in other model systems, to result in strong inhibition of PPase activities and this has been proposed as a possible contributing mechanism to O2 toxicity and O2 sensing (Kerepesi et al. 1984; Sheth et al. 1997; Nyunoya et al. 2005). Therefore, if baseline PTP activity was depressed to begin with by the high O2 environment in the baths, it would dampen or distort the response to exogenous H2O2. The lack of response of this group to DTT that is shown in Fig. 2B is misleading and is partly a reflection of the normalization to controls, also treated with DTT. Absolute values of activity were significantly elevated in all groups following DTT exposure.
The above explanation does not explain why baseline PTP activities were so high in the DMNQ experiments. The diaphragm strips were exposed to the same 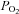 . A plausible explanation is that they were also exposed to 3 mm DMSO throughout incubation. There is strong supporting evidence for a protective influence of low concentrations of DMSO on PPase activities (Misch & Misch, 1967; Fjeld et al. 2000; Santos et al. 2002). The mechanism for this curious effect is not well understood. Though it may relate to the poorly defined antioxidant function of low concentrations of DMSO, it more likely reflects a ‘chemical chaperone’ effect that has been shown to influence the stability of the active sites of the enzymes (Fjeld et al. 2000). As little as 4% DMSO in cell culture elevates lysate phosphatase activity by >2-fold (Santos et al. 2002), results that parallel those observed with 3 mm exposures here. We speculate that DMSO protected the active site of PTP from the influence of high
. A plausible explanation is that they were also exposed to 3 mm DMSO throughout incubation. There is strong supporting evidence for a protective influence of low concentrations of DMSO on PPase activities (Misch & Misch, 1967; Fjeld et al. 2000; Santos et al. 2002). The mechanism for this curious effect is not well understood. Though it may relate to the poorly defined antioxidant function of low concentrations of DMSO, it more likely reflects a ‘chemical chaperone’ effect that has been shown to influence the stability of the active sites of the enzymes (Fjeld et al. 2000). As little as 4% DMSO in cell culture elevates lysate phosphatase activity by >2-fold (Santos et al. 2002), results that parallel those observed with 3 mm exposures here. We speculate that DMSO protected the active site of PTP from the influence of high 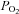 in the baths but did not protect it from the stronger effects of acute oxidant exposure. Understanding this phenomenon in future experiments could have far reaching implications regarding the poorly understood pharmacological influences of DMSO and also the potential role of phosphatases in cellular responses to
in the baths but did not protect it from the stronger effects of acute oxidant exposure. Understanding this phenomenon in future experiments could have far reaching implications regarding the poorly understood pharmacological influences of DMSO and also the potential role of phosphatases in cellular responses to 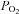 .
.
An important limitation of the approach used in this study was the homogenization and isolation procedures which are likely to have excluded many phosphatases associated with membranes, the nucleus or contractile elements. There are entire classes of receptor-like tyrosine phosphatases that play extremely important biological functions (reviewed in Chiarugi, 2005) and many important Ser/Thr PTases are membrane associated as well (e.g. Begum, 1995). Therefore, further work will be required to determine the extent to which these proteins are redox senstitive and the extent to which methods used here can address the redox sensitivity of these pools of phosphatases.
Effects of oxidants on protein phosphorylation state
Our results demonstrate that oxidant exposure results in subtle but consistent increases in the phosphorylation state over a wide range of proteins. This observation cannot be attributed solely to phosphatase inhibition, as many kinases are also known to be redox sensitive, but in contrast to phosphatases, the majority are activated by oxidation (Chiarugi, 2005; Maher, 2006). In fact, oxidant stimulation of kinases and inhibition of phosphatases appear to be a well coordinated system within many cell signalling networks (Chiarugi, 2005). Though there are several common mechanisms responsible for the redox sensitivity of kinases, one of the primary mechanisms is through phosphatase inhibition (Chiarugi, 2005; Maher, 2006). Kinases often operate within amplification networks, where kinase-kinases are responsible for activating downstream kinases. Inhibition of a corresponding phosphatase at any stage within the chain of reactions will prolong net kinase activity and amplify the activity of downstream kinases.
The overall responses of phosphorylated proteins to DMNQ appeared to affect proteins in more specific molecular weight regions, whereas the responses to 50 μm H2O2 were consistent across a broader range of molecular weights. The more unique pattern for the DMNQ response is not surprising, since it is likely to reflect the localized formation of ROS at the mitochondrial membranes by DMNQ (Tchivilev et al. 2008). Therefore many phosphatases or kinases within other cellular compartments could have been compartmentally restricted from ROS exposure. On the other hand, the more generalized pattern of responses to 50 μm H2O2 represent a stimulus that is probably sufficiently distributed across the sample by diffusion and sustained at concentrations sufficient to broadly influence protein oxidation. The lack of a significant response at 5 μm may reflect the fact that within 20 min of exposure, there was no measurable H2O2 in the muscle chambers (Fig. 4), leaving at least 10 min for the phosphorylation state of proteins to return to an equilibrium status.
Critique of oxidant exposure
Application of oxidants or oxidant-producing agents such as DMNQ is not directly physiological but has the potential of mimicking physiology when they are given at a low concentration. The levels of exogenous oxidant agents were chosen to test a wide dynamic range of exposure and still avoid gross distortion of the tissue. With the possible exception of the 500 μm H2O2, we believe we accomplished this. The absolute concentrations of H2O2 and O2•− that are expected in resting, proliferating, stimulated or stressed cells are not well understood and it is likely that the concentrations of various ROS species are highly localized in the cells and highly dependent on cell type and conditions. Nevertheless, concentrations of H2O2 in normal proliferating cells during normal signalling cycles can reach as high as 0.7 μm (Antunes & Cadenas, 2000) and with an expected 7- to 10-fold drop in [H2O2] from the extracellular space, extracellular exposures in isolated cells of 5–10 μm are likely to fall in a roughly physiological range (reviewed in Stone & Yang, 2006). In agreement with this, in previous studies of dihydrofluorescein oxidation as an assay for oxidant formation in isolated rodent diaphragm, it required exposure of 10 μm H2O2 in the superfusate to approximate the intracellular ROS formed in the transition to hypoxia (Zuo & Clanton, 2005). Another consideration is that intact superfused skeletal muscle such as diaphragm is quite different from isolated cell culture or isolated myofibres because of the relatively large diffusion distances (250–300 μm half diaphragm thickness) and by thin layers of protective cells on both peritoneal and pleural surfaces. Therefore, we would expect that the average intracellular doses of H2O2 for a given extracellular dose to be greatly reduced in this experiment compared to similar treatments of cultured cells. The same diffusion gradient issues exist for 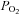 in this preparation. Though in lightly stimulated, thin, isolated muscle such as the diaphragm, it is likely that O2 supplies are adequate in the core of the muscle (Barclay, 2005), O2 gradients imposed by the thickness of the muscle could affect underlying redox state of cells at various layers.
in this preparation. Though in lightly stimulated, thin, isolated muscle such as the diaphragm, it is likely that O2 supplies are adequate in the core of the muscle (Barclay, 2005), O2 gradients imposed by the thickness of the muscle could affect underlying redox state of cells at various layers.
A number of previous studies have evaluated the influence of H2O2 on contractile function in skeletal muscle. Reid et al. (1993) demonstrated a small stimulating effect of H2O2 on diaphragm twitch force in doses from 100 μm to 10 mm and mentioned that the higher concentrations tended to degrade the muscle. Later work by Andrade, Reid and Westerblad in isolated flexor digitorum brevis fibres (a fast fibre type) confirmed the stimulatory effect of brief exposures to 0.1 to 10 μm H2O2 (Andrade et al. 2001). These contractile effects diminished over a 30 min exposure time and higher concentrations (100–200 μm) significantly increased force only for brief periods but caused eventual degradation within 10 min (Andrade et al. 1998). The doses of H2O2 used in this study were selected by direct experimentation to avoid such decrements or stimulations in rat diaphragm muscle force over 30 min. It is not surprising that we did not see either the stimulatory or the degradation effects that were seen with single fast fibres (Andrade et al. 1998, 2001) as single fast fibres are likely to have a much lower antioxidant capacity than oxidative fibres and would presumably be exposed to a much higher intracellular H2O2 concentration because they would be surrounded by a uniform H2O2 in the bathing medium.
Significance and conclusions
We have described a general response to oxidant exposure in skeletal muscle that could influence broad signalling events in the muscle. What would be the overall impact on cell and organ physiology? The results support the concept that localized oxidant formation lowers the barriers for successful phosphorylation events to occur within the region of the cell where the oxidants are being produced. As illustrated schematically in Fig. 10, it is well known that there is considerable chemical inertia that any given kinase must overcome to sustain a successful phosphorylation event on its target protein(s). This barrier is largely due to the fact that the average phosphatase activity has ∼10-fold advantage with respect to potency of its dephosphorylation reaction compared with the corresponding kinase (Maher, 2006). This strong background phosphatase activity can be thought of as a powerful ‘phosphatase tone’. Its function must be to minimize noise within the signalling system. That is, it reduces the probability that random protein phosphorylation events occur. However, strong phosphatase activity is also likely to be responsible for the dynamic elements of many signalling cascades. That is, like rapidly adapting receptors in the nervous system, it ensures that a given signalling network will respond to ‘changes’ in its environment and return to steady state after a change has passed. Oxidants in this context lower the barriers for the response of the system and, therefore, elevate the probability that a given signalling network will emerge from the steady-state for a prolonged enough period to successfully move the signal forward through signal transduction. The localized and transient nature of the oxidants ensures that this effect is localized to the area of stress exposure or receptor signalling and that it does not move beyond the localized environment where it is not needed. This concept has been applied convincingly as a factor in receptor tyrosine kinase function (e.g. Xu et al. 2002; Rhee, 2006), but it has not been applied in a more general sense to a much broader class of oxidant forming events and signalling pathways of cells or tissues.
Figure 10. A schematic diagram of the proposed generalized role of oxidants in facilitating phosphorylation events.

Under steady state conditions, successful protein phosphorylation events must overcome considerable barriers due to strong dephosphorylation activity of phosphatases. Oxidants lower the barriers for successful and lasting phosphorylation activity to occur. This can occur from combinations of PPase inhibition and kinase activation due to ROS.
Is there a down side to such a generalized oxidant response system? In the short term, the consequences are few because the strong antioxidant defences and the powerful reducing capacity of the cytosol ensure that the cell will return to equilibrium in a short time period. However, if the oxidant exposure is too intense, too prolonged, or comes from multiple exposures to stress or to simultaneous receptor activations, the net effect would lead to signalling chaos, low signal to noise, and difficulty for the system to respond with fidelity to any specific stimulus. Perhaps signalling chaos is the true consequence of oxidative stress.
Acknowledgments
The work was supported by NIH R01HL053333. Thanks to S. Ryan Oliver and Anthony Payne for their helpful comments and reviews of the manuscript.
Glossary
Abbreviations
- DifMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- DMNQ
2,3-dimethoxy-1-naphthoquinone
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- PPase
phosphatase
- PTP
protein tyrosine phosphatase
- PPM
a class of serine/threonine phosphatases dependent on Mg2+/Mn2+
- PPP
phosphoprotein phosphatases, a class of serine/threonine phosphatases containing Fe2+/Zn2+
- PTP1B
an important protein tyrosine phosphatase in extracellular receptor kinase signalling
- ROS
reactive oxygen species
- Ser/Thr-PPase
serine/threonine phosphatase
Author contributions
V.W. performed the majority of the experiments and first level analyses. She also participated in the writing, editing and study design. P.R. participated in and directed the protein phosphorylation experiments and data collection, participated in the writing, in the conceptual themes and in the editing of the manuscript. T.C. provided the initial ideas and study design for the project and performed most of the initial and final writing, image analysis, statistical analysis and editing. The experimental procedures were performed at The Ohio State University. All data analysis and writing were performed at the University of Florida.
References
- Agbas A, Hui D, Wang X, Tek V, Zaidi A, Michaelis EK. Activation of brain calcineurin (Cn) by Cu-Zn superoxide dismutase (SOD1) depends on direct SOD1-Cn protein interactions occurring in vitro and in vivo. Biochem J. 2007;405:51–59. doi: 10.1042/BJ20061202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almo SC, Bonanno JB, Sauder JM, Emtage S, Dilorenzo TP, Malashkevich V, Wasserman SR, Swaminathan S, Eswaramoorthy S, Agarwal R, Kumaran D, Madegowda M, Ragumani S, Patskovsky Y, Avarado J, Ramagopal UA, Faber-Barata J, Chance MR, Sali A, Fiser A, Zhang X, Lawrence DS, Burley SK. Structural genomics of protein phosphatases. J Struct Funct Genomics. 2007;8:121–140. doi: 10.1007/s10969-007-9036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998;509:565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade FH, Reid MB, Westerblad H. Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox-modulation. FASEB J. 2001;15:309–311. doi: 10.1096/fj.00-0507fje. [DOI] [PubMed] [Google Scholar]
- Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–126. doi: 10.1016/s0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- Barclay CJ. Modelling diffusive O2 supply to isolated preparations of mammalian skeletal and cardiac muscle. J Muscle Res Cell Motil. 2005;26:225–235. doi: 10.1007/s10974-005-9013-x. [DOI] [PubMed] [Google Scholar]
- Begum N. Stimulation of protein phosphatase-1 activity by insulin in rat adipocytes. Evaluation of the role of mitogen-activated protein kinase pathway. J Biol Chem. 1995;270:709–714. doi: 10.1074/jbc.270.2.709. [DOI] [PubMed] [Google Scholar]
- Caselli A, Marzocchini R, Camici G, Manao G, Moneti G, Pieraccini G, Ramponi G. The inactivation mechanism of low molecular weight phosphotyrosine protein phosphatase by H2O2. J Biol Chem. 1998;273:32554–32560. doi: 10.1074/jbc.273.49.32554. [DOI] [PubMed] [Google Scholar]
- Cheng W, Li B, Kajstura J, Wolin MS, Sonnenblick EH, Hintze TH, Olivetti G, Anversa P. Stretch-induced programmed myocyte cell death. J Clin Invest. 1995;96:2247–2259. doi: 10.1172/JCI118280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi P. PTPs and PTKs: the redox side of the coin. Free Radic Res. 2005;39:353–364. doi: 10.1080/10715760400027987. [DOI] [PubMed] [Google Scholar]
- Chiarugi P, Buricchi F. Protein tyrosine phosphorylation and reversible oxidation: two cross-talking posttranslation modifications. Antioxid Redox Signal. 2007;9:1–24. doi: 10.1089/ars.2007.9.1. [DOI] [PubMed] [Google Scholar]
- Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- Diaz PT, She ZW, Davis WB, Clanton TL. Hydroxylation of salicylate by the in vitro diaphragm: evidence for hydroxyl radical production during fatigue. J Appl Physiol. 1993;75:540–545. doi: 10.1152/jappl.1993.75.2.540. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa A, Garcia A, Hartel S, Hidalgo C, Jaimovich E. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. J Biol Cem. 2009;284:2568–2575. doi: 10.1074/jbc.M804249200. [DOI] [PubMed] [Google Scholar]
- Falk DJ, DeRuisseau KC, Van Gammeren DL, Deering MA, Kavazis AN, Powers SK. Mechanical ventilation promotes redox status alterations in the diaphragm. J Appl Physiol. 2006;101:1017–1024. doi: 10.1152/japplphysiol.00104.2006. [DOI] [PubMed] [Google Scholar]
- Felty Q, Xiong WC, Sun D, Sarkar S, Roy D. Estrogen-induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry. 2005;44:6900–6909. doi: 10.1021/bi047629p. [DOI] [PubMed] [Google Scholar]
- Fjeld CC, Rice AE, Kim Y, Gee KR, Denu JM. Mechanistic basis for catalytic activation of mitogen-activated protein kinase phosphatase 3 by extracellular signal-regulated kinase. J Biol Chem. 2000;275:6749–6757. doi: 10.1074/jbc.275.10.6749. [DOI] [PubMed] [Google Scholar]
- Goosens V, Grooten J, De Vos K, Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:8115–8119. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht D, Zick Y. Selective inhibition of protein tyrosine phosphatase activties by H2O2 and vandate in vitro. Biochem Biophys Res Comm. 1992;188:773–779. doi: 10.1016/0006-291x(92)91123-8. [DOI] [PubMed] [Google Scholar]
- Heffetz D, Bushkin I, Dror R, Zick Y. The insulinomimetic agents H2O2 and vandadate stimulate tyrosine phosphorylation in inact cells. J Biol Chem. 1990;265:2896–2902. [PubMed] [Google Scholar]
- Honkanen RE, Golden T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem. 2002;9:2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- Kerepesi T, Rady P, Balla G, Dauda G. Maturation of the fetal lung II. Effect of hyperoxia on phosphatidic acid phosphatase, pyruvate kinase and superoxide dismutase activity in the newborn rat lung. Acta Paediatr Hung. 1984;25:247–254. [PubMed] [Google Scholar]
- Lee S-R, Kwon K-S, Kim S-R, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- Lee J-E, Kim H, Jang H, Cho E-J, Youn H-D. Hydrogen peroxide triggers the proteolytic cleavage and the inactivation of calcineurin. J Neurochem. 2007;100:1703–1712. doi: 10.1111/j.1471-4159.2006.04340.x. [DOI] [PubMed] [Google Scholar]
- Liu R-M, Shi MM, Giulivi C, Forman HJ. Quinones increase gamma-glutamyl transpeptidase expression by multiple mechanisms in rat lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 1998;274:L330–L336. doi: 10.1152/ajplung.1998.274.3.L330. [DOI] [PubMed] [Google Scholar]
- Maher P. Redox control of neural function: background, mechanisms, and signficance. Antioxid Redox Signal. 2006;8:1941–1970. doi: 10.1089/ars.2006.8.1941. [DOI] [PubMed] [Google Scholar]
- Martins AS, Shkryl VM, Nowycky MC, Shirokova N. Reactive oxygen species contribute to Ca2+ signals produced by osmotic stress in mouse skeletal muscle fibres. J Physiol. 2008;586:197–210. doi: 10.1113/jphysiol.2007.146571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng T-C, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signalling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- Meng T-C, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molecular Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- Misch DW, Misch MS. Dimethyl sulfoxide: activation of lysosomes in vitro. Proc Natl Acad Sci U S A. 1967;58:2462–2467. doi: 10.1073/pnas.58.6.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalibet J, Skorey KI, Kennedy BP. Protein tyrosine phosphatase: enzymatic assays. Methods. 2005;35:2–8. doi: 10.1016/j.ymeth.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Namgaladze D, Hofer HW, Ullrich V. Redox control of calcineurin by targeting the binuclear Fe2+-Zn2+ centre at the enzyme active site. J Biol Chem. 2002;277:5962–5969. doi: 10.1074/jbc.M111268200. [DOI] [PubMed] [Google Scholar]
- Namgaladze D, Shcherbyna I, Kienhofer J, Hofer HW, Ullrich V. Superoxide targets calcineurin signalling in vascular endothelium. Biochem Biophys Res Commun. 2005;334:1061–1067. doi: 10.1016/j.bbrc.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Nyunoya R, Monick MM, Powers LS, Yarovinsky TO, Hunninghake GW. Macrophages survive hyperoxia via prolonged ERK activation due to phosphatase down-regulation. J Biol Chem. 2005;280:26295–26302. doi: 10.1074/jbc.M500185200. [DOI] [PubMed] [Google Scholar]
- Pastula C, Johnson I, Beechem JM, Patton WF. Development of fluorescence-based selective assays for serine/threonine and tyrosine phosphatases. Comb Chem High Throughput Screen. 2003;6:341–346. doi: 10.2174/138620703106298590. [DOI] [PubMed] [Google Scholar]
- Peterson GL. Determination of total protein. Methods Enzymol. 1983;96:95–119. doi: 10.1016/s0076-6879(83)91014-5. [DOI] [PubMed] [Google Scholar]
- Pieri L, Dominici S, Del Bello B, Maellaro E, Comporti M, Paolicchi A, Pompella A. Redox modulation of protein kinase/phosphatase balance in melanoma cells: the role of endogenous and gamma-glutamyltransferase dependent H2O2 production. Biochim Biophys Acta. 2003;1621:76–83. doi: 10.1016/s0304-4165(03)00048-5. [DOI] [PubMed] [Google Scholar]
- Pyner S, Coney A, Marshall JM. The role of free radicals in the muscle vasodilatation of systemic hypoxia in the rat. Exp Physiol. 2003;88:733–740. doi: 10.1113/eph8802524. [DOI] [PubMed] [Google Scholar]
- Reid MB, Haak KE, Francik KM, Volbertg PA, Kabzik PA, West MA. Reactive oxygen in skeletal muscle I: intracellular oxidant kinetics and fatigue in vitro. J Appl Physiol. 1992;73:1797–1804. doi: 10.1152/jappl.1992.73.5.1797. [DOI] [PubMed] [Google Scholar]
- Reid MB, Khawli FA, Moody MR. Reactive oxygen in skeletal muscle III. Contractility of unfatigued muscle. J Appl Physiol. 1993;75:1081–1087. doi: 10.1152/jappl.1993.75.3.1081. [DOI] [PubMed] [Google Scholar]
- Reid MB, Moody MR. Dimethylsulfoxide depresses skeletal muscle contractility. J Appl Physiol. 1994;76:2186–2190. doi: 10.1152/jappl.1994.76.5.2186. [DOI] [PubMed] [Google Scholar]
- Rhee SG. H2O2 a necessary evil for cell signalling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Duteil S, Wary C, Wray DW, Hoff J, Carlier PG. Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. J Physiol. 2006;571:415–424. doi: 10.1113/jphysiol.2005.102327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmeen A, Andersen JN, Myers MP, Meng T-C, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal. 2005;7:560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- Salsman SJ, Hensley K, Floyd RA. Sensitivity of protein tyrosine phosphatase activity to the redox environment, cytochrome c, and microperoxidase. Antioxid Redox Signal. 2005;7:1078–1088. doi: 10.1089/ars.2005.7.1078. [DOI] [PubMed] [Google Scholar]
- Santos ALS, Souto-Padron T, Alviano CS, Lopes AHCS, Soares RMA, Meyer-Fernandes JR. Secreted phosphatase activity induced by dimethyl sulfoxide in Herpetomonas samuelpessoai. Arch Biochem Biophys. 2002;405:191–198. doi: 10.1016/s0003-9861(02)00403-4. [DOI] [PubMed] [Google Scholar]
- Sheth MV, Goodman BE, Freiese JL, Eyster KM. Protein kinase and phosphatase activity in the lungs of normoxic versus hyperoxic rats. Exp Lung Res. 1997;23:475–494. doi: 10.3109/01902149709039239. [DOI] [PubMed] [Google Scholar]
- Sommer D, Coleman S, Swanson SA, Stemmer PM. Differential susceptibilities of serine/theronine phosphatases to oxidative and nitrosative stress. Arch Biochem Biophys. 2002;404:271–278. doi: 10.1016/s0003-9861(02)00242-4. [DOI] [PubMed] [Google Scholar]
- Stone JR, Yang S. Hydrogen peroxide: a signalling messenger. Antioxid Redox Signal. 2006;3&4:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- Takakura K, Beckman JS, MacMillan-Crow LA, Crow JP. Rapid and irreversible inactivation of protein tyrosine phosphatases PTP1B, CD45, and LAR by peroxynitrite. Arch Biochem Biophys. 1999;369:197–207. doi: 10.1006/abbi.1999.1374. [DOI] [PubMed] [Google Scholar]
- Tchivilev I, Madamanchi NR, Vendrov AE, Niu XL, Runge MS. Identification of a protective role for protein phosphatase 1cγ1 against oxidative stress-induced vacular smooth muscle cell apoptosis. J Biol Chem. 2008;283:22193–22205. doi: 10.1074/jbc.M803452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signalling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- Tonks NK. Redox redux: revisiting PTPs and the control of cell signalling. Cell. 2005;121:670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Welte S, Baringhaus K-H, Schmider W, Müller G, Petry S, Tennagels N. 6,8-Difluoro-4-methylumbiliferyl phosphate: a fluorgenic substrate for protein tyrosine phosphatases. Anal Biochem. 2005;338:32–38. doi: 10.1016/j.ab.2004.11.047. [DOI] [PubMed] [Google Scholar]
- Xu D, Rovira II, Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell. 2002;2:251–259. doi: 10.1016/s1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- Zhang A, Jia Z, Guo X, Yang T. Aldosterone induces epithelial-mesenchymal transition via ROS of mitochondrial origin. Am J Physiol Renal Physiol. 2007;293:F723–F731. doi: 10.1152/ajprenal.00480.2006. [DOI] [PubMed] [Google Scholar]
- Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- Zuo L, Clanton TL. Reactive oxygen formation in the transition to hypoxia in skeletal muscle. Am J Physiol Cell Physiol. 2005;289:C207–C216. doi: 10.1152/ajpcell.00449.2004. [DOI] [PubMed] [Google Scholar]
- Zuo L, Christofi FL, Wright VP, Liu CY, Merola AJ, Berliner LJ, Clanton TL. Intra- and extracellular measurement of reactive oxygen species production during heat stress in skeletal muscle. Am J Physiol Cell Physiol. 2000;279:C1066. doi: 10.1152/ajpcell.2000.279.4.C1058. [DOI] [PubMed] [Google Scholar]