

Abstract
Context: Bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma are two rare causes of ACTH-independent Cushing’s syndrome.
Objective: The aim of the study was to evaluate a 35-yr-old woman with ACTH-independent hypercortisolism associated with both micronodular adrenal hyperplasia and ectopic pararenal adrenocortical adenoma.
Design and Setting: In vivo and in vitro studies were performed in a University Hospital Department and academic research laboratories.
Intervention: Mutations of the PRKAR1A, PDE8B, and PDE11A genes were searched for in leukocytes and adrenocortical tissues. The ability of adrenal and adenoma tissues to synthesize cortisol was investigated by immunohistochemistry, quantitative PCR, and/or cell culture studies.
Main Outcome Measure: Detection of 17α-hydroxylase and 21-hydroxylase immunoreactivities, quantification of CYP11B1 mRNA in adrenal and adenoma tissues, and measurement of cortisol levels in supernatants by radioimmunological assays were the main outcomes.
Results: Histological examination of the adrenals revealed nonpigmented micronodular cortical hyperplasia associated with relative atrophy of internodular cortex. No genomic and/or somatic adrenal mutations of the PRKAR1A, PDE8B, and PDE11A genes were detected. 17α-Hydroxylase and 21-hydroxylase immunoreactivities as well as CYP11B1 mRNA were detected in adrenal and adenoma tissues. ACTH and dexamethasone activated cortisol secretion from adenoma cells. The stimulatory action of dexamethasone was mediated by a nongenomic effect involving the protein kinase A pathway.
Conclusion: This case suggests that unknown molecular defects can favor both micronodular adrenal hyperplasia and ectopic adrenocortical adenoma associated with Cushing’s syndrome.
The unusual association of adrenal micronodular hyperplasia and ectopic adrenocortical adenoma as a cause of ACTH-independent Cushing’s syndrome is discussed.
Primary adrenal cortisol hypersecretion is responsible for 15–20% of all cases of Cushing’s syndrome. In 10–15% of cases, Cushing’s syndrome is due to bilateral adrenal lesions that include micronodular and macronodular adrenal hyperplasias and, more rarely, bilateral adenomas or carcinomas (1,2). Micronodular adrenal hyperplasias are defined by the presence of multiple cortical micronodules, i.e. measuring less than 1 cm in diameter. They are themselves divided into two subtypes depending on the presence of internodular atrophy and nodular pigment. Primary pigmented nodular adrenocortical disease (PPNAD) is characterized by multiple pigmented micronodules usually surrounded by internodular cortical atrophy. PPNAD may be isolated or may occur as part of the Carney complex that also includes spotty skin pigmentation, myxomas, and endocrine neoplasms (3). Most patients with PPNAD, especially when the disease is a component of Carney complex, have germline-inactivating mutations of the PRKAR1A [protein kinase A (PKA) regulatory subunit type 1α] gene. Mutations of the phosphodiesterase (PDE) 11A and PDE8B genes have also been described in patients with PPNAD or nonpigmented variants of the disease (4,5). All these genetic events lead to constitutive activation of the cAMP/PKA pathway that secondarily favors glucocorticoid hypersecretion and adrenocortical hyperplasia (2,3). It has been shown that patients with PPNAD exhibit a paradoxical increase in cortisol secretion in response to Liddle’s test; i.e. administration of dexamethasone at doses of 2 mg/d for 2 d followed by 8 mg/d for 2 d (6). This abnormal cortisol response is now used as a biological criterion for the diagnosis of the disease (7). An increase in urinary free cortisol excretion on the second day of high-dose dexamethasone administration higher than 50% of the basal level supports the diagnosis of PPNAD (6).
In the present study, we report a case of ACTH-independent Cushing’s syndrome associated with a paradoxical increase in urinary cortisol level in response to dexamethasone. The patient was found to have bilateral nonpigmented micronodular adrenocortical hyperplasia and ectopic, i.e. pararenal, adrenocortical adenoma. No germinal mutations of the PRKAR1A, PDE8B, and PDE11A genes were detected. Similarly, molecular studies showed no somatic mutations of these three genes in adrenal gland and ectopic adenoma tissues. Immunohistochemical and quantitative PCR studies revealed the presence of 17α-hydroxylase and 21-hydroxylase immunoreactivities and the expression of CYP11B1 mRNA in adrenal and adenoma tissues. Finally, in vitro studies showed the occurrence of a paradoxical stimulatory effect of dexamethasone on cortisol production from cultured adenoma cells.
Patient and Methods
Patient
A 35-yr-old woman was referred to our department of endocrinology for overt Cushing’s syndrome, i.e. presenting with faciotruncal obesity, facial erythrosis, easy bruising, striae, hypertension, and amenorrhea. Plasma cortisol concentrations were 264 μg/liter [730 nmol/liter; normal: 91–308 μg/liter (250–850 nmol/liter)] in the morning and 241 μg/liter (666 nmol/liter) at midnight. Urinary cortisol concentrations, measured on two independent samples, were 793 μg/d [2190 nmol/d; normal: 20–80 μg/d (55–220 nmol/d)] and 821 μg/d (2266 nmol/d), respectively. The morning plasma ACTH concentration was undetectable [<5 pg/ml (1.1 pmol/liter); normal: 10–80 pg/ml (2.2–17.6 pmol/liter)]. During the Liddle’s test, urinary cortisol increased from 821 to 1024 μg/d (2825 nmol/liter). Administration of cosyntropin (250 μg iv) induced a substantial increase in plasma cortisol levels from 253 μg/liter (698 nmol/liter) to 458 μg/liter (1265 nmol/liter). Adrenal computed tomography (CT) scan revealed the presence of a micronodule measuring 8 mm in diameter of the left gland, whereas the right adrenal appeared normal (Fig. 1). Taken together, the biological and radiological data were highly suggestive of ACTH-independent Cushing’s syndrome due to PPNAD. There were no other clinical, biological, and radiological signs of Carney complex. In particular, skin examination did not reveal any lesion suggestive of the disease; pituitary and thyroid functions were normal; and pituitary magnetic resonance imaging, cardiac echography, and breast, pelvic, and thyroid sonographies did not show any abnormality. The patient was therefore thought to have isolated PPNAD, and bilateral adrenalectomy was performed through percutaneous retroperitoneoscopy. Pathological examination of the two glands confirmed the diagnosis of bilateral adrenocortical micronodular hyperplasia. However, there was neither abnormal pigmentation of the tissues nor hyperplasia of the internodular adrenal cortex that appeared partially atrophic (Fig. 2, A–D). Postoperatively, the patient was supplemented with hydrocortisone (30 mg/d) and fludrocortisone (100 μg/d). Three months later, signs of hypercortisolism had not resolved, and a cosyntropin test (250 μg iv), performed at 0800 h before hydrocortisone intake, showed the maintenance of high plasma cortisol level [249 μg/liter (687 nmol/liter)] reaching 409 μg/liter (1128 nmol/liter) after stimulation, suggesting the persistence of hyperfunctioning adrenocortical tissue. Urinary cortisol, measured after interruption of corticosteroid replacement, was still elevated [645 μg/d (1785 nmol/d)]. Abdominal CT scan revealed the presence of a left pararenal, i.e. close to the renal hilum, tumor measuring 38 mm in diameter and exhibiting a spontaneous attenuation value of 28 Hounsfield units (Fig. 3A). Noriodocholesterol scintigraphy showed intense uptake of the tracer by the tumor (Fig. 3B), highly suggesting the diagnosis of ectopic adrenocortical adenoma. The tumor was removed through retroperitoneoscopy, and pathological examination of the tissue confirmed the diagnosis of adrenocortical adenoma (Weiss score: 0; Fig. 3, C and D). Removal of the tumor was followed by the disappearance of all clinical signs of Cushing’s syndrome under substitutive treatment with hydrocortisone (30 mg/d) and fludrocortisone (100 μg/d). Plasma cortisol levels measured at 0800 h before hydrocortisone intake remained undetectable both in the basal condition and after stimulation with cosyntropin.
Figure 1.
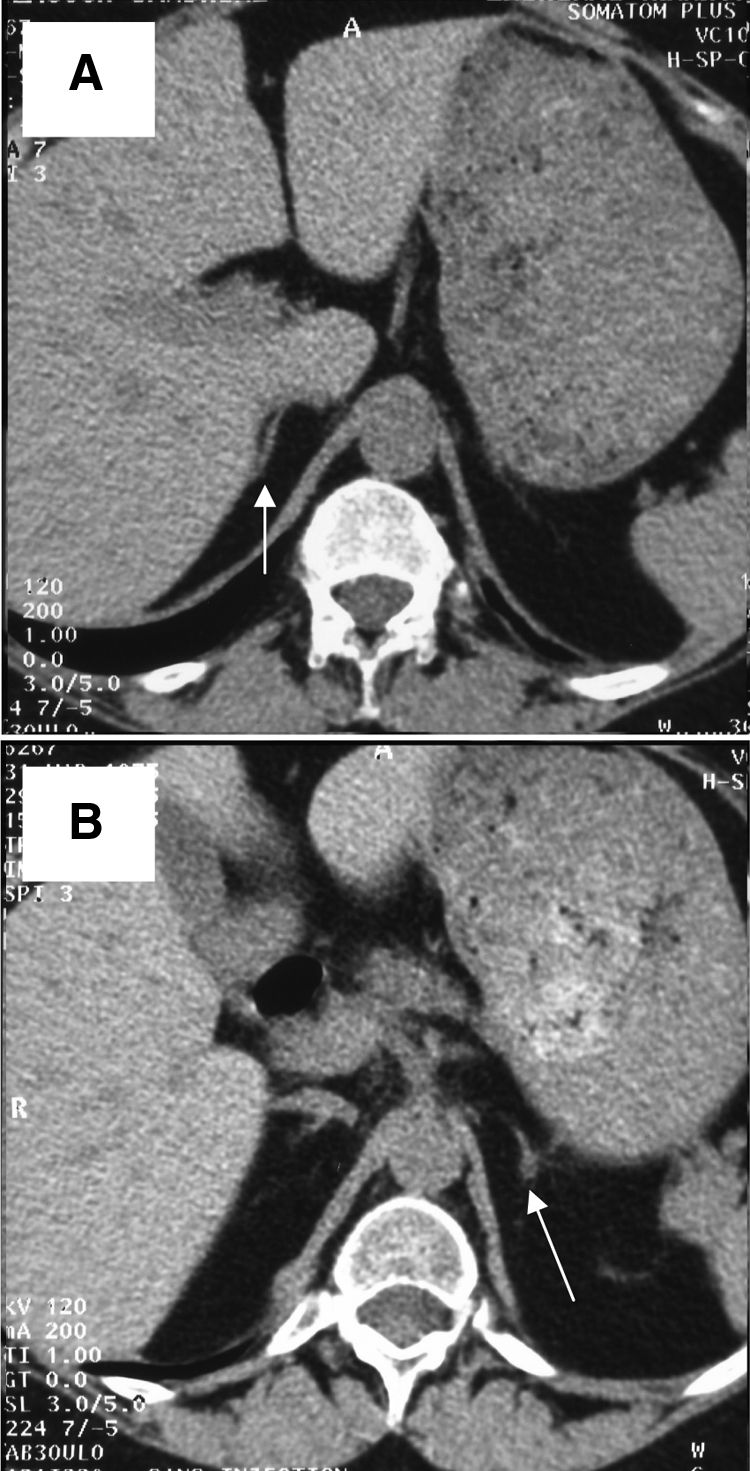
Adrenal CT scan showing normal appearance of the right adrenal gland (arrow; A) and a micronodule of the left adrenal gland (arrow; B) reaching 8 mm in diameter.
Figure 2.

Macroscopic and histological presentations of the left and right adrenal glands. Macroscopic views of the left (A) and right (B) adrenals showing the left micronodule previously visualized at CT scan (arrow). C, Low magnification view (×25) showing the histological structure of the left adrenal micronodule. D, Microphotograph (×25) revealing the presence of several micronodules in the cortex of the right adrenal gland (arrows). E–H, Higher magnification views showing the presence of 17α-hydroxylase (E and F) and 21-hydroxylase (G and H) immunoreactivities in both the micronodules and the internodular cortex in the left and right adrenals (scale bar, 100 μm). G (inset), Microphotograph (×200) of a 21-hydroxylase-positive cell in the left micronodule visualized at CT scan.
Figure 3.

Radiological, scintigraphic, and histological presentations of the ectopic adrenocortical adenoma. A, Abdominal CT scan showing a pararenal tumor measuring 38 mm in diameter with a spontaneous attenuation value of 28 Hounsfield units (arrow). B, Intense uptake of the tracer by the tumor at noriodocholesterol scintigraphy. C, Macroscopic view of the tumor showing multiple pigmented areas. D, Microscopic view of the tumor tissue showing two types of steroidogenic cells, i.e. large spongiocytic lipid-loaded cells (lower left field) and small compact eosinophilic cells (upper right field); magnification, ×80. E and F, Microphotographs showing the presence of 17α-hydroxylase (E) and 21-hydroxylase (F) immunoreactivities in the adenoma tissue. 17α-hydroxylase immunoreactivity was detected in both spongiocytic and compact cells, whereas 21-hydroxylase immunostaining was mainly observed in compact cells (scale bar, 50 μm).
The patient underwent blood sampling for detection of PRKAR1A, PDE8B, and PDE11A germline mutations. In addition, adrenal glands and ectopic adrenocortical adenoma were obtained at surgery and immediately dissected by the pathologist. Adrenal gland and tumor explants were frozen and stored at −80 C until molecular studies or fixed in formalin. Part of the tumor was transported to the laboratory in culture medium for primary culture. The protocol of collection of the tissues and the experimental procedures were approved by the regional ethics committees, and written informed consent was obtained from the patient.
Genetic studies
The screening for germline and somatic base substitutions and small insertions and deletions in the PRKAR1A, PDE8B, and PDE11A genes was performed as previously reported (8,9,10). Briefly, DNA was extracted from peripheral blood leukocytes and adrenal tissues using the Wizard Genomic DNA purification kit (Promega, Charbonnières, France). The 12 exons of PRKAR1A, 22 exons of PDE8B, 20 coding exons of PDE11A (exons 3-23), and all the corresponding flanking intronic sequences were separately PCR amplified using primers and conditions previously described (8,9,10). Both strands of the amplified products were directly sequenced on an automatic sequencer. Nucleotides were numbered in accordance with the reference sequences for PRKAR1A (ENSG00000108946), PDE8B (ENSG00000113237), and PDE11A (ENSG00000128655) genes. To screen for large deletions involving one or more exons of the PRKAR1A gene, DNA from the patient was examined by array-based comparative genomic hybridization using a custom-designed DNA microarray (ExonArrayDx v2.0; GeneDx, Gaithersburg, MD) containing multiple oligonucleotide probes hybridizing to all exons and their flanking intronic regions. Hybridization data were analyzed with Agilent Technologies (Massy, France) software (DNA analytics v4) to evaluate copy number at the exon level.
Immunohistochemistry
Deparaffinized sections from the adrenal and ectopic adrenocortical adenoma tissues were incubated for 1 h at room temperature with polyclonal rabbit antibody to 17α-hydroxylase (1:100; provided by Drs. V. Luu The and G. Pelletier, Laval University Medical Center, Québec, Canada) or goat antibody to human 21-hydroxylase (1:250; Santa Cruz Biotechnology, Santa Cruz, CA). The sections were then incubated with a streptavidin-biotin-peroxidase complex (Dako Corporation, Carpinteria, CA; and Santa Cruz Biotechnology), and the enzymatic activity was revealed with diaminobenzidine. The tissue sections were counterstained with hematoxylin.
Quantitative RT-PCR
Total RNA was extracted using the Tri-Reagent (Sigma- Aldrich, Saint Quentin Fallavier, France) and further purified on RNeasy mini Spin Columns (QIAGEN, Courtaboeuf, France). RNAs were converted into cDNA by using ImProm-II Reverse Transcription System (Promega). PCR amplification was performed in duplicate using the SYBR Green I Master Mix Buffer (Applied Biosystems, Courtaboeuf, France) in an ABI PRISM 7000 Sequence Detector (Applied Biosystems) with the following specific primers for CYP11B1 (NM_000497.3): AGGAGACCTTGCGGCTCTACC and GAACACGCGCACCAATGTC; and glyceraldehyde-3-phosphate dehydrogenase (NM_002046.3): GAGCCAAAAGGGTCATCATC and CCATCCACAGTCTTCTGGGT. The amount of cDNA in each sample was calculated by the comparative threshold cycle method and expressed as 2−ΔCt using GAPDH mRNA as an internal control.
Cell culture experiments
Ectopic adrenocortical adenoma explants were processed for cell culture studies as previously described (11). Tumor cells were cultured at 37 C in 5% CO2. Incubation experiments of cells were conducted in quadruplicate for 24 h after 2 d in culture with fresh DMEM (control experiments) or DMEM with dexamethasone, fluticasone, or ACTH. Cells were also incubated with dexamethasone in the presence of the PKA inhibitor H89 and the glucocorticoid receptor (GR) antagonist RU486. All reagents were obtained from Sigma-Aldrich. Cells were incubated with each secretagogue for 24 h at 37 C. Cortisol concentrations in culture medium were measured using a formerly reported RIA procedure (12).
Statistical analysis
Results are expressed as mean ± se, and statistical significance was assessed by Student’s t test and Bonferroni test after one-way ANOVA.
Results
Screening for germline and somatic PRKAR1A, PDE8B, and PDE11A gene mutations
DNA sequencing revealed no mutation of the PRKAR1A gene either in blood leukocytes or in left and right adrenal glands and ectopic adrenocortical adenoma. In addition, no deletion or duplication was identified by targeted array comparative genomic hybridization analysis with exon level resolution (ExonArrayDx) in the PRKAR1A gene. Similarly, molecular studies did not show any germinal or somatic mutation of the PDE11A and PDE8B genes in the same samples.
Detection of steroidogenic enzymes in the adrenal and ectopic adenoma tissues
Immunohistochemical analysis of the two adrenal glands revealed the presence of 17α-hydroxylase and 21-hydroxylase in both the micronodules and the internodular cortex (Fig. 2, E–H). 17α-Hydroxylase immunostaining was more intense in micronodules, whereas 21-hydroxylase immunostaining was principally detected in the perinodular cortex. In the adenoma tissue, 17α-hydroxylase-like immunoreactivity was visualized in both spongiocytic and compact cells, whereas 21-hydroxylase immunostaining was mainly observed in compact cells (Fig. 3, E and F). PCR product corresponding to CYB11B1 mRNA was detected in the two adrenal glands and the adenoma. Real time RT-PCR showed that the adrenal glands of the patient expressed the CYB11B1 mRNA at lower levels (left, 0.20 ± 0.01; right, 0.34 ± 0.01) than normal adrenals (1.01 ± 0.02; n = 6; P < 0.001). By contrast, higher levels were noticed in the adenoma tissue (i.e. 7.80 ± 0.17; P < 0.001 vs. normal adrenals).
Effect of synthetic glucocorticoids on cortisol production from cultured adrenocortical adenoma cells
Incubation of cultured adrenocortical adenoma cells with graded concentrations of dexamethasone and fluticasone (10−10 to 10−6 m) provoked a dose-dependent stimulation of cortisol production (Fig. 4A). The maximum effects of the two glucocorticoids, 36.0 ± 7.8% for dexamethasone and +27.3 ± 5.7% for fluticasone, respectively, were obtained at a concentration of 10−8 m. Interestingly, dexamethasone (10−8 m) was as efficient as ACTH (10−9 m) to stimulate cortisol release (Fig. 4B). RU486 (10−6 m) failed to modify dexamethasone-induced cortisol secretion. In contrast, H89 (10−5 m) induced a robust decrease (−75 ± 4%) in basal cortisol production and inhibited the cortisol response to dexamethasone (10−8 m).
Figure 4.
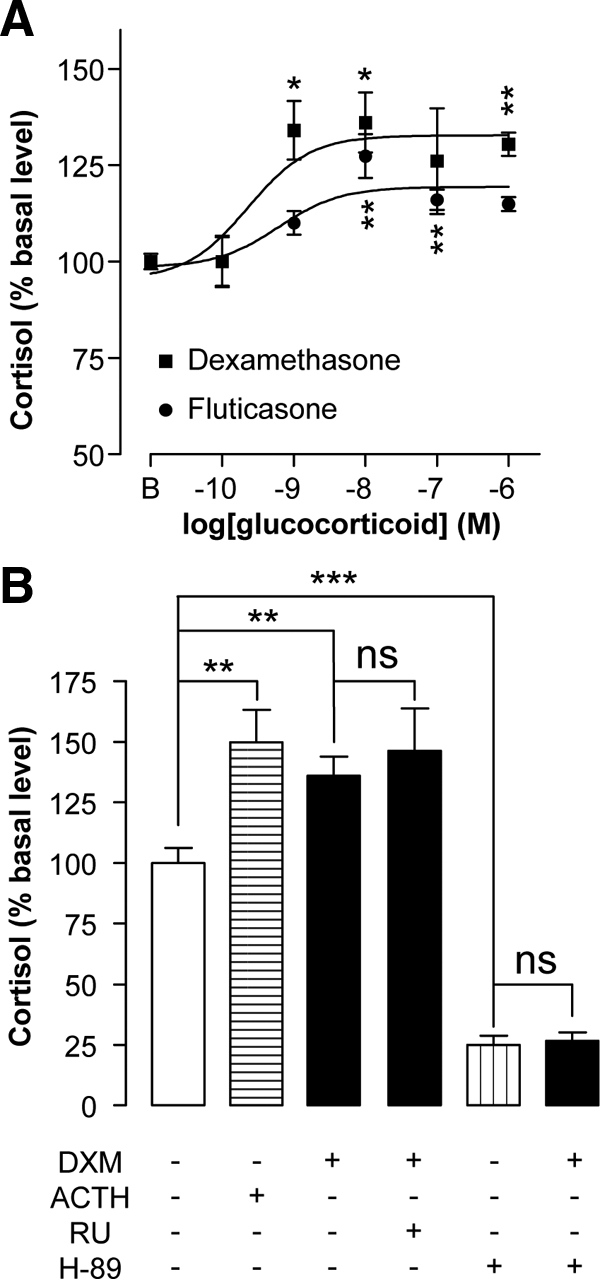
Effects of synthetic glucocorticoids and ACTH on cortisol production by cultured adenoma cells. A, Effects of increasing concentrations (10−10 to 10−6 m) of the synthetic glucocorticoids dexamethasone (▪) and fluticasone (•) on cortisol release. B, Effect of ACTH (10−9 m) on basal cortisol secretion and actions of the GR antagonist RU486 (10−6 m) and the PKA inhibitor H89 (10−5 m) on the cortisol response to dexamethasone (10−8 m). *, P < 0.05; **, P < 0.01; ***, P < 0.0001. ns, Not significant.
Discussion
PPNAD is a rare cause of ACTH-independent Cushing’s syndrome that may be isolated or may occur as part of the Carney complex (1,2). In the present observation, coexistence of ACTH-independent hypercortisolism and micronodular presentation of the left adrenal gland at CT scan was almost pathognomonic of PPNAD. Although dexamethasone-evoked increase in urinary cortisol was lower than 50% (i.e. the cutoff usually used as a diagnostic criterion for the disease; see Ref. 7), the paradoxical response of urinary cortisol excretion to dexamethasone was another finding evocative of PPNAD. However, the absence of pigmentation at histological examination of the adrenal glands led us to exclude this diagnosis. In addition, the lack of hyperplasia of zona fasciculata was not consistent with isolated micronodular adrenocortical disease, according to the new nomenclature recently proposed by Stratakis and Boikos (2). These observations suggest therefore that the patient presented with an unclassified form of micronodular adrenal hyperplasia.
Surprisingly, although the two adrenal glands had been removed in totality, adrenalectomy failed to correct hypercortisolism, indicating the presence of ectopic adrenocortical tissue. Adrenocortical ectopic tissues, also known as adrenal rest tissues, are rather frequent. Because adrenal and gonadal tissues derive from a common embryonic precursor, the urogenital ridge, most of them are located along the migration path of the adrenals and gonads or in the gonads themselves (13). However, extraadrenal adrenocortical tissue is usually not functional and rarely gives rise to ACTH-independent neoplasm. In fact, only nine cases of cortisol-producing ectopic adrenocortical neoplasms with Cushing’s syndrome have been reported so far (14,15,16,17,18,19,20,21). The localizations of the ectopic tumors included the adrenal areas, the liver, the ovary, and, as in the patient described here, the pararenal region. The fact that the first CT scan had been limited to the adrenal areas explains why the tumor, which was located close to the renal hilum, had not been detected. Similarly, the localization of the adenoma as well as the use of retroperitoneoscopy for surgical removal of the adrenals prevented the surgeon from visualizing the tumor during the operation. Unlike several of the published cases (14,15,16,21), the ectopic adrenocortical neoplasm described herein was certainly benign, as indicated by calculation of the Weiss score. The observation that urinary cortisol level decreased only moderately after bilateral adrenalectomy, contrasting with the complete suppression of endogenous cortisol secretion by surgical removal of the adenoma, suggests that hypercortisolism was mainly caused by the ectopic neoplasm. In agreement with this hypothesis, immunohistochemical studies showed an intense staining of adenoma tissues by antibodies to 17α-hydroxylase and 21-hydroxylase, and real-time RT-PCR experiments revealed expression of high levels of CYP11B1 mRNA by the tumor. In addition, adenoma cell incubation studies confirmed the ability of the adenoma tissue to produce substantial amounts of cortisol and showed that two synthetic glucocorticoids, dexamethasone and fluticasone, were able to activate corticosteroidogenesis. This latter result suggests that the paradoxical stimulatory action of dexamethasone on cortisol secretion, noticed in vivo in the preoperative period, was likely the consequence of a direct effect of the drug on adenoma cells, although we cannot exclude that the hyperplastic adrenals may also have been responsive to dexamethasone. It also implies that cortisol may have activated its own production by the tumor tissue through an ultrashort local amplification loop. Interestingly, we observed that the GR antagonist RU486 failed to inhibit the cortisol response to dexamethasone by tumor cells, indicating that the glucocorticoid stimulated cortisol production through a GR-independent and consequently nongenomic mechanism. It is possible that this unexpected effect may be mediated by an unknown membrane receptor and may involve second messengers, as formerly proposed in other tissue models (22). In agreement with this hypothesis, H89, an inhibitor of PKA, was found to decrease basal and dexamethasone-induced cortisol production, suggesting that the action of dexamethasone is mediated by the PKA pathway in adenoma cells, as previously reported in PPNAD cells (23). Paradoxical cortisol responses to dexamethasone have already been observed in patients with cortisol-secreting adrenocortical adenomas and attributed to occurrence of tumor somatic mutations of the PRKAR1A gene (24). However, this mechanism could be excluded in the present case.
Adrenal adenomas responsible for Cushing’s syndrome are usually sensitive to the stimulatory action of ACTH (25). The plasma cortisol response to cosyntropin observed in the patient before surgical removal of its pararenal tumor and the observation that ACTH stimulated cortisol release from cultured tumor cells show that, like eutopic adrenocortical adenomas, ectopic adrenocortical neoplasms can express functional ACTH receptors.
From a physiopathological point of view, the adrenal disease of the patient, that associated bilateral adrenocortical micronodular hyperplasia, ectopic adrenocortical adenoma, and ACTH-independent Cushing’s syndrome, raises two main questions: 1) were the adrenals really involved in hypercortisolism; and 2) could adrenal hyperplasia and tissue ectopia both be explained by a common pathogenic mechanism. First, it is very likely that the two adrenal glands were actually hyperfunctional. Indeed, suppression of circulating ACTH by chronically elevated plasma levels of cortisol originating from the ectopic adenoma should have led to global atrophy of the adrenal cortex. In addition, the fact that bilateral adrenalectomy reduced both urinary cortisol excretion and the cortisol response to cosyntropin by approximately 20% suggests that the hyperplastic glands were partly responsible for hypercortisolism independently of ACTH. This hypothesis is further supported by the observation that the cortex of the two adrenal glands was found to contain 17α-hydroxylase and 21-hydroxylase immunoreactivities and to express 11ß-hydroxylase mRNA, although the expression level of this latter enzyme appeared lower than in the normal adrenal gland. The decreased levels of 11ß-hydroxylase mRNA in the hyperplastic adrenals may indicate that CYP11B1 is mainly, if not exclusively, expressed in micronodules as observed for 17α-hydroxylase. Second, it is conceivable that a single molecular defect occurring during adrenal embryogenesis may have favored both adrenocortical hyperplasia and tissue ectopia with subsequent development of an ACTH-independent neoplasm. Although our molecular studies revealed no somatic mutations of the PRKAR1A, PDE8B, and PDE11A genes in the adrenal glands and adenoma tissue removed from the patient, the involvement of mutations of gene(s) encoding protein(s) of the cAMP signaling pathway in the pathogenesis of the disease is supported by the following observations: the majority of benign adrenocortical lesions seem to be linked to abnormalities of the cAMP/PKA pathway (2); 21-hydroxylase deficiency, a condition that favors chronic elevation of plasma ACTH levels during embryogenesis, is associated with a high frequency of testicular adrenal rests (26); and it is well established that ACTH activates cAMP production in adrenocortical cells (27). Further molecular studies focused on cAMP/PKA pathway-related genes will therefore be useful to characterize the molecular defects involved in the patient’s disease.
In conclusion, we have reported a complex case of ACTH-independent hypercortisolism associating bilateral adrenal micronodular hyperplasia and ectopic adrenocortical adenoma whose secretory activity was stimulated, in vivo and in vitro, by both dexamethasone and ACTH. The molecular mechanism underlying the association of these two rare causes of Cushing’s syndrome remained unknown.
Acknowledgments
We are indebted to H. Lemonnier for technical assistance.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale Unité 413/EA4310, Institut Fédératif de Recherches Multidisciplinaires sur les Peptides 23, the Carney Complex Network (ANR-08-GENOPAT-007), the Centre Hospitalier Universitaire de Rouen, the Réseau COMETE (PHRC AOM 06 179), and the Conseil Régional de Haute-Normandie.
Disclosure Summary: E.L., F.G., R.L., A.H., S.R., J.C., A.R., E.C., P.G., C.S., and J.-M.K. have nothing to declare. J.B. and H.L. received grants from Agence Nationale de la Recherche (ANR-08-GENOPAT-007).
First Published Online November 13, 2009
Abbreviations: CT, Computed tomography; GR, glucocorticoid receptor; PDE, phosphodiesterase; PKA, protein kinase A; PPNAD, primary pigmented nodular adrenocortical disease.
References
- Christopoulos S, Bourdeau I, Lacroix A 2005 Clinical and subclinical ACTH-independent macronodular adrenal hyperplasia and aberrant hormone receptors. Horm Res 64:119–131 [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Boikos SA 2007 Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab 3:748–757 [DOI] [PubMed] [Google Scholar]
- Bossis I, Voutetakis A, Bei T, Sandrini F, Griffin KJ, Stratakis CA 2004 Protein kinase A and its role in human neoplasia: the Carney complex paradigm. Endocr Relat Cancer 11:265–280 [DOI] [PubMed] [Google Scholar]
- Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, Griffin KJ, Stein E, Levine E, Delimpasi G, Hsiao HP, Keil M, Heyerdahl S, Matyakhina L, Libè R, Fratticci A, Kirschner LS, Cramer K, Gaillard RC, Bertagna X, Carney JA, Bertherat J, Bossis I, Stratakis CA 2006 A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet 38:794–800 [DOI] [PubMed] [Google Scholar]
- Horvath A, Mericq V, Stratakis CA 2008 Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med 358:750–752 [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, Chrousos GP, Papanicolaou DA 1999 Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med 131:585–591 [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Kirschner LS, Carney JA 2001 Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 86:4041–4046 [DOI] [PubMed] [Google Scholar]
- Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer B, Zacharieva S, Pignatelli D, Carney JA, Luton JP, Bertagna X, Stratakis CA, Bertherat J 2002 Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet 71:1433–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libé R, Fratticci A, Coste J, Tissier F, Horvath A, Ragazzon B, Rene-Corail F, Groussin L, Bertagna X, Raffin-Sanson ML, Stratakis CA, Bertherat J 2008 Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res 14:4016–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath A, Giatzakis C, Tsang K, Greene E, Osorio P, Boikos S, Libè R, Patronas Y, Robinson-White A, Remmers E, Bertherat J, Nesterova M, Stratakis CA 2008 A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. Eur J Hum Genet 16:1245–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louiset E, Contesse V, Groussin L, Cartier D, Duparc C, Barrande G, Bertherat J, Vaudry H, Lefebvre H 2006 Expression of serotonin 7 receptor and coupling of ectopic receptors to protein kinase A and ionic currents in adrenocorticotropin-independent macronodular adrenal hyperplasia causing Cushing’s syndrome. J Clin Endocrinol Metab 91:4578–4586 [DOI] [PubMed] [Google Scholar]
- Lefebvre H, Contesse V, Delarue C, Feuilloley M, Hery F, Grise P, Raynaud G, Verhofstad AA, Wolf LM, Vaudry H 1992 Serotonin-induced stimulation of cortisol secretion from human adrenocortical tissue is mediated through activation of a serotonin4 receptor subtype. Neuroscience 47:999–1007 [DOI] [PubMed] [Google Scholar]
- Vassiliadi D, Tsagarakis S 2007 Unusual causes of Cushing’s syndrome. Arq Bras Endocrinol Metabol 51:1245–1252 [DOI] [PubMed] [Google Scholar]
- Ney RL, Hammond W, Wright L, Davis WW, Acker J, Bartter FC 1966 Studies in a patient with an ectopic adrenocortical tumor. J Clin Endocrinol Metab 26:299–304 [DOI] [PubMed] [Google Scholar]
- Raith L, Karl HJ 1969 Pregnancy in ectopic adrenal carcinoma. Horm Metab Res 1:149–150 [DOI] [PubMed] [Google Scholar]
- Wallace EZ, Leonidas JR, Stanek AE, Avramides A 1981 Endocrine studies in a patient with functioning adrenal rest tumor of the liver. Am J Med 70:1122–1125 [DOI] [PubMed] [Google Scholar]
- Contreras P, Altieri E, Liberman C, Gac A, Rojas A, Ibarra A, Ravanal M, Serón-Ferré M 1985 Adrenal rest tumor of the liver causing Cushing’s syndrome: treatment with ketoconazole preceding an apparent surgical cure. J Clin Endocrinol Metab 60:21–28 [DOI] [PubMed] [Google Scholar]
- Adeyemi SD, Grange AO, Giwa-Osagie OF, Elesha SO 1986 Adrenal rest tumour of the ovary associated with isosexual precocious pseudopuberty and cushingoid features. Eur J Pediatr 145:236–238 [DOI] [PubMed] [Google Scholar]
- Leibowitz J, Pertsemlidis D, Gabrilove JL 1998 Recurrent Cushing’s syndrome due to recurrent adrenocortical tumor—fragmentation or tumor in ectopic adrenal tissue? J Clin Endocrinol Metab 83:3786–3789 [DOI] [PubMed] [Google Scholar]
- Ayala AR, Basaria S, Udelsman R, Westra WH, Wand GS 2000 Corticotropin-independent Cushing’s syndrome caused by an ectopic adrenal adenoma. J Clin Endocrinol Metab 85:2903–2906 [DOI] [PubMed] [Google Scholar]
- Jain SH, Sadow PM, Nosé V, Dluhy RG 2008 A patient with ectopic cortisol production derived from malignant testicular masses. Nat Clin Pract Endocrinol Metab 4:695–700 [DOI] [PubMed] [Google Scholar]
- Song IH, Buttgereit F 2006 Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol Cell Endocrinol 246:142–146 [DOI] [PubMed] [Google Scholar]
- Louiset E, Stratakis CA, Perraudin V, Griffin KJ, Libé R, Cabrol S, Fève B, Young J, Groussin L, Bertherat J, Lefebvre H 2009 The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. J Clin Endocrinol Metab 94:2406–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, Papageorgiou T, Bourdeau I, Kirschner LS, Vincent- Dejean C, Perlemoine K, Gicquel C, Bertagna X, Stratakis CA 2003 Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res 63:5308–5319 [PubMed] [Google Scholar]
- Mancini T, Kola B, Mantero F, Arnaldi G 2003 Functional and nonfunctional adrenocortical tumors demonstrate a high responsiveness to low-dose adrenocorticotropin. J Clin Endocrinol Metab 88:1994–1998 [DOI] [PubMed] [Google Scholar]
- New MI 2006 Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab 91:4205–4214 [DOI] [PubMed] [Google Scholar]
- Penhoat A, Jaillard C, Saez JM 1989 Corticotropin positively regulates its own receptors and cAMP response in cultured bovine adrenal cells. Proc Natl Acad Sci USA 86:4978–4981 [DOI] [PMC free article] [PubMed] [Google Scholar]