

Abstract
Background: Carney complex (CNC) is a familial multiple neoplasia syndrome frequently associated with primary pigmented nodular adrenocortical disease (PPNAD), a bilateral form of micronodular adrenal hyperplasia that leads to Cushing’s syndrome (CS). Germline PRKAR1A mutations cause CNC and only rarely isolated PPNAD.
Patients and Methods: PRKAR1A mutation analysis in two large families with CS and no other CNC manifestations demonstrated a M1V germline mutation; a total of 21 asymptomatic individuals were screened, and mutation carriers were evaluated for CNC. The mutation was expressed in vitro and functionally tested for its effects on protein kinase A function.
Results: Presymptomatic testing identified five first-degree relatives who were M1V carriers and who were all diagnosed with subclinical, mild CS at ages ranging from 20–56 yr. There were no other signs of CNC. In a cell-free system, we detected a shorter compared with the wild-type type 1α regulatory subunit of protein kinase A (PRKAR1A) protein (43 kDa). This was not identified in cell lines from the patients or in transfection experiments in HEK293 cells that showed no detectable PRKAR1A protein from the M1V-bearing constructs. In these cells, the mutant mRNA was expressed in a 1:1 ratio.
Conclusion: In two large families, the M1V PRKAR1A mutation resulted in a PPNAD-only phenotype with significant variability both in terms of age of onset and clinical severity. Expression studies showed a unique effect of this sequence change. This study has implications for genetic counseling of carriers of this PRKAR1A mutation and patients with CNC and PPNAD and for the study of PRKAR1A-related tumorigenesis.
Mutation in the initiation codon of PRKAR1A leads to a mild Carney complex phenotype.
Carney complex (CNC; MIM no. 160980) is an autosomal dominant multiple neoplasia syndrome, characterized by cardiac and extracardiac myxomas and other tumors along with spotty skin pigmentation and endocrine overactivity (1,2). The most frequent endocrine manifestation of CNC is ACTH-independent Cushing’s syndrome (CS) caused by primary pigmented nodular adrenocortical disease (PPNAD), a form of micronodular bilateral adrenocortical hyperplasia (3). Mutations of the gene coding for the protein kinase A (PKA) regulatory subunit PRKAR1A [the type 1α regulatory subunit (RIα) protein], cause the disease in most CNC patients; few apparently sporadic PPNAD patients also carry PRKAR1A mutations (4,5).
Scant information is available for PRKAR1A genotype and PPNAD/CNC phenotype correlation. We recently reported the first PRKAR1A mutation c.709(−7−2)del6 that was almost exclusively associated with mild CS due to PPNAD; c.709(−7−2)del6 was particularly prevalent among the patients with this phenotype, being present in approximately 20% of them (6). We now report the second PRKAR1A mutation (c.1A→G/p.M1V) that appears to be associated almost exclusively with PPNAD. This observation has implications for genetic counseling and for the investigations related to RIα’s role in adrenal function and tumor formation.
Subjects and Methods
Informed consent was obtained as part of a protocol approved by the institutional review boards of the participating institutions. The index patients described in this study were referred for the investigation of CS; their relatives were recruited after the identification of the mutation (see supplemental Subjects and Methods, published as supplemental data on The Endocrine Society’s Journals Online web site at http://jcem.endojournals.org).
DNA was extracted from peripheral blood leukocytes using the Wizard genomic DNA purification kit (Promega, Madison, WI), and the 12 exons and the flanking intronic sequences of the PRKAR1A gene were amplified using the primers and the conditions described previously (6,7).
Lymphocyte cell lines from patients or control subjects were established and treated as previously described (6,7) (see supplemental Subjects and Methods).
To study the functional effect of M1V on PRKAR1A expression, a PCR-based cloning method was used to generate both the wild- type and mutant expression constructs, according to previously published methods (8). The effect of mutated PRKAR1A on PKA activity was determined as previously described (8).
To assess the expression of the M1V-bearing PRKAR1A gene in the absence of cellular factors, we used a transcription and translation cell-free system (TNT T7 coupled reticulocyte lysate system; Promega) with l-[35S]methionine (EasyTag; PerkinElmer Life and Analytical Sciences, Norwalk, CT), according to the manufacturer’s instructions.
To study the lack of the M1V protein in the cell-based system, a proteasome proteolytic activity inhibitor, clasto-lactacystin β-lactone, was applied to transfected cells as described (9). This treatment functions to inhibit the degradation of the proteins by the proteasome.
Results
We studied two families with the M1V mutation with PPNAD only (Fig. 1A). The proband of the first (CAR 19.01) was first reported in 1957 (10) and is now deceased from, reportedly, complications of pneumonia. Neither his father nor his mother was affected by history. Three of the proband’s daughters (CAR 19.03, 19.05, and 19.06), were M1V carriers, and all presented with CS due to PPNAD. To date, they have been followed each for more than 15 yr after bilateral adrenalectomies and have not had any other CNC manifestations.
Figure 1.

Pedigrees of families CAR 19 (A) and CAR 51 (B). The genotype of the individuals available for analysis is shown above each symbol; the stars indicate heterozygoous presence of M1V substitution; the arrows indicate the probands.
The index patient (CAR 51.01) of the second family was diagnosed with CS at age 12 yr. Clinical signs were present since the age of 7, and his growth declined after the age of 10. After the identification of the M1V mutation in PRKAR1A, family screening identified three more carriers of the mutation: the patient’s father and two siblings (Fig. 1B). Her father (CAR 51.04) had been diagnosed with severe CS at the age of 36, after being admitted for a traumatic femur fracture and multiple abnormalities of the spinal vertebrae. Family pictures revealed a plethoric face since 1945, his asthma had disappeared since 1944, and hypertension was diagnosed in 1948. He has no facial or other pigmented lesions. His second daughter (CAR 51.10) to this date remains completely asymptomatic but with biochemically confirmed hypercortisolemia. Finally, a son (brother of the proband, CAR 51.13), had mild, subclinical ACTH-independent CS, but other CNC manifestations, again, were absent. We also studied 10 relatives of the father of the proband and did not find the mutation there, suggesting that it most likely occurred de novo in the father.
Western blot of extracts from patient cell lines carrying the M1V mutation showed only the normal PRKAR1A protein, as we have demonstrated elsewhere (4) (data not shown). To study the expression of the M1V PRKAR1A isoform, we used first a cell-free transcription and translation TNT T7 coupled reticulocyte lysate system in experiments controlled with the wild-type protein (Fig. 2A). We detected expression of both wild-type and mutant constructs; the wild type corresponded to the expected size (49 kDa), and the mutant was shorter (approximately 43 kDa).
Figure 2.
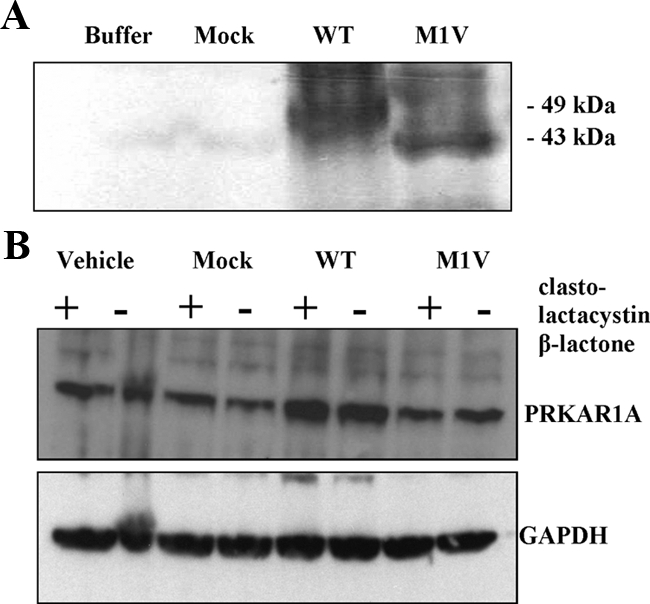
A, Autoradiography after cell-free transcription and translation in TNT T7 coupled reticulocyte lysate system. A shorter (43 kDa) compared with the wild-type PRKAR1A (49 kDa) was detected after transfection with vectors bearing the M1V substitution. B, Western blot analysis of the expression of PRKAR1A in HEK293 cells after transfection with the wild-type and the M1V PRKAR1A constructs; parallel experiments were done with (+) and without (−) clasto-lactacystin β-lactone, a proteasome proteolytic activity inhibitor.
To assess the expression of the M1V-harboring PRKAR1A isoform in a cell-based system, we performed parallel transfection of HEK293 cells with the wild-type and the M1V-bearing construct. Similarly to other nonsynonymous substitutions in PRKAR1A, the M1V cDNA was expressed, as confirmed by sequencing (8). Western blot analysis, however, demonstrated presence of only the wild-type size PRKAR1A (Fig. 2B), after transfection with both the wild-type and the mutant constructs. The protein expression levels from the M1V-transfected cells were lower compared with the wild type and similar to the PRKAR1A levels after transfection with an empty vector (mock). This observation suggested that the detected protein most probably corresponds to the endogenous PRKAR1A in the HEK293 cells.
We then analyzed the effects of the transfection with M1V bearing constructs on the cellular PKA activity. The results of our transfection experiments are presented in the Supplementary Data and Supplementary Figure 1. Briefly, higher PKA activity and reduced binding to cAMP was measured after the transfection with M1V bearing construct–a result consistent with an overall decrease in PRKAR1A protein levels.
Discussion
This study of two large CNC families demonstrated that a mutation in the initiation codon of PRKAR1A, M1V, resulted in a specific phenotype characterized by PPNAD only and often mild atypical CS. The identified mutation carriers, who underwent extensive and detailed clinical screening, all developed CS but no other classical CNC manifestations, like myxomas, schwannomas, or spotty skin pigmentation. The M1V PRKAR1A mutation has never been seen in normal controls; in addition, more than 2000 chromosomes have been tested in our laboratory over the last 10 yr.
Although the mutant mRNA is found to be expressed equal to the wild-type levels in carriers of the mutation, the mutant protein does not seem to be expressed in cells. Notably, in a cell-free system, we detected a shorter PRKAR1A of approximately 43 kDa. This protein corresponds to an alternative translation product starting from a surrogate initiation site-in-frame ATG in the context of the Kozak sequence located 141 bp downstream from the original initiation codon. However, when we transfected the same vectors into HEK293 cells, we were not able to detect shorter PRKAR1A, with or without proteasome proteolytic activity inhibition, suggesting that there is a mechanism different from proteolytic degradation that prevents the expression of M1V in the cell. This observation raises the question of the specific phenotype observed among the carriers of M1V as opposed to the majority of CNC patients, in whom no genotype-phenotype correlation has been established.
The majority of the PRKAR1A mutations lead to nonsense RNA; the mutant RNA species are then degraded by nonsense-mediated decay (NMD) (7). Some aspects of the CNC phenotype were reproduced in the Prkar1a+/− and transgenic, prkar1a-antisense-expressing mice (11,12). The latter developed a late-onset, mild form of adrenal hyperplasia and adenomas associated with lack of suppression of corticosterone secretion in response to dexamethasone (12,13). The variability of CS and corticosterone secretion among patients with PPNAD and CNC and transgenic mice, respectively, has been attributed to possibly incomplete NMD or mutations expressed at the protein level (14), loss of heterozygosity (LOH) in PPNAD nodules in contrast to preservation of hemizygosity for the normal allele in surrounding adrenal cortex (13,15), and possibly other factors such as the presence of modifying genetic defects and a female gender (16).
Recently, a small number of mutations were described that were not subject to NMD; they all led to expressed RIα variants that were associated with increased PKA activity in vitro, just like PRKAR1A mutations that undergo NMD (8,17,21). Interestingly, when LOH could be tested in adrenal tissue from patients with expressed RIα variants, there were no allelic losses (14). Thus, deficient control of the PKA catalytic subunits by either NMD of the mutant allele and LOH of the normal allele (complete RIα loss) or expressed RIα variants that have lost the ability to regulate PKA activity (and thus without need for LOH of the normal allele) is what is responsible for increased PKA activity in PPNAD and other CNC-affected tissues (8,17). These laboratory data indicated that alteration of RIα function alone (and not only its complete loss) is sufficient for increasing PKA activity leading to tumorigenesis and CNC; these data also predicted that quantitative differences of PKA activity in adrenal tissue would be associated with variable forms and/or severity of PPNAD and CS in affected patients.
So far, there has been no significant genotype-phenotype correlation, except in the case of the c.709(−7−2) del6 PRKAR1A mutation, which, similar to M1V, was associated with PPNAD only (6). However, this splice site deletion did not lead to an expressed RIα variant and was subject to NMD. In addition, in contrast to M1V, it appeared to be a low-penetrance variant with several of its carriers manifesting no signs of CS or CNC even after complete hormonal and radiological investigations.
Among the M1V mutation carriers, variability was present in age of onset and severity of CS. Whereas the index patients developed symptoms related to CS before the age of 10 yr, the father of the second proband developed severe CS at the age of 36 yr. Her two siblings were presymptomatically tested (at ages 56 and 45), and both appeared to have only a form of CS. In addition to this intra-familial variability, this family clearly demonstrated differences when compared with previously described CNC patients. One of the hallmarks of CNC, spotty skin pigmentation, which is typically found in more than three fourths of the patients, was absent from both M1V-carrying families. Because PPNAD is the only manifestation of CNC in these two families, we may conclude that the adrenal cortex may be particularly sensitive to minor changes of PKA activity and/or PRKAR1A levels.
Mutation p.M1V results from a change of the first codon in exon 1 from ATG to GTG. This change might cause reduced or absent translation of the wild-type protein or expression of an altered PRKAR1A; however, the actual consequences in vivo are not completely clear. In Smith-Lemli-Opitz syndrome, an autosomal recessive multiple congenital malformation and mental retardation syndrome, M1V has also been suggested to be a mild mutation (18). The reason may be that GTG in this position also may function as a start codon (19). Indeed, although never detected in vivo, the second ATG codon in PRKAR1A, located 141 bp downstream, might be used as an alternative translation initiator codon acting synergistically or independently of the normal start site. Such a shorter protein would lack the dimerization/docking domain, which is critical for the binding to kinase-anchoring proteins and, respectively, for the proper cellular localization of PKA (22).
In conclusion, the M1V mutation resulted in a specific phenotype in two large CNC families in which the affected patients were predominantly characterized by PPNAD and mostly mild atypical CS. There is significant, however, intra-familial variability. This observation has implications for genetic counseling and for the investigations related to the role of PRKAR1A in adrenal function and tumor formation.
Supplementary Material
Acknowledgments
We thank the patients and their families who participated in our research studies and donated their time for this investigation.
Footnotes
This work was supported in part by the NICHD, intramural NIH Project Z01-HD-000642-04 to C.A.S.
Disclosure Summary: A.M.P., F.J.H., A.H., S.W., E.G., E.B., A.A., S.B., J.W.S., J.A.R., M.N., and C.A.S. have nothing to disclose.
First Published Online November 13, 2009
Abbreviations: CNC, Carney complex; CS, Cushing’s syndrome; LOH, loss of heterozygosity; NMD, nonsense-mediated decay; PKA, protein kinase A; PPNAD, primary pigmented nodular adrenocortical disease; RIα, type 1α regulatory subunit.
References
- Boikos SA, Stratakis CA 2006 Carney complex: pathology and molecular genetics. Neuroendocrinology 83:189–199 [DOI] [PubMed] [Google Scholar]
- Carney JA 1995 Carney complex: the complex of myxomas, spotty pigmentation, endocrine overactivity, and schwannomas. Semin Dermatol 14:90–98 [DOI] [PubMed] [Google Scholar]
- Stratakis CA 2009 New genes and/or molecular pathways associated with adrenal hyperplasias and related adrenocortical tumors. Mol Cell Endocrinol 300(1–2):152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA 2000 Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the Carney complex. Hum Mol Genet 9:3037–3046 [DOI] [PubMed] [Google Scholar]
- Groussin L, Jullian E, Perlemoine K, Louvel A, Leheup B, Luton JP, Bertagna X, Bertherat J 2002 Mutations of the PRKAR1A gene in Cushing’s syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab 87:4324–4329 [DOI] [PubMed] [Google Scholar]
- Groussin L, Horvath A, Jullian E, Boikos S, Rene-Corail F, Lefebvre H, Cephise-Velayoudom FL, Vantyghem MC, Chanson P, Conte-Devolx B, Lucas M, Gentil A, Malchoff CD, Tissier F, Carney JA, Bertagna X, Stratakis CA, Bertherat J 2006 A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab 91:1943–1949 [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA 2000 Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat Genet 26:89–92 [DOI] [PubMed] [Google Scholar]
- Greene EL, Horvath AD, Nesterova M, Giatzakis C, Bossis I, Stratakis CA 2008 In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum Mutat 29:633–639 [DOI] [PubMed] [Google Scholar]
- Fitzgerald J, Lamandé SR, Bateman JF 1999 Proteasomal degradation of unassembled mutant type I collagen pro-α (I) chains. J Biol Chem 274:27392–27398 [DOI] [PubMed] [Google Scholar]
- Peterman MG 1957 Suprarenal tumor (Cushing’s syndrome). J Pediatr 50:59–65 [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Kusewitt DF, Matyakhina L, Towns 2nd WH, Carney JA, Westphal H, Stratakis CA 2005 A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res 65:4506–4514 [DOI] [PubMed] [Google Scholar]
- Griffin KJ, Kirschner LS, Matyakhina L, Stergiopoulos S, Robinson-White A, Lenherr S, Weinberg FD, Claflin E, Meoli E, Cho-Chung YS, Stratakis CA 2004 Down-regulation of regulatory subunit type 1A of protein kinase A leads to endocrine and other tumors. Cancer Res 64:8811–8815 [DOI] [PubMed] [Google Scholar]
- Griffin KJ, Kirschner LS, Matyakhina L, Stergiopoulos SG, Robinson-White A, Lenherr SM, Weinberg FD, Claflin ES, Batista D, Bourdeau I, Voutetakis A, Sandrini F, Meoli EM, Bauer AJ, Cho-Chung YS, Bornstein SR, Carney JA, Stratakis CA 2004 A transgenic mouse bearing an antisense construct of regulatory subunit type 1A of protein kinase A develops endocrine and other tumours: comparison with Carney complex and other PRKAR1A induced lesions. J Med Genet 41:923–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer B, Zacharieva S, Pignatelli D, Carney JA, Luton JP, Bertagna X, Stratakis CA, Bertherat J 2002 Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet 71:1433–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M, Lippincott-Schwartz J, Stratakis CA, Bossis I 2006 Depletion of type IA regulatory subunit (RIα) of protein kinase A (PKA) in mammalian cells and tissues activates mTOR and causes autophagic deficiency. Hum Mol Genet 15:2962–29671 [DOI] [PubMed] [Google Scholar]
- Horvath A, Giatzakis C, Robinson-White A, Boikos S, Levine E, Griffin K, Stein E, Kamvissi V, Soni P, Bossis I, de Herder W, Carney JA, Bertherat J, Gregersen PK, Remmers EF, Stratakis CA 2006 Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res 66:11571–11575 [DOI] [PubMed] [Google Scholar]
- Horvath A, Bossis I, Giatzakis C, Levine E, Weinberg F, Meoli E, Robinson-White A, Siegel J, Soni P, Groussin L, Matyakhina L, Verma S, Remmers E, Nesterova M, Carney JA, Bertherat J, Stratakis CA 2008 Large deletions of the PRKAR1A gene in Carney complex. Clin Cancer Res 14:388–395 [DOI] [PubMed] [Google Scholar]
- Witsch-Baumgartner M, Clayton P, Clusellas N, Haas D, Kelley RI, Krajewska-Walasek M, Lechner S, Rossi M, Zschocke J, Utermann G 2005 Identification of 14 novel mutations in DHCR7 causing the Smith-Lemli-Opitz syndrome and delineation of the DHCR7 mutational spectra in Spain and Italy. Hum Mutat 25:412 [DOI] [PubMed] [Google Scholar]
- Kozak M 1997 Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotide in positions +5 and +6. EMBO J 16:2482–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratakis CA, Kirschner LS, Carney JA 2001 Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 86:4041–4046 [DOI] [PubMed] [Google Scholar]
- Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, René-Corail F, Stergiopoulos S, Bourdeau I, Bei T, Clauser E, Calender A, Kirschner LS, Bertagna X, Carney JA, Stratakis CA 2009 Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 94:2085–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS 2006 A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell 24:397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.