

Abstract
Some synthetically useful transformations of organosilicon compounds have been developed since the mid 1970s, based on the new concept that the silicon-carbon bonds are activated toward electrophilic cleavage via the formation of penta- and hexa-coordinate species. This review mainly consists of the following aspects: (1) a general concept for the activation of the silicon-carbon bond via penta- and hexa-coordinate species, (2) synthetic application of hexa-coordinate organopentafluorosilicates, and (3) development of the H2O2 oxidation of the silicon-carbon bond and its synthetic applications via the intramolecular hydrosilylation, silicon-tethered intramolecular radical cyclization and Diels-Alder reaction, and some silicon-containing organometallic reagents for nucleophilic hydroxymethylation and hydroxyallylation synthons.
Keywords: organosilicon chemistry, hypercoordination, organopentafluorosilicate, hydrogen peroxide oxidation, intramolecular hydrosilylation
Introduction
The author was awarded the Japan Academy Prize 2007, together with Hisashi Yamamoto, University of Chicago, for the collaborative work, entitled “Exploitation of chemical and physical properties of main-group element compounds based on flexibility for high coordination”. The work is based on the following common features of main-group element compounds. Organic compounds R n E of certain main group elements, especially heavy elements of groups 13 and 14, have electron-accepting molecular orbitals and thus the central element E behaves as a Lewis acidic center to accept a ligand L to form hyper-coordinate species R n EL, increasing both the electrophilicity of the ligand L and the nucleophilicity of the group R; the electronic states and steric environments around the central element E can be tuned by appropriate modification to the R and/or L. While Yamamoto’s work is mainly based on the former concept for the enhanced electrophilicity of the ligand L, the author has been interested in the latter concept for the enhanced nucleophilicity of the group R, especially in organosilicon compounds.
A brief comparison of silicon with carbon in four categories is first made to determine key characteristic features of organosilicon compounds using the chart shown in Fig. 1. Among some similarities and differences between them, only several of the most striking features are mentioned as follows. (1) Size and electronegativity: silicon is larger and more electropositive than carbon, as shown in the central part, (2) reactive intermediates such as anion, radical, cation, and divalent species shown on the left side: among them, the trivalent silicocation can hardly be present as a free stable species in solution1) in which the vacant site is occupied by any basic species, suggesting that nucleophilic substitution reactions at silicon generally proceed not by a dissociative mechanism but by an associative mechanism, 2) (3) high-coordinate species shown on the right side: this is the most important difference that while a penta-coordinate species of carbon corresponds to a high energy structure at a transition state of SN2 substitution reactions, penta- and hexa-coordinate silicon species are stable enough to be isolated in many cases, 3), 4) and (4) unsaturated species, catenation and polymeric materials shown on the top and bottom: silicon-containing unsaturated compounds are so reactive that they can only be isolated through stabilization by introduction of bulky kinetically-protecting groups. 5) Obviously, most of these three features (2), (3) and (4) related to silicon-containing molecules are based on the feature (1) of the silicon atom. In particular, the large and electropositive silicon atom is the origin of the Lewis acid properties of organosilicon compounds to accept Lewis bases, resulting in the formation of hypercoordinate species. Since the mid 1970s, we have been interested in these inherent properties of organosilicon compounds with the objective of their application in organic syntheses.
Fig. 1.

A schematic comparison between carbon and silicon. rc and χ represent, respectively, the covalent radius and the electronegativity. Structures in parentheses are still unknown.
This review summarizes several aspects obtained in our laboratories at Kyoto University where the author worked for 35 years, as listed below.
A general concept for the activation of the silicon-carbon bond via penta- and hexa-coordinate species
Synthetic application of hexa-coordinate organopentafluorosilicates
Development of the H2O2 oxidation of the silicon-carbon bond and its synthetic applications
A general concept for the activation of the silicon-carbon bond via penta- and hexa-coordinate species
A key step in the synthetic application of organosilicon compounds is the electrophilic cleavage of the silicon-carbon bond, 6) where an electrophile links to the carbon center to form the desired product, while the counter nucleophile attacks the silicon atom, as shown in Scheme 1.
Scheme 1.

In principle, there are two extreme cases for activation of the silicon-carbon bond, depending on the timing of the interaction of the electrophilic part with the organic group and the nucleophilic part with the silicon center. 7) One is the use of electron-rich organic groups, such as allyl and vinyl groups, and the other involves the formation of penta- and/or hexa-coordinate silicon species.
In the former case, as shown in Scheme 2, electrophiles may first interact with the π electron moiety to form a carbocation β to silicon which is stabilized by the silyl group via the so-called σ–π conjugation, followed by nucleophilic attack on silicon to cleave the silicon-carbon bond, resulting in the formation of the product. 8) Thus, organosilicon compounds such as allylsilanes and vinylsilanes containing an “activated group” toward electrophiles have been well recognized as versatile synthetic reagents.
Scheme 2.

The latter concept has been developed for the activation of the silicon-carbon bond in simple “non-activated” alkyl-silanes via the formation of hyper-coordinate species. The complete view is summarized in Scheme 3. This is the main theme of our research in this field.
Scheme 3.

In Scheme 3, while the route denoted as Route A corresponds to the former case for the activated organic groups discussed above, the route denoted as Route B is for the latter “non-activated” case (R = simple alkyl). In Route B, electronegative groups X are introduced on silicon to enhance the Lewis acidity of the silicon center and form the penta-coordinate silicon species. The resulting penta-coordinate silicon complex has 10 electrons around silicon, being two electrons more than the ordinary 8-electron state. These excess electrons are distributed onto the five ligands to enhance the electron density on the R group and thus the silicon-carbon (Si-R) bond becomes susceptible toward electrophilic cleavage. The silicon center in the penta-coordinate complex is still Lewis acidic to accept another Lewis base to form a hexa-coordinate silicon complex of 12 electrons, in which the silicon-carbon bond is further activated toward electrophilic cleavage. In this way, the “non-activated” silicon-carbon bond may be activated for electrophiles up to a synthetically useful level. We have confirmed this concept by using hexa-coordinate organopentafluorosilicates as an extreme case since 1978. 9)
Synthetic application of hexa-coordinate organopentafluorosilicates
The silicon-carbon bond in hexa-coordinate organopentafluorosilicates K2[RSiF 5 ] can readily be cleaved by a variety of electrophiles or oxidants including halogens, 9), 10) peracid, 11) copper(II) halides, 12) silver(I) halides, 13) and palladium(II) salts, 14) as summarized in Scheme 4. 15) There are several points to be mentioned. (a) While organopentafluorosilicates were first reported in 1961 by L. Tansjoe 16) and some basic reactions were studied by R. Mueller 17) by the end of the 1960s, our work has shown that the silicon-carbon bonds therein are readily cleaved by a variety of electrophiles to give various functionalized products and carbon-carbon bond forming products up to a synthetically useful level. Worthy of note is that these transformations hardly occur with ordinary tetra-coordinate organosilicon compounds, demonstrating that the silicon-carbon bonds in the hexa-coordinate silicates are indeed highly activated toward electrophiles. (b) The organopentafluorosilicates are readily prepared from the corresponding organotrichlorosilanes by addition to an aqueous solution containing a large excess of potassium fluoride as air-stable, insoluble white powders. The most impressive example is the reaction with halogen or N-bromosuccinimide (NBS) to form the corresponding organic halides; even in a suspension of an organic solvent, the solid silicate exothermically reacts with NBS to form the organic bromide in an almost quantitative yield. Since the inorganic byproducts are also insoluble, simple filtration and concentration afford an almost pure organic bromide. Alcohols are also obtained by the reaction with m-chloroperoxybenzoic acid (MCPBA) in a polar solvent such as DMF. (c) Most important from a mechanistic point of view was the different stereochemical outcomes from the halogen or NBS cleavage 18) and the alcohol synthesis 19) with inversion and retention at the carbon center, respectively, as shown in Scheme 5. Thus, the former must involve the back-side attack, while the latter alcohol synthesis must proceed through the front-side attack.
Scheme 4.

Scheme 5.

Plausible mechanisms are shown in Scheme 6. The halogen cleavage may be initiated by a single electron transfer from the electron rich hexa-coordinate silicate to the halogen molecule; the resulting halide ion attacks the carbon atom from the back side to give the organic halide with inversion of stereochemistry. In this connection, the high electron donating ability of the hexa-coordinate organosilicate has been confirmed by the reaction with tetracyanoethylene (TCNE); even in the solid state, deep-blue TCNE anion radicals were rapidly formed. 20) In contrast, the peroxide oxidation reaction in a polar solvent may proceed through a penta-coordinate silicate formed by fluoride ion dissociation, to which the peracid oxygen atom links as the sixth ligand and within the resulting hexa-coordinate silicate, the organic group migrates from silicon to the coordinating peracid oxygen atom to give an alkoxy-silicate and eventually the alcohol upon hydrolysis. The retention of configuration is explained by the intramolecular front side attack.
Scheme 6.

Obviously, these mechanistic considerations in Scheme 6 are traced back to the ordinary tetra-coordinate organosilicon compounds via the reverse route as discussed in Scheme 3, and thus strongly suggest the possibility for the oxidative cleavage of the silicon-carbon bonds in readily available tetra-coordinate silicon compounds under appropriate reaction conditions, if the silicon center is designed to be reasonably Lewis acidic by introduction of an electronegative group(s).
In 1983, with a strong certainty for this possibility, we started to survey the reaction conditions suitable for the oxidative cleavage reaction with hydrogen peroxide as the most readily available, practical oxidizing agent instead of peracids such as MCPBA. 19)
Development of the H2O2 oxidation of the silicon-carbon bond and its synthetic applications
Discovery of the H2O2 oxidation.
Indeed, we soon found that the silicon-carbon bonds are readily cleaved by 30% H2O2 as the oxidant in the presence of a fluoride ion, as shown in Scheme 7. 21), 22) In the first trials, we used 90% H2O2 with a greatest care for the oxidation, but finally found that the more practical 30% H2O2 was quite effective.
Scheme 7.

Several points deserve comment. First, the presence of at least one heteroatom, such as a fluorine and alkoxy and amino groups on the silicon, is essential for the oxidation. Second, the oxidation is highly accelerated by a fluoride ion, which has a strong affinity to silicon, to convert the silicon species to the activated penta-coordinate state. Third, in control experiments, the silicon-carbon bonds in some isolable penta-coordinate diorganotrifluorosilicates were found to be easily cleaved under similar conditions but without an extra fluoride ion, demonstrating the penta-coordinate organosilicates to be actual reactive species. 23) Fourth, it was confirmed that the H2O2 oxidation also proceeds with retention of configuration at the carbon center 21a) . Thus, a plausible mechanism is shown in Scheme 8.
Scheme 8.

It should be noted that this discovery overturned the established common knowledge of organic chemists that silicon-carbon bonds are fairly resistant to oxidative cleavage.
Synthetic application.
With this new oxidation reaction in hand, we have developed a variety of new synthetic methodologies and new reagents, as summarized in Eqs. 1–16. Some characteristic features deserve more comment.
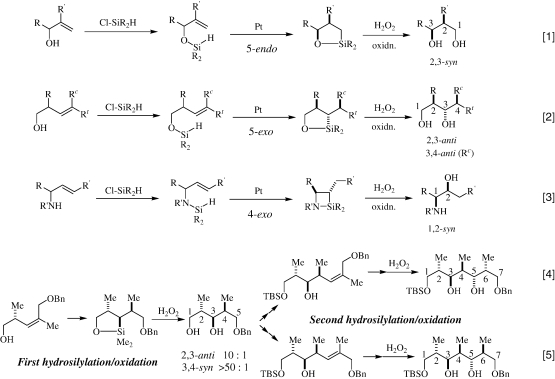 |
Intramolecular hydrosilylation.
The intramolecular hydrosilylation of allyl alcohols and homoallyl alcohols followed by the H2O2 oxidation has provided a new methodology for the regio- and stereo-selective synthesis of 1,3-diol skeletons. 24) In a typical example shown in Eq. 1, 24b)–24d) the hydroxyl group of the 3-hydroxy-2-methyl-1-alkene skeleton is protected by a hydrosilyl group. Subsequent Pt-catalyzed hydrosilylation proceeds in a 5- endo mode to give a five-membered ring product with a high 2,3- syn stereoselectivity. Since the silicon moiety has the oxygen functionality, the resulting silicon-carbon bond is cleaved by the H2O2 oxidation with retention of configuration to form the corresponding 1,3-diol derivative with a high 2,3- syn stereoselectivity.
For homoallyl alcohols, such as the 1-hydroxy-2-methyl-3-alkenes (Eq. 2), 24b) they afford different stereoisomers of 1,3-diol; thus, the intramolecular hydrosilylation proceeds in a 5-exo mode to form a five-membered ring product, in which the 2,3-stereochemistry arising from the entering silyl group is controlled anti to the allylic methyl group and the stereochemistry on the 4-position depends on the olefin geometry of the starting material. These results observed with homoallylic alcohols have also provided two significant aspects from a mechanistic viewpoint: the first example of the direct hydrosilylation to an internal olefin without positional isomerization and the first clear-cut experimental evidence for the cis addition of the Si-H functionality to an olefin.
For the Pt-catalyzed hydrosilylation of allylamines, it proceeds in a 4-exo fashion to finally give a 1,2-aminoalcohol derivative with a high 1,2-syn-stereoselcvitiy, as shown in Eq. 3. 25)
The intramolecular hydrosilylation/oxidation sequence can be repeatedly performed in a stepwise fashion to construct polyols, such as polypropionate skeletons, as shown in Eqs. 4 and 5. 24b)
The catalytic asymmetric intramolecular hydrosilylation of an allylic alcohol is also possible to give an optically active 1,3-diol with a high stereoselectivity (Eq. 6). 26) The intramolecular hydrosilylation of homopropargylic alcohols followed by the H2O2 oxidation provides a new regioselective functionalization of the acetylene group (Eq. 7). 27)
Silicon-tethered intramolecular radical cyclization and Diels-Alder reaction.
The 1-bromovinylsilyl and dichloromethylsilyl groups have been found to be useful as a synthetic equivalent, respectively, to the acetyl radical and the hydroxymethylene diradical, as exemplified by the silicon-tethered radical cyclization/oxidation sequence of allylic alcohols, as shown in Eqs. 8 28) and 9. 29) The latter example demonstrates that the high regio- and stereoselective stepwise radical cyclization can provide four new chiral centers from one chiral center, accompanied by a new carbon-ring annulation.
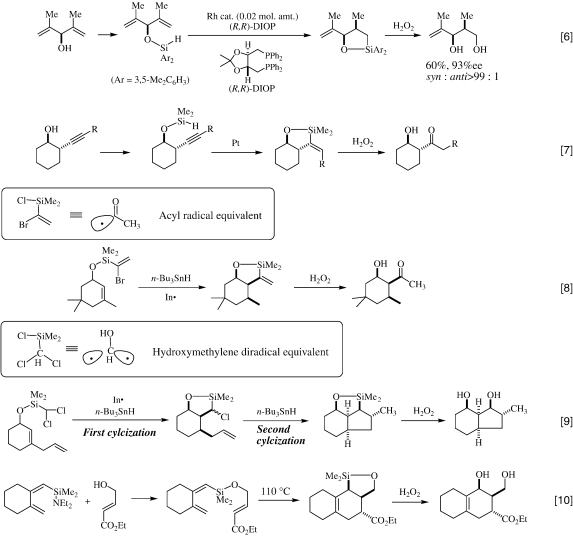 |
Eq. 10 represents the first example of the silicon-tethered intramolecular Diels-Alder reaction, followed by the H2O2 oxidation, as an efficient method for the construction of poly-functionalized cyclohexane derivatives. 30)
Silicon-containing organometallic reagents for nucleophilic hydroxymethylation and hydroxyallylation and as a hydroxide ion equivalent.
One of the interesting silicon reagents is the (isopropoxy)silylmethyl Grignard reagent (i-PrO)Me2SiCH2MgCl, 31) because this reagent contains both a carbon nucleophilic center and a leaving group on silicon, but is quite stable as it can be stored in THF even at room temperature; the isopropoxy group is probably bulky enough to prevent the intermolecular nucleophilic substitution at the silicon. This reagent is used as the nucleophilic hydroxymethylating agent for carbonyl compounds, as shown in Eq. 11.31) The primary product contains a β-hydroxy-silicon moiety which might undergo β-elimination, known as the Peterson elimination, 32) to form the corresponding olefin; however, under the H2O2 oxidation conditions even in the presence of fluoride ions, the oxidative cleavage of the silicon-carbon bond preferentially proceeds, without any such elimination, to form the corresponding 1,2-diol derivative in high yields. In connection with this, it has been reported that the conjugate addition of a hydroxymethyl anion synthon, namely the (allyldimethylsilyl)methyl Grignard reagent, to α,β-enones has also been achieved. 33)
This reagent also undergoes a palladium catalyzed cross-coupling with aromatic halides, such as 3-bromothiophene, to give, after the H2O2 oxidation, the hydroxymethylation products, as shown in Eq. 12. 31) The copper-catalyzed cross-coupling reaction with allylic halides, followed by the H2O2 oxidation, affords the corresponding homoallyl alcohols, as shown by Eqs. 13 and 14. This approach significantly provides the most efficient, regio- and stereo-specific transformation of allylic halides to homoallylic alcohols without scrambling of the olefin geometry and the allylic position, as well as the one-carbon elongation of certain functionalized alcohols. 34)
A metallated (allyl)aminosilane is a practical reagent for the stereoselective α-hydroxyallylation of aldehydes to form erythro-1,2-diol skeletons, as shown in Eq. 15. 35)
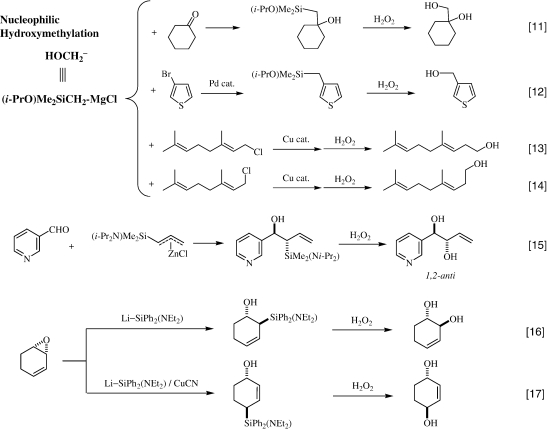 |
Aminosilyllithium reagents, the first stable functionalized silyllithiums which we found in 1992, 36) act as a hydroxide ion synthon, as shown in Eqs. 16 and 17. 37)
Conclusion
In this review, we have concentrated on our own work, but our concept about the activation of the silicon-carbon bond via hypercoordination and the H2O2 oxidative cleavage of the silicon-carbon bond have been widely recognized as a new general concept and a new synthetically useful transformation.
The first point has clearly been described by R. Corriu and his co-workers in their review article 3) as follows: “This was the first demonstration of the synthetic applications of hypercoordinate organosilicon compounds and stimulated other chemists to use the same idea that the silicon-carbon bonds are highly activated by hypercoordination. This idea has now become a widely accepted basic concept in synthetic organic chemistry.”
The second point, i.e., the synthetic usefulness of the H2O2 oxidation of the silicon-carbon bond, was recognized by many synthetic chemists soon after the discovery by our group, together with a similar oxidative cleavage reaction independently developed by I. Fleming. 38) We are proud of the fact that the oxidation reaction is now generally known as the “Tamao oxidation” and/or “Tamao-Fleming oxidation”. As reference data, the citation number of our first paper published in 1983 has reached 300 times by the end of 2007.
All results described in this review are rather old, but the author is convinced that the concept and the information are still very informative for synthetic organic chemists.
Acknowledgements
The research described in this review was performed at the Department of Synthetic Chemistry, Faculty of Engineering, Kyoto University. I would like to express my sincere thanks to all the former co-workers whose names appear in the references.
Profile
Kohei Tamao received his B. Eng., M. Eng., and Ph.D. of Eng. from Kyoto University under the direction of Professor Makoto Kumada in 1961, 1967, and 1971, respectively. He became an Assistant Professor at Kyoto University in 1970 and was promoted to Associate Professor in 1987, and then to Professor in 1993 at Institute for Chemical Research, Kyoto University. He served as Director of the Institute for two years from 2000. In 2005, he was awarded the title of Professor Emeritus at Kyoto University. He is the present Director of the RIKEN Advanced Science Institute. He received the Chemical Society of Japan Award in 1999, The American Chemical Society, Frederic Stanley Kipping Award in 2002, The Toray Science & Technology Prize in 2002, The Asahi Prize in 2003, Medal of Honor with Purple Ribbon in 2000, and The Japan Academy Prize in 2007. He served as a research leader of the Kyoto University COE (Center of Excellence) “Elements Science” supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan (2000–2004). His current research interests are organosilicon chemistry and related elemento-organic chemistry, σ- and π-conjugated electronic materials, reactive intermediates, and transition metal catalysis.

References
- 1).Kim, K.-C., Reed, C. A., Elliott, D. W., Mueller, L. J., Tham, F., Lin, L. and Lambert, J. B. (2002) Crystallographic Evidence for a Free Silylium Ion. Science 297, 825–827 [DOI] [PubMed] [Google Scholar]
- 2).Holmes, R. R. (1990) The Stereochemistry of Nucleophilic Substitution at Tetracoordinated Silicon. Chem. Rev. 90, 17–31 [Google Scholar]
- 3).Chuit, C. C., Corriu, R. J. P., Reye, C. and Young, J. C. (1993) Reactivity of Penta- and Hexa-coordinate Silicon Compounds and Their Role as Reactive Intermediates. Chem. Rev. 93, 1371–1448 [Google Scholar]
- 4).Tamao, K., Hayashi, T., Ito, Y. and Shiro, M. (1992) Electronic and Steric Effects in Anionic Pentacoordinate Diorganotrifluorosilicates: X-ray Structures and 13C NMR Studies for Evaluation of Charge Distribution in Aryl Groups on Silicon. Organometallics 11, 182–191 [Google Scholar]
- 5).Okazaki, R. and West, R. (1996) Chemistry of Stable Disilenes. Adv. Organomet. Chem. 39, 231–274 [Google Scholar]
- 6).Colvin, E. (1981) Silicon in Organic Synthesis. Butterworths, London [Google Scholar]
- 7).Tamao, K. (1985) Silafunctional Compounds in Organic Synthesis. In Organosilicon and Bioorganosilicon Chemsitry (ed. Sakurai, H.). Ellis Horwood, Chichester, UK, pp. 231–242
- 8).a) Hosomi, A. (1988) Characteristics in the Reactions of Allylsilanes and Their Applications to Versatile Synthetic Equivalents. Acc. Chem. Res. 21, 200–206; [Google Scholar]; b) Fleming, I., Dunogues, J. and Smithers, R. (1989) The Electrophilic Substitution of Allylsilanes and Vinylsilanes. Org. React. 37, 57–575; [Google Scholar]; c) Fleming, I., Barbero, A. and Walter, D. (1997) Stereochemical Control in Organic Synthesis Using Silicon-Containing Compounds. Chem. Rev. 97, 2063–2192 [DOI] [PubMed] [Google Scholar]
- 9).Tamao, K., Yoshida, J., Takahashi, M., Yamamoto, H., Kakui, T., Matsumoto, H., Kurita, A. and Kumada, M. (1978) A Novel General and Practical Method for Anti-Markownikoff Hydrohalogenation of Olefins via Organopentafluorosilicates Derived from Hydrosilylation Products. J. Am. Chem. Soc. 100, 290–291 [Google Scholar]
- 10).a) Tamao, K., Yoshida, J., Murata, M. and Kumada, M. (1980) Stereochemistry at Carbon in Cleavage of the Carbon-Silicon Bond in exo- and endo-2-Norbornylpentafluorosilicates by Various Brominating Agents. J. Am. Chem. Soc. 102, 3267–3269; [Google Scholar]; b) Tamao, K., Yoshida, J., Yamamoto, H., Kakui, T., Matsumoto, H., Takahashi, M., Kurita, A., Murata, M. and Kumada, M. (1982) Preparation of Organopentafluorosilicates and Their Cleavage Reactions by Halogens and N-Bromosuccinimide. Synthetic and Mechanistic Aspects. Organometallics 1, 355–368 [Google Scholar]
- 11).Tamao, K., Kakui, T. and Kumada, M. (1978) A Convenient Procedure for Preparing Primary Alcohols from Olefins. A Novel Facile Oxidative Cleavage of Carbon-Silicon Bonds by m-Chloroperoxybenzoic Acid. J. Am. Chem. Soc. 100, 2268–2269 [Google Scholar]
- 12).Yoshida, J., Tamao, K., Kakui, T., Kurita, A., Murata, M., Yamada, K. and Kumada, M. (1882) Copper(II) Oxidation of Organopentafluorosilicates. Organometallics 1, 369–380 [Google Scholar]
- 13).Tamao, K., Matsumoto, H., Kakui, T. and Kumada, M. (1979) A Stereoselective Synthesis of Symmetical (E,E)-1,3-Dienes by Silver(I)-Promoted Homo Coupling of (E)-Alkenylpentafluorosilicates. Tetrahedron Lett. 1137–1140
- 14).Yoshida, J., Tamao, K., Yamamoto, H., Kakui, T., Uchida, T. and Kumada, M. (1982) Organoflurosilicates in Organic Synthesis. Carbon-Carbon Bond Formation Promoted by Palladium Salts. Organometallics 1, 542–549 [Google Scholar]
- 15).Kumada, M., Tamao, K. and Yoshida, J. (1982) Chemistry of Organopentafluorosilicates. J. Organomet. Chem. 239, 115–132 [Google Scholar]
- 16).Tansjoe, L. (1961) On the Reactions of N-substituted Alkyltriaminosilanes. V. Acta Chem. Scand. 15, 1583–1594 [Google Scholar]
- 17).Mueller, R. (1966) Uber Organofluorosilicate und Ihre Verwendung zur Darstellung Metallorganischer Verbindungen. Organometal. Chem. Rev. 1, 359–377 [Google Scholar]
- 18).Tamao, K., Yoshida, J., Murata, M. and Kumada, M. (1980) Stereochemistry at Carbon in Cleavage of the Carbon-Silicon Bond in exo- and endo-2-Norbornylpentafluorosilicates by Various Brominating Agents. J. Am. Chem. Soc. 102, 3267–3268 [Google Scholar]
- 19).Tamao, K., Kakui, T., Akita, M., Iwahara, T., Kanatani, R., Yoshida, J. and Kumada, M. (1983) Oxidative Cleavage of the Silicon-Carbon Bonds in Organosilicon Fluorides to Alcohols. Tetrahedron 39, 983–990 [Google Scholar]
- 20).Yoshida, J., Tamao, K., Kumada, M. and Kawamura, T. (1980) Alkylation of TCNE with Orgnopentafluorosilicates: Implication of One-Electron Transfer Mechanism. J. Am. Chem. Soc. 102, 3269–3270 [Google Scholar]
- 21).a) Tamao, K., Ishida, N., Tanaka, T. and Kumada, M. (1983) Hydrogen Peroxide Oxidation of the Silicon-Carbon Bond in Organoalkoxysilanes. Organometallics 2, 1694–1696; [Google Scholar]; b) Tamao, K. and Ishida, N. (1984) Silyl Groups Synthetically Equivalent to the Hydroxy Group. J. Organomet. Chem. 269, C37–C39; [Google Scholar]; c) Tamao, K., Kumada, M. and Maeda, K. (1984) Hydrogen Peroxide Oxidation of Alkenyl(alkoxy)silanes. Tetrahedron Lett. 25, 321–324 [Google Scholar]
- 22).a) Tamao, K. (1996) Oxidative Cleavage of the Silicon-Carbon Bond: Development, Mechanism, Scope and Limitations. In Advances in Silicon Chemistry (ed. Larson, G. L.). Vol. 3, Jai Press, 1–62; ; b) Jones, G. R. and Landais, Y. (1996) The Oxidation of the Carbon-Silicon Bond. Tetrahedron 52, 7599–7662 [Google Scholar]
- 23).Tamao, K., Hayashi, T. and Ito, Y. (1991) Hydrogen Peroxide Oxidation of the Silicon-Carbon Bond: Mechanistic Studies. In Frontiers of Organosilicon Chemistry (eds. Bassindale, S. R. and Gasper, P. P.). The Royal Society of Chemistry, UK, pp. 197–207 [Google Scholar]
- 24).a) Tamao, K., Tanaka, T., Nakajima, T., Sumiya, R., Arai, H. and Ito, Y. (1986) Intramolecular Hydrosilation of Alkenyl Alcohols: A New Approach to the Regioselective Synthesis of 1,2- and 1,3-Diols. Tetrahedron Lett. 27, 3377–3380; [Google Scholar]; b) Tamao, K., Nakajima, T., Sumiya, R., Arai, H., Higuchi, N. and Ito, Y. (1986) Stereocontrol in Intramolecular Hydrosilation of Allyl and Homoallyl Alcohols: A New Approach to the Stereoselective Synthesis of 1,3-Diol Skeletons. J. Am. Chem. Soc. 108, 6090–6091; [DOI] [PubMed] [Google Scholar]; c) Tamao, K., Nakagawa, Y., Arai, H., Higuchi, N. and Ito, Y. (1988) Intramolecular Hydrosilation of α-Hydroxy Enol Ethers: A New Highly Stereoselective Route to Polyhydroxylated Molecules. J. Am. Chem. Soc. 110, 3712–3713; [Google Scholar]; d) Tamao, K., Nakagawa, Y. and Ito, Y. (1995) Regio- and Stereoselective Intramolecular Hydrosilation of α-Hydroxy Enol Ethers: 2,3-syn-2-Methoxymethoxy-1,3-nonanediol. Org. Synth. 73, 94–109 [Google Scholar]
- 25).Tamao, K., Nakagawa, Y. and Ito, Y. (1990) Platinum-Catalyzed Intramolecular Hydrosilation of Allylamines: Formation of 1-Aza-2-silacyclobutanes and Application to Stereoselective Synthesis of 2-Amino Alcohols. J. Org. Chem. 55, 3438 [Google Scholar]
- 26).Tamao, K., Tohma, T., Inui, N., Nakayama, O. and Ito, Y. (1990) Catalytic Asymmetric Intramolecular Hydrosilation. Tetrahedron Lett. 31, 7333–7336 [Google Scholar]
- 27).Tamao, K., Maeda, K., Tanaka, T. and Ito, Y. (1988) Intramolecular Hydrosilation of Acetylenes: Regioselective Functionalization of Homopropargyl Alcohols. Tetrahedron Lett. 29, 6955–6958 [Google Scholar]
- 28).Tamao, K., Maeda, K., Yamaguchi, T. and Ito, Y. (1989) 1-Silylvinyl Radical Cyclization: Silicon-Mediated Regio- and Stereo-Selective Hydroacylation and Hydrovinylation of Allyl Alcohols. J. Am. Chem. Soc. 111, 4984–4985 [Google Scholar]
- 29).Tamao, K., Nagata, K., Ito, Y., Maeda, K. and Shiro, M. (1994) (Dichloromethyl)dimethylsilyl Group as a Hydroxymethylidene Diradical Equivalent: Radical Annulation of Dienols and Its Application to the Stereoselective Synthesis of cis- and trans-Hydrindan Ring Systems. Synlett, 257–259
- 30).Tamao, K., Kobayashi, K. and Ito, Y. (1989) Nickel(0)-Catalyzed Cyclization of 1,7-Diynes via Hydrosilation: One-Step Synthesis of 1,2-Dialkylidene-cyclohexanes with a Z-Vinylsilane Moiety. J. Am. Chem. Soc. 111, 6478–6479 [Google Scholar]
- 31).a) Tamao, K., Ishida, N. and Kumada, M. (1983) (Diisopropoxymethylsilyl)methyl Grignard Reagents: A New, Practically Useful Nucleophilic Hydroxymethylating Agent. J. Org. Chem. 48, 2120; [Google Scholar]; b) Tamao, K. and Ishida, N. (1984) (Isopropoxydimethylsilyl)methyl Grignard Reagent: A New Nucleophilic Hydroxymethylating Agent for Aldehydes and Ketones. Tetrahedron Lett. 25, 4245–4248; [Google Scholar]; c) Tamao, K., Ishida, N., Ito, Y. and Kumada, M. (1990) Nucleophilic Hydroxymethylation of Carbonyl Compounds: 1-(Hydroxymethyl)cyclohexanol. Org. Synth. 69, 96–105 [Google Scholar]
- 32).Ager, D. J. (1990) The Peterson Olefination Reaction. Org. React. 38, 1–223 [Google Scholar]
- 33).Tamao, K. and Ishida, N. (1984) Conjugate Addition of a Hydroxymethyl Anion Synthon to α,β-Enones. Tetrahedron Lett. 25, 4249–4252 [Google Scholar]
- 34).Tius, M. A. and Fauq, A. (1986) Total Synthesis of (–)-asperdiol. J. Am. Chem. Soc. 108, 6389–6391 [Google Scholar]
- 35).Tamao, K., Nakajo, E. and Ito, Y. (1987) Metalated (Allyl)aminosilanes: A New, Practical Reagent for the Stereoselective α-Hydroxyallylation of Aldehydes to erythro-1,2-Diol Skeletons. J. Org. Chem. 52, 957–968 [Google Scholar]
- 36).a) Tamao, K., Kawachi, A. and Ito, Y. (1992) The First Stable Functional Silyl Anions: (Aminosilyl)lithiums. J. Am. Chem. Soc. 114, 3989–3990; [Google Scholar]; b) Kawachi, A. and Tamao, K. (1997) Preparations and Reactions of the Functionalized Silylithiums. Bull. Chem. Soc. Jpn. 70, 945–955 [Google Scholar]
- 37).Tamao, K., Kawachi, A., Tanaka, Y., Ohtani, H. and Ito, Y. (1996) Synthetic Applications of Functionalized Silyl Anions: Aminosilyl Anions as Hydroxy Anion Equivalent. Tetrahedron 52, 5765–5772 [Google Scholar]
- 38).a) Fleming, I., Henning, R., Parker, D. C., Plaut, H. E. and Sanderson, P. E. (1995) The Phenyldimethylsilyl Group as a Masked Hydroxy Group. J. Chem. Soc., Perkin Trans. 1, 317–337; [Google Scholar]; b) Fleming, I. (1996) Silyl-to-Hydroxy Conversion in Organic Synthesis. Chemtracts, Org. Chem. 9, 1–64 [Google Scholar]