

α-Galactosidase from S. cerevisiae has been purified and crystallized in glycosylated and deglycosylated states. X-ray diffraction data were collected to 1.95 Å resolution from the deglycosylated form.
Keywords: yeast, α-galactosidases, melibiase, glycoside hydrolase family 27, glycosylation
Abstract
Saccharomyces cerevisiae α-galactosidase is a highly glycosylated extracellular protein that catalyzes the hydrolysis of α-galactosidic linkages in various glucids. Its enzymatic activity is of interest in many food-related industries and has biotechnological applications. Glycosylated and in vitro deglycosylated protein samples were both assayed for crystallization, but only the latter gave good-quality crystals that were suitable for X-ray crystallography. The crystals belonged to space group P4212, with unit-cell parameters a = b = 101.24, c = 111.52 Å. A complete diffraction data set was collected to 1.95 Å resolution using a synchrotron source.
1. Introduction
α-Galactosidases (EC 3.2.1.22) catalyse the hydrolysis of α-d-galactosidic linkages present in galacto-oligosaccharides and polymeric galactomannans as well as in glucids such as stachyose, raffinose and melibiose. Transglycosylation activity has also been observed in some α-galactosidases. These enzymes are widely distributed in animals, plants and microorganisms and exhibit different substrate specificities and biochemical characteristics (Dey & Pridham, 1972 ▶). α-Galactosidase activity has biotechnological applications in the beet industry in the hydrolysis of raffinose and other galacto-oligosaccharides and in the processing of soybean and other legume products. Organisms expressing α-galactosidase, or the purified enzyme, are also used for biomass and ethanol production coupled to the degradation of molasses. The enzyme is also used as a nutritional supplement in human diets for the treatment of gastric disorders arising from the utilization of galacto-oligosaccharides by the intestinal microflora. α-Galactosidase is also added to animal feed in order to maximize the energetic conversion of galacto-oligosaccharides by monogastric animals (Sakuraba et al., 2006 ▶; Murphy & Power, 2002 ▶).
Saccharomyces cerevisiae α-galactosidase (ScAGal) is classified into family 27 of the glycosyl hydrolases (Cantarel et al., 2009 ▶) with other α-galactosidases, α-N-acetylgalactosaminidases (EC 3.2.1.49), isomalto-dextranases (EC 3.2.1.94) and β-l-arabinopyranosidase (EC 3.2.1.88). Several α-galactosidases from bacteria are classified into family 36 and both families (together with family 31) are included in clan GH-D. The three-dimensional structures of α-galactosidases from Thermotoga maritima (glycosyl hydrolase family 36; PDB code 1zy9; Joint Center for Structural Genomics, unpublished work), Homo sapiens (Garman & Garboczi, 2004 ▶), Trichoderma reseei (Golubev et al., 2004 ▶) and Oryza sativa (Fujimoto et al., 2003 ▶), and the α-N-acetylgalactosaminidase from Gallus gallus (Garman et al., 2002 ▶) have been solved by X-ray crystallography. These proteins display an (α/β)8 barrel as their main structural motif and catalyze hydrolysis by a double-displacement mechanism with retention of the anomeric configuration of the substrate. All the proteins classified as belonging to family 27 by sequence similarity and biochemical evidence are proposed to have similar folds to those reported for the known structures. However, there are some features regarding substrate recognition and specificity, oligomeric state and protein stability that remain unexplained. To our knowledge, no structural information is available for yeast α-galactosidase. Study of the three-dimensional structure of this enzyme is essential in order to elucidate its catalytic mechanism and to fully understand the processes responsible for substrate recognition and specificity. Resolution of the atomic structure of ScAGal will open new perspectives for biotechnological applications and protein engineering. Here, we report the purification, crystallization and preliminary X-ray diffraction data of ScAGal.
2. Materials and methods
2.1. Cloning, expression and purification
The MEL1 gene (P04824) encoding an α-galactosidase from S. cerevisiae was amplified by the polymerase chain reaction from the pMelα vector (ampr ori CEN6/ARSH4 MEL1 URA3; Melcher et al., 2000 ▶) and cloned into the YEpFLAG plasmid (ampr ori 2m FLAG TRP1; Eastman Kodak Company) by homologous recombination in yeast strain BJ3505 (pep4::HIS3, prb-Δ1.6R HIS3, lys2-208, trp1-Δ101, ura 3–52, gal2, can1; Eastman Kodak Company). The forward primer 5′-CTATATCGTAATACACCAAGCTCGACCTCGCGATGTTTGCTTTCTACTTTCTCA-3′ and reverse primer 5′-GGTCGT ACGGGCCCGGATCCATCGATAGATCCAATCATCAATACTTCTCGCC-3′, used for PCR amplification of the gene, carried 30 nucleotides (shown in bold) of the specific sequence required for homologous recombination with the vector YEpFLAG.
The final construct was expressed in S. cerevisiae BJ3505 cells (Eastman Kodak Company) grown in 1 l YPHSM medium (1% glucose, 3% glycerol, 1% yeast extract and 8% peptone) at 303 K and 250 rev min−1 for 96 h in a 2 l Erlenmeyer flask. Cells were collected by centrifugation (7000 rev min−1 for 10 min at 277 K) and the extracellular protein in the supernatant was concentrated with the Millipore TFF system and then purified with ANTI-FLAG M2 affinity resin (Sigma) packed in 10 ml chromatography columns (Bio-Rad 731-1550). Elution was performed by competition with FLAG peptide following the manufacturer’s recommendations. After elution, 2 mM DTT was added to the sample. Purified protein in 0.02 M Tris–HCl, 0.150 M NaCl, 2 mM DTT was concentrated to 10 mg ml−1 by ultrafiltration with Amicon Ultra-4 (Millipore, UFC 803024). The homogeneity of the purified protein sample was confirmed by SDS–PAGE (Laemmli, 1970 ▶).
2.2. Deglycosylation
After protein purification, deglycosylation was performed using endoglycosidase H (Endo H; New England Biolabs). Protein was required in its native form; therefore, the denaturation step required for total deglycosylation was avoided. Endo H treatment was accomplished in 6 h following the manufacturer’s recommendations. An extra purification step with ANTI-FLAG M2 affinity resin (described above) was performed after deglycosylation in order to eliminate Endo H contamination of the protein sample. The deglycosylated protein was concentrated to 2.5 mg ml−1 by ultrafiltration using Amicon Ultra-4 (Millipore, UFC 803024).
2.3. Crystallization
Initially, crystallization conditions were explored using high-throughput techniques with a NanoDrop robot (Innovadyne Nanodrop I) and commercially available screens. Crystal Screen, Crystal Screen II, Crystal Screen Lite, SaltRx and Index Screen from Hampton Research and Screen Classic from Jena Biosciences were assayed using the sitting-drop vapour-diffusion method at 291 K. Drops consisting of 0.25 µl precipitant solution and 0.25 µl pure ScAGal (6 mg ml−1 in 0.05 M Tris–HCl, 0.150 M NaCl and 0.002 M DTT) were equilibrated against 60 µl reservoir solution in sitting-drop microplates (Innovaplate SD-2). Initial hits were then tested on Cryschem (Hampton Research) sitting-drop plates by mixing 1 µl protein solution with 1 µl precipitant solution and equilibrating against 500 µl reservoir solution. Crystallization trials in the presence of agarose were performed by adding 0.2% agarose to the drop. This was performed using a preheated stock of 0.4% agarose in mother-liquor solution. The screening of crystallization conditions for the deglycosylated sample of ScAGal (2.5 mg ml−1 in 0.05 M Tris–HCl, 0.150 M NaCl and 0.002 M DTT) was performed with the PACT Suite and JCSG+ Suite from Qiagen following the same strategy as before. Crystals grew in several conditions with polyethylene glycol (PEG) 3350 as the main precipitant agent. These conditions were then optimized on Cryschem plates and good-quality crystals with a polyhedrical shape grew in 18–20%(w/v) PEG 3350, 0.1 M bis-tris propane pH 8.5, 0.2 M KSCN. Crystals reached maximum dimensions of approximately 200 × 150 × 150 µm within two weeks.
2.4. X-ray data collection and processing
All crystals were cryoprotected before being cooled to 100 K in liquid nitrogen. The drop solution was substituted by a cryoprotectant solution consisting of mother liquor plus 20%(v/v) glycerol. Diffraction data were collected using synchrotron radiation on beamline ID23-1 at the European Synchrotron Radiation Facility (ESRF, Grenoble) using an ADSC Quantum Q315r detector at a crystal-to-detector distance of 283.5 mm and a wavelength of 0.9792 Å. The exposure time was set to 0.9 s and the oscillation range was 1° per image. The collected diffraction data were processed with MOSFLM (Leslie, 1992 ▶) and scaled using the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
The preliminary search for crystallization conditions of ScAGal only gave phase separation in conditions where PEG was the main precipitant agent and gave clear drops in almost all other conditions. Small plate-shaped crystals or needles grew within two weeks in two drops with low-molecular-weight PEGs and low pH. Based on these initial conditions, we designed more than a thousand crystallization experiments in order to explore different variables individually. We tested different concentrations of low- and medium-molecular-weight PEGs as well as the effect of pH, salt type and concentration, buffer composition and experimental setup. Optimized medium plate-shaped crystals grew in 22–24% PEG 1000, 0.1 M citrate buffer pH 4.5, 0.2 M Li2SO4. We tested the diffraction patterns of more than 40 crystals, but most of them were twinned and showed weak and highly anisotropic diffraction. In an attempt to improve the crystal quality, we added 0.2% agarose to the crystallization drops. This experimental setup slowed crystal growth, leading to more ordered crystals with stronger diffraction patterns than those obtained previously, but they still showed twinning (Fig. 1 ▶).
Figure 1.

The best crystals obtained with the glycosylated sample of ScAGal. The crystals in (b) were grown in the presence of 2% agarose within the drop.
ScAGal is a highly glycosylated extracellular protein. Post-translational modifications in the secretion pathway lead to a protein in which carbohydrates represent 30–40% of the final molecular weight. These modifications usually make protein samples rather heterogeneous and exacerbate the process of crystallization and structural analysis. One way to overcome this crystallization bottleneck is to reduce the N-glycans to single residues by endoglycosydase-H (Endo H) treatment (Lehle et al., 2006 ▶). Endo H cleaves the oligosaccharide moieties, leaving single N-acetylgalactosamine residues at each glycosylation site under ideal experimental conditions (the unfolded state). Since the protein has to be in a native folded state for crystallization purposes, there are some poorly accessible glycosylation sites that remain only partially deglycosylated; nevertheless, this enzymatic treatment significantly reduced the heterogeneity of the sample without affecting the protein folding. We performed initial screening of crystallization conditions for the deglycosylated sample using the commercially available screens PACT suite and JCSG+ suite and with the same plates, drop size and temperature as used previously. Crystals grew in almost every condition of the PACT screen and the crystal morphology varied from plate-shaped crystals to cubes and pyramids. The best crystals were very small and grew with PEG 3350 as the main precipitant agent. The suitable pH ranged from 6.5 to 8.5, but the best crystals grew at pH values between 7.5 and 8.5. After initial optimization of the conditions, visually good crystals were obtained in 18–20% PEG 3550, 0.1 M bis-tris propane pH 7.5–8.5 and 0.2 M KSCN. Although Endo H treatment was decisive in obtaining good-quality crystals of ScAGal, the pH value was also essential to obtain crystals that diffracted well. As shown in Figs. 2 ▶ and 3 ▶, crystals obtained at pH 7.5 presented a highly anisotropic diffraction pattern, while crystals grown above pH 8 were untwinned and their diffraction patterns were of high quality. It is interesting to note that crystals obtained at pH 7.5 were only useful after annealing treatment (Bunick et al., 1998 ▶). The diffraction data obtained after annealing of crystals grown at pH 7.5 were indexed in space group P422 with four molecules in the asymmetric unit and the maximum observed resolution was 3.0 Å; therefore, further analysis was not pursued.
Figure 2.

(a) Crystals of deglycosylated ScAGal grown at pH 7.5. X-ray diffraction pattern from these crystals are shown before (b) and after (c) annealing for 1 s.
Figure 3.

(a) Crystal of deglycosylated ScAGal grown at pH 8.5. Its size is approximately 0.2 × 0.2 × 0.15 mm. (b) X-ray diffraction pattern obtained from this crystal using a synchrotron source. The resolution limit is 2.0 Å (1.5 Å at the edge of the detector).
A full diffraction data set of 180° was collected to a resolution of 1.9 Å on the ESRF ID23.1 beamline from a single crystal grown at pH 8.5 (Fig. 3 ▶). Data-collection statistics are summarized in Table 1 ▶. The unit-cell parameters were a = b = 101.24, c = 111.52 Å. Data processing showed that the crystal belonged to the tetragonal crystal system and analysis after scaling of systematic absences in reflection intensities along h00, 0k0 and 00l indicated that the space group was P4212. Assuming that the protein was deglycosylated and a theoretical molecular weight of the enzyme of 52 kDa, the content of the asymmetric unit was estimated. Only one molecule could fit, with a Matthews coefficient (Matthews, 1968 ▶) of 2.75 Å3 Da−1 and a solvent content of 55.27%. Structural solution was attempted using rice α-galactosidase (PDB code 1uas; Fujimoto et al., 2003 ▶), which shares 38% sequence identity, as a model for molecular replacement using the MOLREP program (Vagin & Teplyakov, 1997 ▶). A single solution containing one molecule in the asymmetric unit was found using reflections within the resolution range 74.96–2.12 Å and a Patterson radius of 40 Å, which after rigid-body fitting led to an R factor of 0.50. Refinement of the ScAGal structural model is currently in progress.
Table 1. Data-collection statistics.
Values in parentheses are for the high-resolution shell.
| Crystal data | |
| Space group | P4212 |
| Unit-cell parameters | |
| a (Å) | 101.25 |
| b (Å) | 101.25 |
| c (Å) | 111.52 |
| Data collection | |
| Beamline | ID23.1 (ESRF) |
| Temperature (K) | 100 |
| Wavelength (Å) | 0.979 |
| Resolution (Å) | 74.96–1.95 (2.06–1.95) |
| Data processing | |
| Total reflections | 455518 (73906) |
| Unique reflections | 42919 (6140) |
| Multiplicity | 10.6 (12.0) |
| Completeness (%) | 100.0 (100.0) |
| I/σ(I) | 7.6 (1.7) |
| Mean I/σ(I) | 33.3 (7.1) |
| Rmerge† (%) | 6.7 (45.7) |
| Rp.i.m.‡ (%) | 2.2 (13.7) |
| Molecules per ASU | 1 |
| Matthews coefficient (Å3 Da−1) | 2.75 |
| Solvent content (%) | 55.27 |
R
merge = 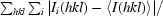
 , where I
i(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements.
, where I
i(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements.
R
p.i.m. = 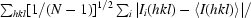 , where N is the redundancy of reflection hkl.
, where N is the redundancy of reflection hkl.
Acknowledgments
RFL received a FPU fellowship from Ministerio de Educación y Ciencia. APR received a María Barbeito fellowship from Xunta de Galicia. Research at Universidade da Coruña was supported by grant 07TAL010103PR from Xunta de Galicia co-financed by FEDER (CEE). General support to the laboratory during 2008–2009 was funded by Programa de axudas para a consolidación e a estruturación de unidades de investigación competitivas da Consellería de Educación e Ordenación Universitaria (Xunta de Galicia). Research at Instituto de Química-Física Rocasolano was supported by grant BIO2007-67708-C04-04 from Dirección General de Investigación. This is a product of the project ‘Factoría de Cristalización’ Ingenio/Consolider 2010.
References
- Bunick, G., Harp, J., Timm, D. & Hanson, L. (1998). Rigaku J.15, 6–13.
- Cantarel, B., Coutinho, P., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res.37, D233–D238. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dey, P. & Pridham, J. (1972). Adv. Enzymol. Relat. Areas Mol. Biol.36, 91–130. [DOI] [PubMed]
- Fujimoto, Z., Kaneko, S., Momma, M., Kobayashi, H. & Mizuno, H. (2003). J. Biol. Chem.278, 20313–20318. [DOI] [PubMed]
- Garman, S. C. & Garboczi, D. N. (2004). J. Mol. Biol.337, 319–335. [DOI] [PubMed]
- Garman, S. C., Hannick, L., Zhu, A. & Garboczi, D. N. (2002). Structure, 10, 425–434. [DOI] [PubMed]
- Golubev, A., Nagem, R., Brandao Neto, J., Neustroev, K., Eneyskaya, E., Kulminskaya, A., Shabalin, K., Savel’ev, A. & Polikarpov, I. (2004). J. Mol. Biol.339, 413–422. [DOI] [PubMed]
- Laemmli, U. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- Lehle, L., Strahl, S. & Tanner, W. (2006). Angew. Chem. Int.45, 6802–6818. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Matthews, B. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Melcher, K., Sharma, B., Ding, W. V. & Nolden, M. (2000). Gene, 247, 53–61. [DOI] [PubMed]
- Murphy, R. & Power, R. (2002). J. Ind. Microbiol. Biotechnol.28, 97–102. [DOI] [PubMed]
- Sakuraba, H. et al. (2006). J. Hum. Genet.51, 341–352. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.